
Abstract
Background
Single-cell RNA sequencing (scRNA-seq) strives to capture cellular diversity with higher resolution than bulk RNA sequencing. Clustering analysis is critical to transcriptome research as it allows for further identification and discovery of new cell types. Unsupervised clustering cannot integrate prior knowledge where relevant information is widely available. Purely unsupervised clustering algorithms may not yield biologically interpretable clusters when confronted with the high dimensionality of scRNA-seq data and frequent dropout events, which makes identification of cell types more challenging.
Results
We propose scSemiAAE, a semi-supervised clustering model for scRNA sequence analysis using deep generative neural networks. Specifically, scSemiAAE carefully designs a ZINB adversarial autoencoder-based architecture that inherently integrates adversarial training and semi-supervised modules in the latent space. In a series of experiments on scRNA-seq datasets spanning thousands to tens of thousands of cells, scSemiAAE can significantly improve clustering performance compared to dozens of unsupervised and semi-supervised algorithms, promoting clustering and interpretability of downstream analyses.
Conclusion
scSemiAAE is a Python-based algorithm implemented on the VSCode platform that provides efficient visualization, clustering, and cell type assignment for scRNA-seq data. The tool is available from https://github.com/WHang98/scSemiAAE.
Similar content being viewed by others

Introduction
With the boom in sequencing techniques, single-cell transcriptome sequencing quantifies gene expression levels at the resolution of individual cells, providing new insights into the internal heterogeneity of cellular tissues [1]. Clustering is a key link in single-cell transcriptional profiling and plays an important role in revealing cell subtypes, dividing gene sequences, and inferring cell lineages. Traditional clustering methods are mainly divided into density-based clustering, neural network, ensemble learning, k-means, mixture model, graph-based clustering and hierarchical clustering [1]. Responding to the dimensional catastrophe [2] and the explosive growth of sample volumes caused by scRNA-seq [3,4,5], early clustering studies often combined PCA [6], t-SNE [7, 8], UMAP [9] and other dimensionality reduction methods to complete cell grouping and visualization, including pcaReduce [10], TooManyCells [11] and Seurat [12], etc. However, scRNA-seq data is often sparse and noisy [13] due to a complex combination of biological variability and technological reasons. Traditional clustering methods ignore the extreme sparsity of gene expression in single-cell transcriptome sequences, thus cannot achieve ideal clustering results with basic dimensionality reduction methods alone [3].
Relying on single-cell sequencing technology, researchers have access to large-scale sample data, which provides a unique development opportunity for the application of deep learning. scDeepCluster [14, 15] employs an autoencoder with the Zero-Inflated Negative Binomial (ZINB) [16] distribution to simultaneously reduce dimensionality and denoise the data, and then uses a deep embedding clustering algorithm to identify cell types for the data in the bottleneck layer. scGAE [17] provides a new perspective for exploiting the information between cells and genes by building K-Nearest Neighbor (KNN) [18] graphs, considering count matrices and adjacency matrices as the input to the autoencoder. However, the huge computational effort involved in erecting the adjacency matrix makes this approach stretch in applications with large-scale data. scDSC [19] combines Convolutional Neural Network (CNN) [20, 21] and an autoencoder in a self-supervised manner to further explore the fusion of spatial structure of cells and intergenic information. Variational Autoencoders (VAEs) [22] apply the Kullback-Leibler (KL) divergence penalty to impose a prior distribution (usually a Gaussian distribution) on the hidden code vectors of the autoencoder. scDHA [23] is a stacked Bayesian self-learning network based on VAEs that projects data into multiple low-dimensional spaces. Although VAEs perform well in generative models, the regularization based on the KL loss restricts the setting of the prior distribution. Generative Adversarial Networks (GANs) [24] firstly introduce adversarial training to directly shape the output distribution of the network through Generative Moment Matching Network (GMMN) [25], which allows for alternative choices for the prior distribution. scDEC [26] models the data based on a symmetric GAN structure, jointly optimizing latent feature representation learning and cell clustering. Recently, Adversarial Autoencoders (AAEs) [27] have been proposed, which combine the advantages of probabilistic autoencoders and generative adversarial networks to perform variational inference by matching the aggregated posteriors of the hidden code vectors of the autoencoder to an arbitrary prior distribution. It has been shown that this approach achieves better results in reconstructing samples and image classification [27, 28].
Many downstream analyses, such as differential expression, trajectory inference, etc., rely on the initial clustering results, which require the clustering results to be biologically interpretable. Due to the lack of support from prior information, unsupervised clustering sometimes fails to yield meaningful clusters consistent with prior knowledge. Consequently, the user often needs to repeatedly adjust the cluster parameters manually until a satisfactory cluster is found. We note that prior knowledge is widely available in many cases. A considerable number of cell type information have been published, such as Montoro [29], Puram [30]. Taking advantage of the prior information can avoid suboptimal or illogical clustering results to some extent.
