
Abstract
Background
Genetic relatedness is currently estimated by a combination of traditional pedigree-based approaches (i.e. numerator relationship matrices, NRM) and, given the recent availability of molecular information, using marker genotypes (via genomic relationship matrices, GRM). To date, GRM are computed by genome-wide pair-wise SNP (single nucleotide polymorphism) correlations.
Results
We describe a new estimate of genetic relatedness using the concept of normalised compression distance (NCD) that is borrowed from Information Theory. Analogous to GRM, the resultant compression relationship matrix (CRM) exploits numerical patterns in genome-wide allele order and proportion, which are known to vary systematically with relatedness. We explored properties of the CRM in two industry cattle datasets by analysing the genetic basis of yearling weight, a phenotype of moderate heritability. In both Brahman (Bos indicus) and Tropical Composite (Bos taurus by Bos indicus) populations, the clustering inferred by NCD was comparable to that based on SNP correlations using standard principal component analysis approaches. One of the versions of the CRM modestly increased the amount of explained genetic variance, slightly reduced the ‘missing heritability’ and tended to improve the prediction accuracy of breeding values in both populations when compared to both NRM and GRM. Finally, a sliding window-based application of the compression approach on these populations identified genomic regions influenced by introgression of taurine haplotypes.
Conclusions
For these two bovine populations, CRM reduced the missing heritability and increased the amount of explained genetic variation for a moderately heritable complex trait. Given that NCD can sensitively discriminate closely related individuals, we foresee CRM having possible value for estimating breeding values in highly inbred populations.
Similar content being viewed by others


Background
“All genomes are equal, but some genomes are more equal than others Footnote 1” with apologies to George Orwell (1903–1950).
Accurate measures of genetic relationships among individuals are needed to accelerate artificial selection for genetic improvement [1] and to refine methods for gene discovery [2]. By accounting for patterns of relatedness, particularly within but also between families, relationships among individuals lay the foundation for robustly connecting genotype to phenotype. Genetic relatedness is currently estimated by traditional pedigree-based approaches (NRM for numerator relationship matrices) [3], augmented by molecular information [genomic relationship matrices (GRM)] [4]. Because meiotic recombination is stochastic and pedigree information is not always available or error free, GRM can give more precise estimates of genetic relatedness than basic pedigree information since the latter makes simplifying assumptions [4]. For example, while we predict that full-sibs and half-sibs share approximately 50 and 25 % of their DNA, respectively, simple pedigree information is unable to account for the exact percentage shared, or indeed which DNA segments have been inherited. Moreover, because of linkage disequilibrium (LD) and linkage, associations of DNA markers with quantitative trait loci (QTL) are expected to erode during successive meioses at a slower rate than pedigree relationships, which increases their utility across generations [5]. Overall, these advantages of marker-based relationships have increased the attractiveness of single nucleotide polymorphism (SNP) chips in genetic improvement programs.
GRM are essentially computed by genome-wide SNP genotype similarities (e.g. correlations) among all pair-wise combinations of individuals [4]. These correlations exploit SNP genotypes that are shared between two individuals, one SNP at a time. However, it is an open question whether correlation is the best way to relate SNP genotype data given that (1) any non-linear relationships are poorly characterised or undetected by correlations and (2) it is not immediately obvious what is the best approach when assessing whole genomes that have been abstracted to long complex numerical systems described by a small three letter SNP alphabet (0,1,2). We hypothesize that there is unexplored potential to characterise alternative and/or complementary measures of relatedness, for example through pattern recognition approaches sensitive to the information contained in complex patterns. One such alternative is normalised compression distance (NCD), which has previously been used to successfully cluster various data, including musical compositions into genres [6]. The basic principle of NCD as it applies to genomics is that patterns in the SNP genotypes from one individual can be used to describe similar patterns in the SNP genotypes from a second individual. The ability of one individual to describe another individual can be quantified mathematically by data compression, approximated via real-world compressors like the gzip application tool of UNIX systems (http://www.gzip.org). If the data compression relating the two genomes is strong, then they are deemed to be closely related and are awarded a short distance. Applying this process systematically across a genotyped population can be used to build a compression relationship matrix (CRM), analogous to a GRM. In a preliminary study, we explored the application of NCD to two sheep populations, one that included multiple breeds [7] and another with known sire groups and a half-sib population structure (unpublished data). We found that the method had merit in recovering both breed history and sire group structure.
