
Abstract
Background
Single Nucleotide Variants (SNVs), including somatic point mutations and Single Nucleotide Polymorphisms (SNPs), in noncoding cis-regulatory elements (CREs) can affect gene regulation and lead to disease development. Several approaches have been developed to identify highly mutated regions, but these do not take into account the specific genomic context, and thus likelihood of mutation, of CREs.
Results
Here, we present SMuRF (Significantly Mutated Region Finder), a user-friendly command-line tool to identify these significantly mutated regions from user-defined genomic intervals and SNVs. We demonstrate this using publicly available datasets in which SMuRF identifies 72 significantly mutated CREs in liver cancer, including known mutated gene promoters as well as previously unreported regions.
Conclusions
SMuRF is a helpful tool to allow the simple identification of significantly mutated regulatory elements. It is open-source and freely available on GitHub (https://github.com/LupienLab/SMURF).
Similar content being viewed by others


Background
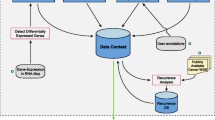
With the advent of next-generation sequencing technologies, a growing catalogue of genome-wide datasets has become available. This includes whole-genome sequencing to detect single nucleotide variants (SNVs) in diseased tissue (eg: TCGA Research Network: http://cancergenome.nih.gov/) as well as maps of histone variants and chromatin accessibility [1]. Using these datasets, numerous cis-regulatory elements (CREs) have been identified as recurrently mutated in cancer and other diseases. A notable example is the TERT promoter in glioma, melanoma, medulloblastoma, hepatocellular carcinoma, lung adenocarcinoma, thyroid and bladder cancers [2]. The mutations in this promoter create new transcription factor binding sites [3, 4], leading to increased TERT expression and ultimately immortalization and genomic instability [5]. Enhancers and anchors of chromatin interaction can also display recurrent mutation, such as the PAX5 enhancer in chronic lymphocytic leukemia [6, 7] and CTCF binding sites in colorectal cancer [8].
Others have previously developed methods to identify important clusters of somatic point mutations based on proximity [9] or an enrichment compared to the local background [10]. However, the mutation rate of a CRE is impacted by its chromatin accessibility and the binding of transcription factors, as demonstrated by a lower rate of mutation in open compared to closed chromatin [11]. Therefore, recurrently mutated CREs should be identified against a background of other regulatory elements with a matched chromatin accessibility in the same cell or tissue type. To achieve this, SMuRF receives a user-defined set of regions of interest as the input rather than relying on a proximity clustering of SNVs and provides a user-friendly tool to identify, filter, and annotate significantly mutated genomic regions.
Implementation
SMuRF consists of two main steps. The first filters, counts, annotates, and intersects the list of SNVs with the set of genomic coordinates, using a custom Bash script and the BEDTools suite [12]. The second consists in running a binomial test in R followed by a mutation rate filter to determine which genomic intervals are significantly enriched in SNVs and producing output figures as well as files for downstream analyses.
Input processing
The SNVs in BED or vcf format, are optionally filtered for known SNPs. This will remove either all known SNPs or only those with a minor allele frequency above 1% to preserve potentially interesting acquired SNVs that also occur as extremely rare polymorphisms in the population.
Subsequently, the input genomic regions are annotated as either gene promoter regions or as distal regulatory elements. This is done by overlapping those genomic intervals with a catalogue of gene promoters, derived from Gencode transcription start site annotations [13].
Finally, the input SNVs and genomic intervals are intersected to map all SNVs to unique genomic intervals, and the resulting data structure forms the starting point of the statistical analysis for mutation enrichment.
All of the above filtering and annotating can be achieved with data from any genome for which the required annotation files are available. Those for human builds hg19 and hg38 are supplied with the tool for convenience.
Identifying significantly mutated regions
The binomial test used by SMuRF to determine whether a given genomic region is significantly enriched for mutations requires an expected mutation rate. Depending on the sample cohort, the user can choose how this mutation rate is calculated. For each sample, the average number of mutations per base pair in input regions is calculated first. The “allsamples” option uses the average of those individual mutation rates across the entire sample cohort. However, if a subset of samples is more or less mutated than the rest, this could lead to biased results when a particular region contains mutations from that subset. For example, if a subset of samples is hypermutated relative to the rest of the cohort, this would artificially raise the background mutation rate, in effect reducing the number of significantly mutated elements identified. In these cases, the “regionsamples” option can be used, and the expected mutation rate when testing a particular region will be the average of the mutation rates for the individual samples mutated within that region only.