Recently, some semi-supervised clustering algorithms have been proposed, such as scDCC [31], scSemiAE [32], and ItClust [33] etc. scDCC converts partial prior knowledge into pairwise constraints and adds them as additional terms in the loss to guide the deep learning model to better learn latent representations. However, this algorithm constructs soft pairwise constraints with some subjectivity and needs more prior information to define each cell one by one. scSemiAE predicts cell types through a classifier and transfers partial labels information to an autoencoder for fine tuning. It is not difficult to find that the performance of the classifier has a great influence on learning features, and the latent space has no regularization constraints, which may cause overfitting. ItClust performs supervised learning and cell classification on scRAN-seq data, exploiting cell type-specific gene expression information from the source data. However, the quality and quantity of the reference data can highly affect the training and clustering results of the target dataset.
Here, we propose a more flexible framework of semi-supervised clustering, scSemiAAE, which carefully designs a ZINB loss-based autoencoder architecture that inherently integrates adversarial training and semi-supervised modules in the latent space. The study indicates that we can guide the model to obtain a better latent representation by a small portion of label information, and then get more accurate clustering results. A series of experiments on spanning multiple datasets show that scSemiAAE outperforms published unsupervised and semi-supervised clustering tools of clustering accuracy and scalability.
Methods
Workflow of the scSemiAAE model
Dataset information
The proposed scSemiAAE method is evaluated on nine real scRNA-seq datasets. Table 1 provides an overview of the specific properties of these datasets. The 10X PBMC [34], Human kidney cells [35], Human liver [36], Tabula Muris [37] and Karagiannis [38] datasets are provided by the 10X Genomics scRNA-seq platform. For the Worm neuron cells [39] dataset, the author analyzes approximately 50,000 cells from L2 larval stage of Caenorhabditis elegans and identified cell types. The CITE-seq PBMC [40] dataset is divided into 15 clusters by cluster analysis and gene differential expression analysis. The Shekhar mouse retina cells [41] and Baron(human) [42] datasets are provided by the Drop-seq and inDrop platforms, respectively.
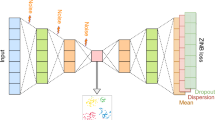
The scSemiAAE for scRNA-seq data analysis is mainly consists of three computational modules (Fig. 1). The first section is an autoencoder based on the ZINB model, which provides a low-dimensional representation. The second module constructs cross-entropy [34] that introduces label information into the latent space for semi-supervised learning. The third part builds a discriminator network to distinguish between “real” and “fake”samples. Moreover, we use the encoder of the autoencoder as a generator network for making "fake" samples. The details of each step are described as follows.

The illustration of scSemiAAE model. A The scRNA-seq count matrix X is preprocessed through gene filtering, screening of highly variable genes, and normalization. Next, it is divided into m_ and m depending on whether it contains true labels. B The encoder receives m_ and m to generate the corresponding latent variables z_ and z, respectively. C The SoftMax layer transforms the latent vector z_ into the pseudo-label c, which is then combined with the partial true label y_ to create a cross-entropy loss. D The decoder reconstructs the potential representation z with a zero-inflated negative binomial loss constraint. E Simultaneously, the latent feature z is fed to the discriminator for adversarial training, comprising the discriminator loss. F After completing training process, all the latent z and labels c are concatenated, and the final clustering results are given by a Gaussian mixture model
Data preprocessing
Raw single cell RNA sequence count data \(X\), where rows indicate cells and columns point to genes, is preprocessed by the Python package SCANPY [43]. Firstly, genes that are not counted in the cell are filtered. Secondly, the size factor is calculated, and the read counts are normalized. We denote the total number of RNA molecules per cell as \(n_{i}\), and its corresponding median as \({\text{med}}\left( {n_{i} } \right)\), thus the size factor of the cell \(i\) is \({\text{sf}}_{i} = n_{i} /{\text{med}}(n_{i} )\). Let the \({\text{j}}_{th}\) gene expression value of the \(i_{th}\) cell of the input matrix \(X\) be \(x_{ij}\) and the normalized expression be \(x^{^{\prime}}_{ij} = x_{ij} /{\text{sf}}_{i}\). Finally, to prevent domination by highly expressed genes and features, we log-transform the value \(l_{ij} = \log_{2} (x^{^{\prime}}_{ij} + 1)\), and scale the counts \(m_{ij} = (l_{ij} - {\text{mean}}_{j} )/{\text{std}}_{j}\). Here, \({\text{mean}}_{j}\) and \({\text{std}}_{j}\) are the mean and standard deviation of the logarithmic expression values of \({\text{j}}_{th}\) gene in all cells, respectively. The processed matrix \(M\), consisting of elements \(m_{ij}\), is fed to subsequent cluster analysis.
Autoencoder based on ZINB model
In this paper, we apply a denoising autoencoder based on ZINB loss to capture the features of single cell RNA sequences. We first corrupt the processed matrix with random Gaussian noise, then map the read counted input to the embedding space for clustering.