Prior to that, we explored a basic measure of within-genome compression efficiency (CE) by expressing the SNP genotype file sizes in bits before and after data compression by the gzip tool. Plotting these CE values relative to heterozygosity [8] yielded clusters of individuals that are similar to those produced by population differentiation such as F statistics (F ST), and consistent with phylogeography. Genome-wide CE can be considered as reflecting patterns in both allele order and allele proportion that are known to differ systematically between breeds, but to be shared among closely related individuals. These shared patterns among individuals could include, but not be limited to, genome-wide heterozygosity and runs of homozygosity [9]. Similar to GRM correlation, CE is a hypothesis-free pattern recognition tool. It can exploit very complex shared patterns that do not need to be defined a priori. The utility of inferred relationship matrices can be validated in the normal manner—that is, by using them to predict genetic merit for complex phenotypes using best linear unbiased prediction (BLUP) and evaluating their accuracy.
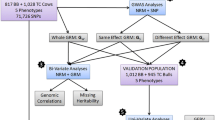
Here, we studied two animal populations of commercial relevance to Australian agricultural production that have matching phenotype data for yearling weight, a complex phenotype of moderate heritability, one Brahman (BB) and one Tropical Composite (TC). These populations have recorded pedigrees that show the presence of both full-sib and half-sib individuals. Furthermore, these populations represent historically admixed populations that were founded from contributions of both Bos indicus and Bos taurus progenitors, which are sub-species that arose from independent domestication events and last shared a common ancestor more than 200,000 years ago [10]. For the first time, we compared compression-based best linear unbiased predictions (CBLUP) with genomic (GBLUP) and pedigree-based (PBLUP) predictions for yearling weight. We present the outcome of the clustering, the proportion of missing heritability [11] and the prediction accuracies for yearling weight that were obtained by using different approaches to estimate the relationship between individuals.
Methods
Animal resources and SNP genotyping platforms
Animals, phenotypes and genotypes used in this study were a subset of those used in [12]. Briefly, we used data on 816 Brahman (BB) and 1028 Tropical Composite (TC) cows genotyped using either the BovineSNP50 [13] or the BovineHD (Illumina Inc., San Diego, CA, USA) that includes more than 770,000 SNPs. For animals that were genotyped with the lower density array, genotypes were imputed to higher-density based on the genotypes of relatives based on pedigree, as described previously [14]. The imputation was performed using 30 iterations of BEAGLE [15] within breeds, using 519 Brahman and 351 Tropical Composite animals genotyped using the BovineHD as reference. From the resulting 729,068 SNP genotypes per individual, we extracted the genotypes from 71,726 SNPs that were highly polymorphic in Bos indicus cattle (GGP Indicus HD Chip; http://www.neogeneurope.com/Agrigenomics/pdf/Slicks/NE_GeneSeekCustomChipFlyer.pdf).
Animal clustering by genotype
Genome-wide CE
First, we computed animal-to-animal relationships that could be ascertained from genotype data using the basic CE approach corrected for heterozygosity (CEh), as described in [8]. This approach computes the CE for the genotype file of each individual and then expresses it against heterozygosity (Het) across the whole genome:
where SB and SA indicate the genotype file size expressed in bits before and after compression by gzip, respectively. The underlying principle of CEh is the same as for any genetic clustering method: the closer the match in the numerical patterns present in two genotype files is, the closer is the inferred genetic relationship between them. No attempt was made to discriminate DNA segments that were identical by descent from those that were identical by state.