In both cases, the resulting p-value is then adjusted for multiple testing and the final set of regions is further filtered to include only those that pass a mutation rate threshold. This threshold is defined for each cohort by ranking the mutation rates for each region and identifying the inflection point, as previously described [14].
A number of output files are generated and these are detailed within the manual; they include a list of genes whose promoters are significantly mutated for use in gene ontology analyses, as well as a bed-formatted list of mutated regions annotated as distal regulatory elements to allow the user to associate them to target genes through GREAT [15] or C3D [16]. The main output figure is a scatter plot of -log10(q-value) against the number of unique samples mutated in the region, and color-coded to distinguish gene promoters from distal regulatory elements.
Results and discussion
To illustrate the above steps, we used publicly available acquired SNVs from 88 liver cancer samples [17] and chromatin accessibility data from HepG2 [1] that provides a reference set for CREs. The total number of SNVs per sample used in the analysis after filtering ranged from 1344 to 25,121 (Fig. 1a), with an average of 1.2% falling within one of the 278,135 CREs (Fig. 1b) as identified in HepG2. While the input SNV numbers covered a wide range, no subset of patients was abnormally hyper or hypomutated, so we selected the “allsamples” mode to calculate the background mutation rate for each CRE. In total, 9485 individual CREs contained at least one mutation, of which 72 (6 promoters and 66 distal regulatory elements) were found to be significantly enriched for mutations (q-value ≤0.05 and peak mutation rate ≥ threshold) (Fig. 2 and Additional file 1: Table 1). These regulatory elements were each recurrently mutated in 2–5 samples.

Overview of SNVs and their genomic distribution. a) The total number of SNVs in each sample considered in the analysis after filtering. They range from 1344 to 25,012. b) Percentage of SNVs falling within HepG2 open chromatin regions. Despite the range of total SNV numbers, the fraction that fall within the input genomic regions remains stable across the dataset, at 1.2% on average

Significantly mutated regions identified by SMuRF. Each of the 72 genomic intervals that passed the significance (q-value ≤0.05) and mutation rate filters are represented. The negative log of the q-value calculated from the binomial test for each region is plotted against the number of unique samples with a mutation within that region. The most frequently and most significantly mutated regions include the promoters of both known and novel genes of interest in liver cancer
Among the highly mutated promoters were those for the TERT, TP53, ACSM1, TNFRSF8, and PCGF5 genes, all previously reported recurrently mutated regions in liver cancer [18]. Also significantly mutated, however, was the promoter of a gene with unknown function, RP11-484D2.2, highlighting the potential of this type of analysis for uncovering novel regions of interest.
To further assess the ability of this approach to identify mutated regulatory elements that are relevant to the samples of interest, we compared the number of significantly mutated CREs identified in HepG2 to those found in other tissue types when using the same liver cancer mutation data. Chromatin accessibility data from eight ENCODE cell lines [1], including HepG2, was randomly sampled five times, matching for peak number and peak length, and SMuRF was run on each iteration using the same settings detailed above (Fig. 3). Significantly fewer (Mann-Whiney U test p-value range: 0.007–0.012) mutated CREs were identified in each of the seven other cell lines compared to HepG2.

Assessing the sample specificity of SMuRF. SMuRF was run on matched chromatin accessibility data from seven other tissue types. Each peak set was randomly sampled 5 times and SMuRF was run on each iteration. SK-N-SH_RA had the lowest peak number and was not sampled. The selected peak sets were also matched to the HepG2 dataset for peak length. The number of significantly mutated CREs identified by SMuRF in each run are shown as green diamonds, with the height of the bar for each tissue corresponding to the average CRE number
Conclusions
Whole-genome sequencing and chromatin accessibility data sets in numerous normal and diseased tissues are becoming more commonly available. SMuRF aims to help further our understanding of the importance of non-coding elements in disease initiation and progression, by highlighting those regulatory elements most likely to have a functional importance due to their high burden of mutation.
Abbreviations
- CRE:
-
Cis-regulatory element
- SNP:
-
Single nucleotide polymorphism
- SNV:
-
Single nucleotide variant
References
ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74.
Vinagre J, Almeida A, Pópulo H, Batista R, Lyra J, Pinto V, Coelho R, Celestino R, Prazeres H, Lima L, Melo M, da Rocha AG, Preto A, Castro P, Castro L, Pardal F, Lopes JM, Santos LL, Reis RM, Cameselle-Teijeiro J, Sobrinho-Simões M, Lima J, Máximo V, Soares P. Frequency of TERT promoter mutations in human cancers. Nat Commun. 2013;4:2185.