Typically, the matrix \(M\) is corrupted by \(C(M) = M + \lambda \delta\), \(\delta\) denotes random Gaussian noise, \(\lambda\) corresponds to its coefficient. Each layer of the encoder \(E\) function can be expressed as \(f_{E} (m) = {\text{mW}}_{E} + b_{E}\), and each layer of the decoder \(D\) function can be represented as \(f_{D} \left( z \right) = zW_{D} + b_{D}\), where \(W\) is the weight matrix and \(b\) is the bias vector. The encoder represents the data in a low-dimensional space and thus gets the latent layer \(z\), while the decoder tries to reconstruct from the compressed data. Theoretically, optimizing this procedure can lead to a condensed version of the primitive high-dimensional form. Unlike conventional autoencoders, the decoder does not perform reconstruction and only gives the ZINB distribution parameters \(P\). In this regard, we attach three separate fully connected layer on the last hidden layer \(f_{D^{\prime}} \left( {z^{\prime}} \right)\) of the decoder.
where \(sf\) refers to the size factor, \(D\) indicates the decoder function, \(sigmod\) means the activation function, \(W_{M}\),\(W_{\pi }\) and \(W_{\theta }\) represent the parameters weights to be learned in the last three fully connected layers, respectively.
A negative binomial distribution, with mean \(u\), the dispersion \(\theta\), and the additional coefficient of the zero-probability point quality weight \(\pi\) (probability of dropout events), parameterizes the ZINB loss. Notably, the distribution is calculated using the original gene count matrix \(X\).
Semi-supervised module
Cross-entropy [44] originates from Shannon's information theory. It is often represented as the difference between the predicted probability distribution and the true probability distribution in model learning. The smaller the value of cross-entropy, the more accurate the prediction of the model. Considering the proportion of labeled samples, the clustering approaches can be chosen flexibly. When only a small portion of the label information is available, the labeled samples \(m_{ - }\) are first separated, corrupted, and then fed into the encoder to produce the corresponding latent variables \(z_{ - }\). Subsequently, the partial latent features are passed through the SoftMax layer to generate the pseudo-labels \(c\), which construct the cross-entropy loss with the true labels \(y_{\_}\). Here, we adopt 20%-25% of the true label information to guide the model learning parameters.
The number of label information used for semi-supervision \(S\), the true cell labels \(y_{\_}\), and the pseudo-labels \(c\), constitute the cross-entropy loss. Note that the true cell labels \(y_{\_}\) are not cell types, but simply represent information about which class a cell belongs to.
Discriminator and Generator Networks
Generative adversarial network (GAN) usually iteratively training the generative model \(g\varphi \left( {t|s} \right)\) and the discriminative model \(d\chi \left( t \right)\) to realize the adversarial training [9]. We feed samples from the generator (“fake” samples) and the target distribution (“true” samples) into a discriminative model for training to correctly predict whether a sample is “true” or “fake”. The generative model takes \(s\) as input, extracted from the selected prior distribution \(p\left( s \right)\). To fool the discriminative model [24], it continues training until the generated samples \(t\) are indistinguishable from the target samples \(t\). The following minimax function [9] can accomplish the target:
It turns out that, for ideal discriminative models, optimizing the generator equal to minimizing the Jensen-Shannon divergence between the generative distribution and the target distribution [9]. Overall, it is rational to presume the discriminator rapidly reaches optimal performance during training [9]. Further, we could bypass the complicated Jensen-Shannon divergence calculation and thus learn the distribution easily.
To prevent the model from overfitting, we impose regularization constraints on the latent space by adversarial training. A discriminative network is trained to divide potential samples from \(p\left( z \right)\) and \(q_{\varphi } \left( {z|m} \right)\).The latter is both a probabilistic encoder in the autoencoder and a generative model in the adversarial framework. The loss for training the discriminator \(d_{\chi } \left( z \right)\) is:
where \(z_{{true_{i} }} = 0:n - 1 \sim p\left( z \right)\), \(z_{{fake_{j} }} = n:2n - 1 \sim q_{\varphi } \left( {z|m} \right)\), \(d\) represents the discriminator, and n is the size of the training batch, \(p(z) = N(u,\Sigma )\).
We consider the encoder of the autoencoder as the generator and the latent vectors \(z\) as the generated samples. A set of vectors with the same dimension are drawn from a multivariate Gaussian distribution as the true samples, and the generator loss is constructed as follows:
where \(z_{{fake_{i} }} = 0:n - 1 \sim q_{\varphi } \left( {z|m} \right)\), \(n\) refers to the training batch size. As the two losses continue to optimize, the distribution of generated samples is constantly moving to the target distribution of the generative model. In this case, the discriminator is maximally puzzled and cannot differentiate between "true" and "fake" samples.
We summarize the training procedure of the algorithm in Table 2 to ensure the completeness and readability of the algorithm.
Metric of performance evaluation
The paper compares different clustering methods based on multiple metrics such as Adjusted Rand Index (ARI) [45], Normalized Mutual Information (NMI) [46] and Accuracy (ACC) [14].