Normalised compression distance computation
The main weakness of CEh is that two genotype strings can have the same CE and heterozygosity despite being different (e.g. 0000000000 and 2222222222). It is not clear how common this phenomenon is in real population genetic data, but it has the potential to confound some of the observed clustering. To address this, we used normalised compression distance (NCD) [6] to develop alternative measures of relationship. NCD is a way of measuring the similarity between two objects. NCD is obtained by approximating a non-computable similarity metric called normalized information distance (NID). The principle is that NCD will award short distances to highly related sequences, on the grounds that shared patterns result in compression gain when two similar files are concatenated, but not when two dissimilar files are concatenated. In other words, a short distance is awarded when the information in the first genotype file can be used to describe the information in the second genotype file. We used the gzip application tool of UNIX systems (http://www.gzip.org) as our real world compressor. The application gzip is based on the lossless data compression algorithm DEFLATE that was originally described by [16]. NCD only clusters those genotype files that compress the same for the same reasons. That is, in contrast to CEh, 0000000000 will now only cluster with 0000000000 and not with 2222222222.
As previously reported [7], the formula for the computation of NCD between two individuals x and y based on their respective SNP genotype sequence is:
where Z(xy) represents the size of the compressed file that contains both SNP genotype sequences to be compared and Z(x) and Z(y) are the sizes of the compressed file with the separate SNP genotypes for x and y, respectively.
Building a compression relationship matrix from the NCD values
Relationship matrices are based on estimates of similarity, but through NCD we have computed ‘distance’ not similarity. Therefore, the construction of the CRM from all pair-wise NCD values first requires conversion of the compression distance to an equivalent similarity measure. While distance and similarity share commonalities they are not equivalent. In practice, there are numerous ways of inter-relating them. Here, we explored two approaches, producing two different CRM (CRM1 and CRM2) from the same NCD input.
The first method (CRM1) made use of the universal distance (d) to similarity (s) conversion law of Shepard [17]. We used \(s_{i,j} = 2.5e^{{ - 5d_{i,j} }}\), which was selected in an ad hoc fashion by confirming that the resulting similarity between individuals i and j (s i,j ) covered the 0–1 interval observed for correlation (and therefore for all GRM), where d i,j is the NCD between the i and j individual pair. This method proved to have a scaling issue which can bias estimates of genetic parameters. This problem was overcome through computation of CRM2 (described below).
The second method (CRM2) attempted to better ground the NCD in established genetics—that is, an expectation of relatedness of 1 for self–self pairs, 0.5 for full sibs and 0.25 for half-sibs. This expectation is governed by the laws of inheritance and the likely molecular outcomes of meiosis when applied to a diploid mammalian genome. CRM2 used a linear conversion method defined as follows:
This linear method has the appealing feature of yielding an average value of 1 for self–self pairs, including a spread around 1 that reflects inbreeding. This result shows that the CRM2 matrix is scaled in a manner more suitable for the estimation of genetic parameters. The remaining values approximate 0.5 for full sibs, 0.25 for half-sibs and so on. Similar to GRM but unlike NRM, these values were not pre-defined by pedigree expectation but derived from the SNP genotype data. Therefore, they are likely to more accurately capture the observed variability that is inherent to the process of meiosis that gives rise to each individual compared to its relatives.
The third compression-based relationship matrix (CRM3) was entirely independent of the NCD approach described above. The aim of CRM3 was to produce another set of genetically sensible relatedness values, through application of a two-step process: an initial window-based CE step and a subsequent correlation step. To achieve this, we produced a matrix with as many columns as sets of 50 consecutive SNPs that can be built from the genotype file, and as many rows as animals in the analysis. Individual cells in this matrix contain the CE value for each window for each animal. This matrix was then used as input for a correlation analysis so that, for each pair of animals, the correlation across their respective CE values was used as a measure of relatedness. Animals that had sets of SNPs that compressed in the same way along their respective genomes were awarded high correlations.