Horn S, Figl A, Rachakonda PS, Fischer C, Sucker A, Gast A, Kadel S, Moll I, Nagore E, Hemminki K, Schadendorf D, Kumar R. TERT promoter mutations in familial and sporadic melanoma. Science. 2013;339:959–61.
Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA. Highly recurrent TERT promoter mutations in human melanoma. Science. 2013;339:957–9.
Chiba K, Lorbeer FK, Shain AH, DT MS, Schruf E, Oh A, Ryu J, Darzacq X, Bastian BC, Hockemeyer D. Mutations in the promoter of the telomerase gene TERT contribute to tumorigenesis by a two-step mechanism. Science. 2017;357:1416–20.
Cobaleda C, Schebesta A, Delogu A, Busslinger M. Pax5: the guardian of B cell identity and function. Nat Immunol. 2007;8:463–70.
Puente XS, Beà S, Valdés-Mas R, Villamor N, Gutiérrez-Abril J, Martín-Subero JI, Munar M, Rubio-Pérez C, Jares P, Aymerich M, Baumann T, Beekman R, Belver L, Carrio A, Castellano G, Clot G, Colado E, Colomer D, Costa D, Delgado J, Enjuanes A, Estivill X, Ferrando AA, Gelpí JL, González B, González S, González M, Gut M, Hernández-Rivas JM, López-Guerra M, Martín-García D, Navarro A, Nicolás P, Orozco M, Payer ÁR, Pinyol M, Pisano DG, Puente DA, Queirós AC, Quesada V, Romeo-Casabona CM, Royo C, Royo R, Rozman M, Russiñol N, Salaverría I, Stamatopoulos K, Stunnenberg HG, Tamborero D, Terol MJ, Valencia A, López-Bigas N, Torrents D, Gut I, López-Guillermo A, López-Otín C, Campo E. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature. 2015;526(7574):519–24.
Katainen R, Dave K, Pitkänen E, Palin K, Kivioja T, Välimäki N, Gylfe AE, Ristolainen H, Hänninen UA, Cajuso T, Kondelin J, Tanskanen T, Mecklin J-P, Järvinen H, Renkonen-Sinisalo L, Lepistö A, Kaasinen E, Kilpivaara O, Tuupanen S, Enge M, Taipale J, Aaltonen LA. CTCF/cohesin-binding sites are frequently mutated in cancer. Nat Genet. 2015;47:818–21.
Weinhold N, Jacobsen A, Schultz N, Sander C, Lee W. Genome-wide analysis of noncoding regulatory mutations in cancer. Nat Genet. 2014;46:1160–5.
Wadi L, Uuskula-Reimand L, Isaev K, Shuai S, Huang V, Liang M, Thompson D, Li Y, Ruan L, Paczkowska M, Krassowski M, Dzneladze I, Kron K, Murison A, Mazrooei P, Bristow RG, Simpson JT, Lupien M, Wilson MD, Stein LD, Boutros PC, Reimand J: Candidate cancer driver mutations in super-enhancers and long-range chromatin interaction networks. bioRxiv . 2017: 236802.
Polak P, Karlić R, Koren A, Thurman R, Sandstrom R, Lawrence M, Reynolds A, Rynes E, Vlahoviček K, Stamatoyannopoulos JA, Sunyaev SR. Cell-of-origin chromatin organization shapes the mutational landscape of cancer. Nature. 2015;518:360–4.
Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–2.
Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S, Barnes I, Bignell A, Boychenko V, Hunt T, Kay M, Mukherjee G, Rajan J, Despacio-Reyes G, Saunders G, Steward C, Harte R, Lin M, Howald C, Tanzer A, Derrien T, Chrast J, Walters N, Balasubramanian S, Pei B, Tress M, Rodriguez JM, Ezkurdia I, van Baren J, Brent M, Haussler D, Kellis M, Valencia A, Reymond A, Gerstein M, Guigó R, Hubbard TJ. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 2012;22:1760–74.
Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, Young RA. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–19.
CY ML, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. 2010;28:495–501.
Mehdi T, Bailey SD, Guilhamon P, Lupien M. C3D: A tool to predict 3D genomic interactions between cis-regulatory elements. Bioinformatics, bty717 https://doi.org/10.1093/bioinformatics/bty717.