The Rand index [47] measures the agreement between two cluster assignments, while the ARI corrects for the lack of a constant value when cluster assignments are randomly chosen. The ARI values are in the range [−1, 1]. A value of one indicates perfect grouping. A value of zero shows a random assignment of samples to groups, and negative values point to wrong cluster assignments. We define the following four quantities (1) p : the count of target pairs in the same sets in \(P\) but varied groups in \(Q\) (2) q : The count of target pairs in the same sets in \(Q\) but varied groups in \(P\)(3) m : the count of target pairs in the same sets in both \(P\) and \(Q\) (4) n : the count of target pairs in varied groups in both \(P\) and \(Q\).
Assume \(P\) and \(Q\) are true, and predict label assignments given \(N\) data points with \(U_{P}\) and \(U_{Q}\) clusters, respectively. Given two cluster assignments \(P\) and \(Q\), with \(U_{P}\) and \(U_{Q}\) clusters on \(N\) data points respectively, the NMI is defined as the mutual information between \(P\) and \(Q\), divided by the entropy of the \(P\) and \(Q\) clusters. Here, \(C_{n}^{2} \,\) is the number of combinations of two elements taken from \(n\) elements. The combination is unordered, and it is calculated by \(C_{n}^{2} = n\left( {n - 1} \right)/2\).
ACC denotes the best match between the predicted cluster and the true cluster. Let \(\hat{k}_{i}\)and \(k_{i}\) be the prediction of the clustering methods and the true label of the data point, ACC is expressed as follows:
Implementation
scSemiAAE is implemented in Python 3 (version 3.8.13) using PyTorch [48, 49] (version 1.11.0). In the ZINB model-based autoencoder, the size of the hidden layer is set to (256, 128, 64, 64, 128), where the size of the bottleneck layer is 64. Each layer of the autoencoder adds a dropout of 0.2 and a standard deviation of Gaussian random noise is 1.0. The number of neurons of the discriminator is set to (64, 128, 256, 1).
In the data pre-training stage, the learning rate is set to 0.001, the number of training is 100, the batch size is 128, and the optimizer is Adam [50]. After getting the initialized weights, in the training phase, the algorithm regards the encoder of the autoencoder as the generator, the optimizer selects Adadelta [51], and the parameters are set to rho = 0.95 , lr = 0.01. The discriminator offers Adam as the optimizer, and its parameters are set as the initial learning rate β1 = 0.9, β2 = 0.999, lr = 0.0001. The batch size is set to 128 and the number of training epochs is 100. To coordinate the learning capabilities of the generator and discriminator, we set the discriminator to learn every 5 epochs of training. Cell label usage is set to 20% or 25% for different datasets (Fig. 3C). In addition, for Gaussian mixture clustering to get clustering labels, we give validation results based on experiments (Additional file 1: Figure S2). All experiments are performed on RTX 3060 (16G). The acquisition and implementation of baseline methods and other parameters sensitivity analyses are provided in the Additional file 1.
Results
Visualization and accuracy evaluation of different methods
To evaluate the performance of scSemiAAE to distinguish between different cell subpopulations and identify cell types, the research tests on the real scRNA-seq datasets with diverse cell types and numbers. The clustering performances of these methods are evaluated on the basis of (1) whether cell subpopulations could be clearly distinct in the latent space, and (2) whether the clustering results can accurately infer the true cell types. To address the first issue, we apply t-Distributed Stochastic Neighbor Embedding (t-SNE) to project the bottleneck layer into a 2D space to visualize the latent features learned by different methods for scRNA-seq data. To assess the clustering results of different strategies for the second problem, this paper adopts three common metrics, Normalized Mutual Information (NMI), Adjusted Rand Index (ARI), and Accuracy (ACC), based on true cell labels.
As shown in Fig. 2, we select five real datasets of varying complexity for visualization. It is not hard to find that scSemiAAE can achieve ideal separation and clear boundaries for datasets with 8, 10 and 15 different cell subtypes. In differentiation, other methods tend to mix distinct cell subtypes. On the Human kidney dataset, scDHA can also distinguish different types of clusters sparsely compared to scSemiAAE. However, the identified clusters are scattered and incomplete. Cells of various types are mixed in scSemiAE, and it is hard to get satisfactory boundaries between clusters. For example, orange cells and red cells are connected, and the clusters are not dispersed enough. For scDSC, red indicates that cells are distributed over the entire plot on Human liver dataset. We have the identical observation on the 10X_PBMC, Worm neuron cells, and CITE_PBMC datasets. Compared with other clustering methods, scSemiAAE can identify all different cell types, clarify the boundaries between clusters, and ensure the dispersion between clusters.