GRM computation
The construction of the GRM was based on the correlation between genotypes and was computed according to the methodology developed in [4]:
where Z is the (number of animals by number of SNPs) matrix of genotypes and p i is the frequency in the population of the B allele for the ith SNP. ZZ T represents the number of shared SNP alleles between all pairs of individuals and the division of ZZ T by \(2\mathop \sum \nolimits p_{i} (1 - p_{i} )\) aims at scaling the GRM to make it analogous to the numerator relationship matrix (NRM) based on pedigree. This is the standard approach for genomic prediction of breeding values [4].
The computations of NRM, GRM, CRM1, CRM2 and CRM3 are schematically summarised in Fig. 1 using toy examples. The UNIX scripts for these examples are in Additional file 1. We begin with a genotype file (a in Fig. 1) that comprises 30 SNPs and five animals. The genotype profiles of Animals 1, 2, 3, 4 and 5 are encrypted by {10 “0” + 10 “1” + 10 “2”}, {3 “2” + 10 “0” + 10 “1” + 7 “2”}, {3 * {3 “0” + 3 “1” + 3 “2”} + “0,1,2”}, {3* {3 “0” + 2 “1” + 3 “2” + “1”} + “0,1,2”}, and {10 * “0,1,2”}, respectively. It becomes immediately apparent that the profiles of Animals 1 and 2 and those of Animals 3 and 4 are very similar. On closer inspection, we also find a similarity between the profiles of Animals 3 and 5 because each trio of identical genotypes (e.g. “000”) in Animal 3 is matched by a trio of “0,1,2” in Animal 5.

Given a genotype file (a) and a plausible pedigree (b), one can compute an NRM (c) and a GRM (d). One can also compute an NCD matrix (e) which in turn can be transformed into CRM1 (f) and CRM2 (g) given two different distance to similarity transformations. A sliding window-based version of the CE analysis (h) can be used to generate a correlation matrix which underpins the computation of CRM3 (i)
The encrypted genotype profiles are awarded the following genome-wide CE; 30, 27, 23, 20 and 32 %, respectively for Animals 1 to 5. On the one hand, Animals 1 and 5 have the most regular genotypes, resulting in the highest CE and on the other hand, Animals 3 and 4 have the most irregular genotypes, resulting in the lowest CE. A plausible pedigree graph is given in b of Fig. 1, which also contains two un-genotyped animals, U1 and U2, and where arrows indicate direction from parent to offspring. Based on this pedigree and these genotypes, we can compute the NRM (c in Fig. 1) and the GRM (d in Fig. 1), respectively.
The NRM reveals that Animal 1 is the sole inbred animal since it results from a parent-offspring mating. The GRM reveals that Animals 2 and 4 are the most inbred and captures the strong (parent-offspring) relationship between Animals 1 and 2 (GRM value = 0.35) and Animals 3 and 4 (GRM value = 0.67). The GRM also captures the most distant relationships (GRM value = −0.67), i.e. of Animal 2 with Animals 3 and 4, which are unrelated based on the NRM.
Based on this genotype file, we can also compute the NCD matrix (e in Fig. 1), which shows the shortest distances on the diagonals (i.e. self–self comparisons). Consistent with the GRM and the NRM, the longest distances observed in the NCD matrix (NCD values = 0.625 and 0.591) are between Animal 2 and Animals 3 and 4. Two distance-to-similarity transformations were used to generate CRM1 (f in Fig. 1) and CRM2 (g in Fig. 1). These two CRM differ in that CRM1 has un-scaled diagonal values and unbounded off-diagonal values, whereas CRM2 has scaled diagonal values such that they average to 1 and off-diagonal values bounded between 0 and 0.75.