Alexandrov LB, Nik-Zainal S, Wedge DC, SAJR A, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale A-L, Boyault S, Burkhardt B, Butler AP, Caldas C, Davies HR, Desmedt C, Eils R, Eyfjörd JE, Foekens JA, Greaves M, Hosoda F, Hutter B, Ilicic T, Imbeaud S, Imielinski M, Imielinsk M, Jäger N, DTW J, Jones D, Knappskog S, Kool M, Lakhani SR, López-Otín C, Martin S, Munshi NC, Nakamura H, Northcott PA, Pajic M, Papaemmanuil E, Paradiso A, Pearson JV, Puente XS, Raine K, Ramakrishna M, Richardson AL, Richter J, Rosenstiel P, Schlesner M, Schumacher TN, Span PN, Teague JW, Totoki Y, ANJ T, Valdés-Mas R, van Buuren MM, vant Veer L, Vincent-Salomon A, Waddell N, Yates LR, Australian Pancreatic Cancer Genome Initiative, ICGC Breast Cancer Consortium, ICGC MMML-Seq Consortium, ICGC PedBrain, Zucman-Rossi J, Futreal PA, Mc Dermott U, Lichter P, Meyerson M, Grimmond SM, Siebert R, Campo E, Shibata T, Pfister SM, Campbell PJ, Stratton MR. Signatures of mutational processes in human cancer. Nature. 2013;500:415–21.
Fujimoto A, Furuta M, Totoki Y, Tsunoda T, Kato M, Shiraishi Y, Tanaka H, Taniguchi H, Kawakami Y, Ueno M, Gotoh K, Ariizumi S-I, Wardell CP, Hayami S, Nakamura T, Aikata H, Arihiro K, Boroevich KA, Abe T, Nakano K, Maejima K, Sasaki-Oku A, Ohsawa A, Shibuya T, Nakamura H, Hama N, Hosoda F, Arai Y, Ohashi S, Urushidate T, Nagae G, Yamamoto S, Ueda H, Tatsuno K, Ojima H, Hiraoka N, Okusaka T, Kubo M, Marubashi S, Yamada T, Hirano S, Yamamoto M, Ohdan H, Shimada K, Ishikawa O, Yamaue H, Chayama K, Miyano S, Aburatani H, Shibata T, Nakagawa H. Whole-genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nat Genet. 2016;48:500–9.
Acknowledgements
The authors would like to thank Seyed Ali Madani Tonekaboni and Parisa Mazrooei for their comments and suggestions in the development of this tool and the preparation of the manuscript.
Funding
Research supported by SU2C Canada Cancer Stem Cell Dream Team Research Funding (SU2C-AACR-DT-19-15) provided by the Government of Canada through Genome Canada and the Canadian Institutes of Health Research, with supplemental support from the Ontario Institute for Cancer Research through funding provided by the Government of Ontario. Stand Up To Cancer Canada is a program of the Entertainment Industry Foundation Canada. Research Funding is administered by the American Association for Cancer Research International - Canada, the scientific partner of SU2C Canada. This work was also supported by Prostate Cancer Canada; Canadian Cancer Society, Movember Foundation (grant number RS2014–04), and the Princess Margaret Cancer Foundation. M.L. holds an Investigator Award from the Ontario Institute for Cancer Research; a Canadian Institutes of Health Research (CIHR) New Investigator Award; and a Movember Rising Star Award from Prostate Cancer Canada. P. G is supported by a CIHR Fellowship (MFE 338954).
Availability of data and materials
Project name: SMuRF.
Project home page: https://github.com/LupienLab/SMURF
Operating system (s): Unix/Linux.
Programming language: Bash (≥4.1.2), R (≥3.3.0).
Other requirements: Bash (≥4.1.2), R (≥3.3.0) and BEDTools (≥2.26.0). It requires the following R packages: GenomicRanges, gtools, gplots, ggplot2, data.table, psych, and dplyr.
License: GNU GPLv3.
Any restrictions to use by non-academics: none.
The datasets generated and/or analysed during the current study are available in the following manuscripts:[1] and [17].
Author information
Authors and Affiliations
Contributions
PG wrote the software and performed the analyses with input from ML. PG and ML wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
SMuRF output for the 72 significantly mutated CREs in liver cancer. (TXT 13 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Guilhamon, P., Lupien, M. SMuRF: a novel tool to identify regulatory elements enriched for somatic point mutations. BMC Bioinformatics 19, 454 (2018). https://doi.org/10.1186/s12859-018-2501-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12859-018-2501-y