Latent representation visualization. The images base on embedded representations of the 10X_PBMC, Human kidney cells, Worm neuron cells, Human liver and CITE_PBMC datasets. Each dot indicates a cell, and the different colors of the dots point to the predicted labels
In terms of consistency of the clustering results with the true labels, we compare scSemiAAE with ten baseline methods, including scDeepCluster [14], scDEC [24], scDSC [17], scDHA [21], SC3 [52], scGAE [16], scDCC [29], Itclust [33],scAL [46], scSemiAE [30]. Notice that the first six ones are unsupervised methods, and the remaining ones are semi-supervised clustering algorithms. Figure 3A and B show the two partitioning techniques, respectively. Apparently, our model significantly surpasses current deep clustering methods on the 10X_PBMC, Human kidney cells, Worm neuron cells, Shekhar mouse retina raw cells, and CITE_PBMC datasets, and slightly better on the Human Liver dataset. For the Worm neuron dataset, scSemiAAE significantly raises ACC by 6.12%, NMI by 7.08%, and ARI by 11.20% compared to the suboptimal metrics of all algorithms. On the 10X_PBMC dataset, scSemiAAE greatly improves by 7.99% on ACC, 2.03% on NMI, and 5.76% on ARI in contrast to the suboptimal metrics of all approaches. The details of the datasets, complete cluster images and indicator comparison are shown in the Additional file 1

Benchmarking results on real scRNA-seq datasets. Clustering performances of scDeepCluster, scDSC, scDEC, scDHA, SC3, scGAE, scDCC, scAL, Itclust, scSemiAE and scSemiAAE, measured by ACC, NMI and ARI. The first six ones are unsupervised methods, and the remaining ones are semi-supervised clustering algorithms. A Comparison with semi-supervised clustering approaches on three datasets with the top 2000 highly scattered genes. B The results of unsupervised clustering algorithms. C scSemiAAE uses different proportions of labels on seven real datasets, measured by NMI
Robustness of scSemiAAE on highly dispersed genes
Most single cell analysis pipelines apply gene filtering strategies to select low variance genes and only keep high dispersion genes (eg. SCANPY). Selecting genes that are highly scattered can enlarge differences between cells but lose critical information between cell populations. To assess the robustness of scSemiAAE to highly scattered genes, we conduct experiments on the top 2000 highly scattered genes in three datasets (Fig. 3A), and then reveal the performance of scSemiAAE and the baseline methods. As the diagram displays, scSemiAAE consistently exceeds other semi-supervised clustering models using full datasets.
Scalability of scSemiAAE for large-scale datasets
The large-scale sample size is one of the main characteristics of single-cell sequencing technology applications, and whether it can handle large-scale data is an important consideration for current clustering algorithms. The experiments on three larger datasets - Shekhar mouse retina raw data, Tabula Muris, and Karagiannis - demonstrate that scSemiAAE is effective for clustering large-scale single-cell transcriptome data. For example, on the Shekhar dataset with 27,466 samples, our algorithm achieved an Adjusted Rand Index (ARI) value of 0.8 or higher compared to the "reference" labels, indicating high consistency (Fig. 3B). Similarly, on the Tabula Muris dataset with 54,439 cells and 40 cell types across 20 organs and tissues, the clustering NMI metric by the method is 0.7456; on the Karagiannis dataset with 72,914 cells, the ACC and NMI metrics also reached above 0.7 (Fig.4B). Overall, these results demonstrate that scSemiAAE performs well on large datasets. In comparison, neither scGAE nor SC3 can handle datasets with more than 8000 samples under the same memory conditions.

Model performance analysis of scSemiAAE. A Comparing the scalability of different algorithms on the real datasets by ARI and NMI metrics. B Clustering effects based on large-scale datasets. C Differential expression analysis bases on Baron (human) data
Furthermore, we plot boxplots of ARI and NMI metrics for 11 different clustering algorithms on six real datasets to compare the scalability of models. scSemiAAE demonstrates desirable agreement with reference cell labels on different scRNA datasets with sample sizes ranging from thousands to tens of thousands. The highest ARI value exceeds 0.8, and the lowest ARI value is also above 0.6 (Fig. 4A left panel). Relatively, the box lengths of other baseline methods are obviously longer, and their average clustering accuracy is much lower than our algorithm. The NMI boxplot (Fig. 4A right panel) also displays the same characteristics, with the shortest box length and the highest average clustering accuracy.
Maker genes identification
Gene expression matrix and cluster labels can be used to identify the differentially expressed genes (DEGs) in each cluster. Here, we choose the Baron (human) dataset to extract its gene markers and analyze the relationship between cell groups and cells. For the dataset, the author collects the transcriptomes of beyond 12,000 single pancreatic cells from two mouse strains and four human donors. Cell clusters could correspond to previously identified cell types, including four types of immune cells, exocrine cell types, activated and quiescent stellate cells , rare epsilon cells, Schwann cells and vascular cells [42]. Figure 4C shows the first 2 marker genes of each cluster. As can be seen from the chart, most of the differentially expressed genes selected according to the scSemiAAE cluster labels are involved in significant expression differences between clusters.
Discussion
Single-cell transcriptome clustering can identify disease-relevant cell types and subpopulations from heterogeneous samples, contributing to further unravel the physiological mechanisms of cells. Among current clustering tools, unsupervised methods are still dominant. However, when the final number of cell classes is not known, it is possible that unsupervised algorithms fail to produce biologically consistent cell clusters. This requires the user to manually iterate the clustering parameters to achieve satisfactory performance. Not surprisingly, for some datasets, we do not always find the right parameters to adjust the results [3, 53].