The genotype file can also be divided in windows (or genomic regions) of consecutive SNPs and CE can then be computed for each window by animal combination. Here, we have four windows of 7, 7, 7, and 9 consecutive SNPs giving rise to the CE matrix h of Fig. 1. Windows with seven identical genotypes, such as the first window in Animal 1 and the third window in Animal 2, yield a high CE of 71.4 %. Column-wise, a correlation matrix based on CE values for all pairs of animals is computed to generate CRM3 (i in Fig. 1). In this case, Animals 4 and 5 are identical according to CRM3, while the relationship between Animals 1 and 2 has disappeared.
Comparing CRM and GRM
Variance components estimates
We used mixed-model equations and the Qxpak.5 software [18] for the estimation of genetic parameters and the prediction of breeding values for yearling weight in the two populations. The general model was as follows:
where y is the vector of yearling weight observations, X in an incidence matrix relating observation in y with the vector of fixed effects in β (i.e., contemporary group comprised of sex, year and location, and the covariates of age of dam, indicine percent and age of measurement) [12]. The summation goes for r, the number of random components fitted in the model. Z is an incidence matrix relating observations in y with the vector of random additive effects in u r which are assumed to be normally distributed with zero mean and variance \({\text{V}}\left( {{\mathbf{u}}_{r} } \right) = {\mathbf{C}}_{r} \sigma_{{{\text{u}}r}}^{2}\), where C r is the relationship matrix based on either the pedigree (NRM) or markers (GRM, CRM1, CRM2 or CRM3), and \(\sigma_{{{\text{u}}r}}^{2}\) is the additive genetic variance associated with u r . Finally, e is the vector of random residual effects assumed to be normally distributed with zero mean and variance \({\text{V}}\left( {\mathbf{e}} \right) = {\mathbf{I}}\sigma_{\text{e}}^{2}\), where I denotes an identity matrix and \(\sigma_{\text{e}}^{2}\) is the residual variance.
Twelve models were explored: one each (i.e., four) with a single additive effect from either relationship matrix (NRM, GRM, CRM1 and CRM2), and then an informative subset of combinations of the above models. These 12 models are defined in Tables 1 and 2 for BB and TC, respectively.
Using yearling weight, we compared the performances of NRM, GRM, CRM1, CRM2 and CRM3 according to the resultant genetic parameters, EBV and prediction accuracies. For the computation of accuracy, 20 % of phenotypes were randomly set to missing values. The reported accuracy was the average of 20 random splits of the data (i.e. 80 % calibration versus 20 % validation). Estimates of breeding values based on NRM, GRM, CRM1, CRM2 and CRM3 were compared and the accuracy of the resulting predictions was computed from the correlation between the EBV and the adjusted phenotypes (Table 3).
Models 5, 6 and 7 (i.e. GRM, CRM1 and CRM2) can estimate the fraction of missing heritability (C miss) using the formulae of [11]:
where \(\sigma_{u}^{2}\) is the variance due to the genotype data (i.e. either GRM or CRM1 or CRM2 in our context) and \(\sigma_{a}^{2}\) is the estimate of the additive genetic variance based on pedigree (i.e. the NRM in our context). NRM, GRM, CRM2 and CRM3 have scaled values with self–self pairs close to or equal to 1. This implies that any differences between their genetic parameter estimates are unlikely to be a simple artefact of scaling.
Signatures of selection
In order to detect signatures of selection and regions of evolutionary interest, we applied a sliding window version of CE, as previously described in [8]. This approach exploits the sensitive pattern recognition capability of CE to identify haplotype blocks that occur in one population but not in the other. Briefly, the CE of non-overlapping windows at the population level was computed for both populations and corrected for heterozygosity (CEh). The correction for heterozygosity de-emphasizes simple patterns that are enforced by runs of homozygosity (ROH). We computed 1435 windows of 50 consecutive SNPs across the 71 K SNPs. For comparison purposes, the F ST [19] in the BB vs. TC contrast was computed for each SNP. Then, the F ST of the whole window was estimated based on the average F ST of all SNPs contained in the window.