Therefore, it is particularly important to incorporate prior knowledge into clustering models. Notably, the priori information here can be partial cell types, cell labels, number of classes, marker genes, protein restrictions, etc. In addition, multi-omics sequencing data can equally serve as the prerequisite, such as CITE-seq [54] (simultaneous analysis of single-cell transcriptome and surface proteins) and single-cell ATAC-seq [55]. Researchers choose to introduce different background knowledge depending on the experimental purpose and algorithm design. In this paper, the proposed scSemiAAE employs partially real cell labels as the priori. We give the details of the data and the source of the label information and place these in the Additional file 1.
Furthermore, this paper presents several directions for improving scSemiAAE. First, we can try different adversarial losses when training the discriminator. The conditional adversarial loss (CGAN) [56] concatenates label information and latent variables, and then send them to the discriminator for training. Considering that this loss can make full use of the remaining pseudo-labels, we believe that Gaussian mixture clustering can be removed, and the pseudo-labels can be used as the final clustering results. This simplifies the model on the one hand and integrates latent features generation and clustering on the other hand. Second, if genes and regulatory elements (REs) were added to the scSemiAAE, it might help to further improve the clustering performance. Third, some studies have developed packages for batch effect correction due to the limitations of sequencing technologies, such as SCALEX [57] and Harmony [58]. It makes sense to explore how this data integration analysis can be incorporated into the scSemiAAE model.
With scSemiAAE, researchers can perform scRNA-seq analysis on cell types or tissues of interest, further revealing the biological meaning behind the features. We hope that scSemiAAE will help discover new cell types and contribute to the understanding of different cell populations.
Conclusion
In this study, we propose scSemiAAE that adopts a deep generative model to accurately characterize cellular subpopulations for scRNA-seq data. scSemiAAE inherently integrates adversarial training and semi-supervised clustering by carefully designing a ZINB adversarial autoencoder-based architecture. It is a strong and effective tool for scRNA-seq data, including potential layers visualization, cell clustering, differential expression analysis.
A series of experiments show that scSemiAAE could acquire better performance compared to current clustering techniques, since it can capture ideal latent characteristics to promote cell type identification. The studies also prove that scSemiAAE can handle large-scale datasets and shows robustness and noise resistance on genes with high dispersion. In addition, scSemiAAE can well identify differentially expressed genes, which helps to further explain the biological significance of cell type assignment.
Availability of data and materials
The datasets in the paper are available at https://github.com/WHang98/scSemiAAE.
Abbreviations
- scRNA-seq:
-
Single-cell RNA sequencing
- ZINB:
-
Zero-inflated negative binomial
- PCA:
-
Principal component analysis
- t-SNE:
-
T-distributed Stochastic Neighbor Embedding
- UMAP:
-
Uniform manifold approximation and projection
- KNN:
-
K-nearest neighbor
- CNN:
-
Convolutional neural network
- KL:
-
Kullback-Leibler
- VAEs:
-
Variational autoencoders
- GANs:
-
Generative adversarial networks
- GMMN:
-
Generative moment matching network
- AAEs:
-
Adversarial autoencoders
- GMM:
-
Gaussian mixture
- ARI:
-
Adjusted rand index
- NMI:
-
Normalized mutual information
- ACC:
-
Accuracy
- DEGs:
-
Differentially expressed genes
- CGAN:
-
Conditional adversarial network
- REs:
-
Regulatory elements
References
Petegrosso R, Li Z, Kuang R. Machine learning and statistical methods for clustering single-cell RNA-sequencing data. Brief Bioinform. 2020;21(4):1209–23.
Stegle O, Teichmann SA, Marioni JC. Computational and analytical challenges in single-cell transcriptomics. Nat Rev Genet. 2015;16(3):133–45.
Kiselev VY, Andrews TS, Hemberg M. Challenges in unsupervised clustering of single-cell RNA-seq data. Nat Rev Genet. 2019;20(5):273–82.
Kolodziejczyk AA, Kim JK, Svensson V, Marioni JC, Teichmann SA. The technology and biology of single-cell RNA sequencing. Mol Cell. 2015;58(4):610–20.
Shapiro E, Biezuner T, Linnarsson S. Single-cell sequencing-based technologies will revolutionize whole-organism science. Nat Rev Genet. 2013;14(9):618–30.
Yang J, Zhang D, Frangi AF. Yang J-y: Two-dimensional PCA: a new approach to appearance-based face representation and recognition. IEEE Trans Pattern Anal Mach Intell. 2004;26(1):131–7.
Van Der Maaten L: Learning a parametric embedding by preserving local structure. In: Artificial intelligence and statistics: 2009. PMLR: 384-391.
Van der Maaten L, Hinton G: Visualizing data using t-SNE. J Mach Learn Res 2008, 9(11).
McInnes L, Healy J, Melville J: Umap: Uniform manifold approximation and projection for dimension reduction. arXiv preprint arXiv:180203426 2018.
Yau C. pcaReduce: hierarchical clustering of single cell transcriptional profiles. BMC Bioinform. 2016;17(1):1–11.