Results
Clustering animals by genotype
In Fig. 2, each point in the scatter represents either a single BB (top panel) or TC (bottom panel) animal. Two animals that cluster together can be assumed to share more genotype patterns than two animals that are further apart and thus are more likely to be related by descent. The consequence of using the new Indicine SNP chip was higher heterozygosity (particularly in the BB population), coupled with greater CE when compared to genotyping the same population using the Bovine HD BeadChip. It is likely that the increase in heterozygosity observed using the Indicine SNP chip resulted from a reduction in SNP ascertainment bias [20] that is associated with genotyping Bos indicus populations using the HD BeadChip that was designed for Bos taurus. It is less clear why the CE also increased but this may reflect the Indicine SNP chip’s greater ability to exploit regularities at common runs of heterozygous sites. Like any other form of regularity, runs of heterozygosity (strings of 1s) are a possible source of compressible patterns in genomes with high heterozygosity. Based on its improved performance, all remaining analyses were performed using the Indicine SNP chip.

Comparison of CEh using different genotyping platforms. A comparison of CEh for BB (top panel) and TC (bottom panel) cows genotyped using both the HD chip (red dots) with 750 K SNPs and the new 71 K Indicus SNP chip (black dots). Each point represents a single animal
Relatedness between animals using NRM, GRM and NCD
Tables 4, 5 and 6 show summary data that relate NRM with GRM, NCD and CRM. The pedigree-based NRM [1] was computed recursively after tracing back three generations of ancestors. No inbreeding was detected based on pedigree and the self–self relationships for the 816 BB individuals averaged at close to 1 for GRM, CRM2 and CRM3 (Table 6).
For relationships corresponding to NRM values of 0.25 (i.e. those existing between half-sibs or between grand-parent and grand-offspring), the average GRM were equal to 0.196 and 0.201 for BB and TC cattle, respectively. Self–self relationships were equal to 0.997 and 0.987 for BB and TC cattle, respectively.
The relationship between GRM and NCD for each pair of individuals is plotted in Fig. 3 for the main bulk of the data. The full parameter space including self–self pairs is in Additional file 2: Figure S1. Figure 3 reveals a population sub-structure in both populations that is more complex than that obtained by analysing either GRM or NCD, separately. This suggests that these two metrics operate synergistically and together provide a more complete understanding of relationships in the population. Highlighting half-sibs (as defined by pedigree information) yielded a cluster that was centred on a GRM of 0.25. Half-sibs that had a GRM of ~0 probably represent pedigree errors or mistakes in DNA handling.

Comparison of GRM and NCD. Each point represents a pair of BB (BB; a) and TC cows (TC; b). We have plotted the parameter space showing all pairs except the self–self pairs. An outlier animal with an unusual Zebu contribution was identified by NCD but not by GRM for both populations (red dots). Half-sibs in a pedigree form a main cluster around the expected GRM of 0.25
A distinct cluster with an increased NCD was observed in the BB population (Fig. 3). This was also observed for the TC animals but was less pronounced. In both populations, the pairs in these clusters had a given individual in common. For the BB population, the Zebu genetic contribution of this individual (#1100) was much smaller (i.e. 0.528 compared to greater than 0.829 for all the other animals in the population), while for the TC population, the Zebu genetic contribution of the individual in question (#1582) was substantially larger (i.e. 0.652 compared to less than 0.56 for all the other animals in the population). Surprisingly, GRM was not able to identify the markedly different Zebu contribution of these individuals in either population.
Next, we attempted to identify genome properties that were responsible for the similarities and differences between the GRM and NCD measures of relatedness. To answer this question, we overlaid the average Zebu contribution (based on a principal component analysis that also included Angus and Nelore data) [12] of each pair (see Additional file 3: Figure S2). It is clear that, for both populations, GRM and NCD resulted in measures of relationships that were much more similar to each other for pairs of individuals that were relatively purebred than for cross-bred animals. Thus, for the BB population, the linear part of the plot is enriched with BB pairs that have an average Zebu contribution of more than 0.96, while for the TC population, it is enriched in TC pairs that have an average Zebu contribution of less than 0.1. Thus, the correlation-based (GRM) and compression-based (CRM2) measures of genetic similarity tend to agree on estimates of relatedness for purebred pairs of individuals.
The impact of the two NCD mapping approaches (CRM1 and CRM2) on the estimate of similarity from the same NCD value is in Fig. 4. Both versions of CRM were negatively related to NCD, because similarity is inversely related to distance. CRM2 resulted in a linear relationship whereas CRM1 resulted in a non-linear exponential relationship. The linear relationship for CRM2 explains its higher accuracy in computing genetic parameters. This is because the similarity values are more consistent with biological expectation. In other words, the similarities produced by CRM2 resemble more closely the expected genetic relationships between self–self pairs (1), full-sibs (0.5), half-sibs (0.25) and other relatives. Consequently, CRM2 can be expected to estimate genetic parameters more accurately.

Relationship between NCD and CRM1 (black) and NCD and CRM2 (red). BB population on top panel, TC population on bottom panel. CRM1 bears a quadratic relationship to NCD, whereas CRM2 has a linear relationship. Trend lines were added to illustrate the difference between CRM1 and CRM2
Computational performance
We explored the computing time required for each population. For the BB population and once the relationship matrices have been built, the mixed model that contained a single additive effect took 31 s for the NRM, 4 min and 6 s for the GRM and 4 min and 10 s for the CRM2. The longer time taken for the models with either GRM or CRM2 reflects the higher density of these marker-based relationship matrices. However, the computing of NCD, which is required for CRM1 and CRM2, consumed substantially more time than the pair-wise correlations required for GRM. In our implementation, it took 102.6 and 162.4 h to compute the NCD matrices for BB and TC, respectively. The reason being the DEFLATE algorithm included in the gzip tool was not incorporated into the UNIX script, so we had to resort to numerous input and output operations that call on the compression function of GZIP externally.
Computational demands aside, the relationship matrices based on NCD were highly related to the GRM based on the strong negative correlation between pairs of individuals (high correlations corresponded to small NCD distances) (Tables 4, 5). Furthermore, we have previously documented the broad similarity in clustering produced by the GRM and NCD dimension reduction plots for sheep breeds [7].
Estimating genetic parameters and accuracies of predicted breeding values
Overall, estimates of genetic parameters were quite similar for GRM and CRM2 (Tables 1, 2). It should be noted that the point estimate for CRM2 explained more genetic variation (Vc2) than the NRM and GRM for both the BB and TC populations, although the absolute difference was small and likely not statistically significant. Furthermore, in the light of the equivalent cross-validation accuracies (perhaps even better for the BB population) that were obtained by using CRM2 (Table 3), it is tempting to speculate that implementing CRM2 for these populations and for this phenotype (YWT, yearling weight) using the latest Indicus 71 K SNP chip might lead to slightly better breeding decisions. Estimates of variance components based on CRM3 are in Table 7. CRM3 did not result in as much as an increase in the genetic variance explained compared to NRM and GRM as observed for CRM1 and CRM2. For both populations and in agreement with the accuracy results, the missing heritability was lowest for CRM2, which implies that it outperformed the GRM by a small margin (Table 8).
Signatures of selection
The comparison between F ST and CEh revealed a high positive relationship (r = 0.75), which confirmed the relevance of using CE to detect putative selected genomic regions in cattle. Genomic windows on the X chromosome consistently displayed high values for both metrics, which confirms previous results observed for populations that include Bos indicus and Bos taurus ancestry [21]. A likely explanation is that the level of sequence divergence between the X chromosomes of these two sub-species is greater than that between autosomal chromosomes [21].