Schwartz GW, Zhou Y, Petrovic J, Fasolino M, Xu L, Shaffer SM, Pear WS, Vahedi G, Faryabi RB. TooManyCells identifies and visualizes relationships of single-cell clades. Nat Methods. 2020;17(4):405–13.
Satija R, Farrell JA, Gennert D, Schier AF, Regev A. Spatial reconstruction of single-cell gene expression data. Nat Biotechnol. 2015;33(5):495–502.
Eling N, Morgan MD, Marioni JC. Challenges in measuring and understanding biological noise. Nat Rev Genet. 2019;20(9):536–48.
Xie J, Girshick R, Farhadi A: Unsupervised deep embedding for clustering analysis. In: International conference on machine learning: 2016. PMLR: 478-487.
Guo X, Gao L, Liu X, Yin J: Improved deep embedded clustering with local structure preservation. In: Ijcai: 2017. 1753-1759.
Eraslan G, Simon LM, Mircea M, Mueller NS, Theis FJ. Single-cell RNA-seq denoising using a deep count autoencoder. Nat Commun. 2019;10(1):1–14.
Luo Z, Xu C, Zhang Z, Jin W: scGAE: topology-preserving dimensionality reduction for single-cell RNA-seq data using graph autoencoder. bioRxiv 2021.
Mucherino A, Papajorgji PJ, Pardalos PM: K-nearest neighbor classification. In: Data mining in agriculture. Springer; 2009: 83-106.
Gan Y, Huang X, Zou G, Zhou S, Guan J. Deep structural clustering for single-cell RNA-seq data jointly through autoencoder and graph neural network. Briefings Bioinform. 2022;23(2):bbac018.
LeCun Y, Bottou L, Bengio Y, Haffner P. Gradient-based learning applied to document recognition. Proc IEEE. 1998;86(11):2278–324.
Krizhevsky A, Sutskever I, Hinton GE. Imagenet classification with deep convolutional neural networks. Commun ACM. 2017;60(6):84–90.
Pu Y, Gan Z, Henao R, Yuan X, Li C, Stevens A, Carin L: Variational autoencoder for deep learning of images, labels and captions. Adv Neural Inf Process Syst 2016, 29.
Tran D, Nguyen H, Tran B, La Vecchia C, Luu HN, Nguyen T. Fast and precise single-cell data analysis using a hierarchical autoencoder. Nat Commun. 2021;12(1):1–10.
Goodfellow I, Pouget-Abadie J, Mirza M, Xu B, Warde-Farley D, Ozair S, Courville A, Bengio Y. Generative adversarial networks. Commun ACM. 2020;63(11):139–44.
Li Y, Swersky K, Zemel R: Generative moment matching networks. In: International conference on machine learning: 2015. PMLR: 1718-1727.
Liu Q, Chen S, Jiang R, Wong WH. Simultaneous deep generative modelling and clustering of single-cell genomic data. Nature Mach Intell. 2021;3(6):536–44.
Makhzani A, Shlens J, Jaitly N, Goodfellow I, Frey B: Adversarial autoencoders. arXiv preprint arXiv:151105644 2015.
Creswell A, Bharath AA. Denoising adversarial autoencoders. IEEE Trans Neural Netw Learn Syst. 2018;30(4):968–84.
Montoro DT, Haber AL, Biton M, Vinarsky V, Lin B, Birket SE, Yuan F, Chen S, Leung HM, Villoria J. A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature. 2018;560(7718):319–24.
Puram SV, Tirosh I, Parikh AS, Patel AP, Yizhak K, Gillespie S, Rodman C, Luo CL, Mroz EA, Emerick KS. Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell. 2017;171(7):1611-1624. e1624.
Tian T, Zhang J, Lin X, Wei Z, Hakonarson H. Model-based deep embedding for constrained clustering analysis of single cell RNA-seq data. Nat Commun. 2021;12(1):1–12.
Dong J, Zhang Y, Wang F. scSemiAE: a deep model with semi-supervised learning for single-cell transcriptomics. BMC Bioinformatics. 2022;23(1):1–13.
Hu J, Li X, Hu G, Lyu Y, Susztak K, Li M. Iterative transfer learning with neural network for clustering and cell type classification in single-cell RNA-seq analysis. Nature Mach Intell. 2020;2(10):607–18.
Zheng GX, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J. Massively parallel digital transcriptional profiling of single cells. Nat Commun. 2017;8(1):1–12.
Young MD, Mitchell TJ, Vieira Braga FA, Tran MG, Stewart BJ, Ferdinand JR, Collord G, Botting RA, Popescu D-M, Loudon KW. Single-cell transcriptomes from human kidneys reveal the cellular identity of renal tumors. Science. 2018;361(6402):594–9.
MacParland SA, Liu JC, Ma X-Z, Innes BT, Bartczak AM, Gage BK, Manuel J, Khuu N, Echeverri J, Linares I. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat Commun. 2018;9(1):1–21.
Schaum N, Karkanias J, Neff NF, May AP, Quake SR, Wyss-Coray T, Darmanis S, Batson J, Botvinnik O, Chen MB. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris: the Tabula Muris consortium. Nature. 2018;562(7727):367.
Karagiannis TT, Cleary JP Jr, Gok B, Henderson AJ, Martin NG, Yajima M, Nelson EC, Cheng CS. Single cell transcriptomics reveals opioid usage evokes widespread suppression of antiviral gene program. Nat Commun. 2020;11(1):2611.
Cao J, Packer JS, Ramani V, Cusanovich DA, Huynh C, Daza R, Qiu X, Lee C, Furlan SN, Steemers FJ. Comprehensive single-cell transcriptional profiling of a multicellular organism. Science. 2017;357(6352):661–7.
Mimitou EP, Cheng A, Montalbano A, Hao S, Stoeckius M, Legut M, Roush T, Herrera A, Papalexi E, Ouyang Z. Multiplexed detection of proteins, transcriptomes, clonotypes and CRISPR perturbations in single cells. Nat Methods. 2019;16(5):409–12.
Shekhar K, Lapan SW, Whitney IE, Tran NM, Macosko EZ, Kowalczyk M, Adiconis X, Levin JZ, Nemesh J, Goldman M. Comprehensive classification of retinal bipolar neurons by single-cell transcriptomics. Cell. 2016;166(5):1308-1323. e1330.
Baron M, Veres A, Wolock SL, Faust AL, Gaujoux R, Vetere A, Ryu JH, Wagner BK, Shen-Orr SS, Klein AM. A single-cell transcriptomic map of the human and mouse pancreas reveals inter-and intra-cell population structure. Cell Syst. 2016;3(4):346-360 e344.
Wolf FA, Angerer P, Theis FJ. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018;19(1):1–5.
De Boer P-T, Kroese DP, Mannor S, Rubinstein RY. A tutorial on the cross-entropy method. Ann Oper Res. 2005;134(1):19–67.
Hubert L, Arabie P. Comparing partitions. J Classif. 1985;2(1):193–218.
Strehl A, Ghosh J: Cluster ensembles---a knowledge reuse framework for combining multiple partitions. Journal of machine learning research 2002, 3(Dec):583-617.
Rand WM. Objective criteria for the evaluation of clustering methods. J Am Stat Assoc. 1971;66(336):846–50.
Paszke A, Gross S, Chintala S, Chanan G, Yang E, DeVito Z, Lin Z, Desmaison A, Antiga L, Lerer A: Automatic differentiation in pytorch. 2017.
Paszke A, Gross S, Massa F, Lerer A, Bradbury J, Chanan G, Killeen T, Lin Z, Gimelshein N, Antiga L: Pytorch: an imperative style, high-performance deep learning library. Adv Neural Inf Process Syst 2019, 32.
Loshchilov I, Hutter F: Decoupled weight decay regularization. arXiv preprint https://arxiv.org/abs/1711.05101 2017.
Zeiler MD: Adadelta: an adaptive learning rate method. arXiv preprint https://arxiv.org/abs/1212.5701 2012.
Kiselev VY, Kirschner K, Schaub MT, Andrews T, Yiu A, Chandra T, Natarajan KN, Reik W, Barahona M, Green AR. SC3: consensus clustering of single-cell RNA-seq data. Nat Methods. 2017;14(5):483–6.
Xing E, Jordan M, Russell SJ, Ng A: Distance metric learning with application to clustering with side-information. Adv Neural Inf Process Syst 2002, 15.
Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H, Satija R, Smibert P. Simultaneous epitope and transcriptome measurement in single cells. Nat Methods. 2017;14(9):865–8.
Cao J, Cusanovich DA, Ramani V, Aghamirzaie D, Pliner HA, Hill AJ, Daza RM, McFaline-Figueroa JL, Packer JS, Christiansen L. Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science. 2018;361(6409):1380–5.
Mirza M, Osindero S: Conditional generative adversarial nets. https://arxiv.org/abs/1411.1784, 2014.
Xiong L, Tian K, Li Y, Ning W, Gao X, Zhang QC. Online single-cell data integration through projecting heterogeneous datasets into a common cell-embedding space. Nat Commun. 2022;13(1):1–17.
Korsunsky F, Fan J, Slowikowski K, Zhang F, Wei K, Baglaenko Y, Brenner M, Loh P, Raychaudhuri S: Fast, sensitive, and flexible integration of single cell data with Harmony. bioRxiv, 461954. 2018.
Acknowledgements
This work is supported by the open fund of Information Materials and Intelligent Sensing Laboratory of Anhui Province (Grant No. IMIS202105), the Xinjiang Autonomous Region University Research Program (No. XJEDU2019Y002), the National Natural Science Foundation of China (No. U19A2064, 61873001).
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
ZW developed the model and wrote the manuscript, Dr. HW guided the model construction, and Associate Professor JZ and Professor CZ gave revisions to the manuscript. All authors unanimously approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
The details of the real datasets, implementation of baseline methods and additional tables and plots.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, Z., Wang, H., Zhao, J. et al. scSemiAAE: a semi-supervised clustering model for single-cell RNA-seq data. BMC Bioinformatics 24, 217 (2023). https://doi.org/10.1186/s12859-023-05339-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12859-023-05339-4