To identify autosomal genomic windows with markedly different genetic patterns between BB and TC cattle, we examined the genome-wide distribution of CEh in each population (Fig. 5). The average CEh for BB and TC populations was plotted against F ST and yielded a positive relationship (Fig. 6a), which suggests that the approach has merit in population differentiation. The difference between the CEh for the BB and TC populations was plotted against F ST (Fig. 6b) to identify population-specific outliers. Detailed investigation of the extreme windows in Fig. 6b allowed us to identify population-specific sweeps. The autosomal windows with the most extreme differences between the two populations are in Table 9. Nine of the ten regions had high CEh values for the BB population and much lower values for the TC population (Table 9). The gene content of each region was determined using reference genome built Btau UMD3.1 (see Additional file 4: Table S1) and then compared with published data on selection sweeps in cattle.

Genome-wide view of compression peaks in BB (red profile) versus TC cows (blue profile). Heterozygosity corrected CE (CEh) was estimated separately for BB (blue) and TC cattle (red) and plotted in genomic order. Windows with extreme population differences in CEh are identified in Table 9. Chromosome number is indicated above the plot

a Relationship between average CEh and F ST. Each dot represents a window of 100 consecutive SNPs. Windows in the top left quadrant are identified as important by CEh but not F ST. Blue dots are windows on the X chromosome. Windows in the top left quadrant are identified as important by CEh but not F ST. b Relationship of the difference in CEh between populations (TC-BB) with F ST
These regions carry genes that are involved in bovine reproduction (NCOA2), immune function (BCL2) and fatness (ATP5H) (Table 9). It is also important to note that there were two separate instances where two highly functionally related proteins were identified in independent genomic regions: (1) monocarboxylate transporter coded by SLC16A5 on BTA19 and its paralog coded by SLC16A4 on BTA3 and (2) two subunits of the mitochondrial ATP synthase, the F1 catalytic core complex coded by ATP5B on BTA5 and the membrane-spanning F0 complex coded by ATP5H on BTA19 (see Additional file 4: Table S1).
Discussion
Inference of genomic relationships
It is well established that shared patterns of allele composition can be used to infer genomic relationships between individuals. Since both GRM and CRM are based on genome-wide similarity, it is not surprising to detect a close relationship between these two approaches. NCD shows merit as an alternative or complementary measure of genomic relatedness to GRM. Overall, short NCD distances reflect high co-sharing of genome-wide heterozygosity, runs of homozygosity and compositionally complex haplotypes, which may not be identified by other approaches. Collectively, these genomic features have implications for inbreeding, population structure and the identification of signatures of selection.
The quadratic relationship between NCD and GRM implies that NCD is particularly well adapted to discriminate closely related individuals. This conclusion is borne out by current and past data. Previously, we found that NCD can separate some full-sibs from half-sibs in cases where GRM cannot [7]. This observation may be consistent with that of [22] who found that a haplotype-based method uncovered Mendelian inconsistencies between second degree relatives more effectively than single-SNP approaches. Furthermore, in a recent application of the NCD method, we examined its application to a high-density sheep SNP dataset and found that it was able to differentiate two Poll Merino sire groups from each other and that Poll Merino individuals could be distinguished from Merino individuals by NCD but not by GRM [7]. In the data presented here, NCD clearly differentiated an individual with an unusual Zebu contribution in both the BB (separating an individual that is genetically more like a TC) and the TC (separating an animal that is genetically more like a BB) populations. This sensitive discrimination may reflect that NCD relies on ‘distance’, which enforces separation, versus the correlation’s use of ‘similarity’, which establishes connection.
Any measure of genomic similarity (whether correlation, CE, or other) needs to be clearly grounded in the biology of meiosis for it to provide meaningful estimates of genetic parameters. That is, it must yield the expected relationship values of ~1 for self–self pairs, ~0.5 for full-sibs and ~0.25 for half-sibs. These values are implicit (they emerge naturally) for correlation-based measures of relationships, but not for NCD. In this regard, the linear transformation that we used for CRM2 was superior to the quadratic transformation [17] of CRM1. The validation of CRM2 was apparent through the higher estimate of genetic variance, the corresponding slight reduction in missing heritability and the modest increase in accuracy of predictions of phenotype when compared to GRM and NRM.
Other authors have explored various options for optimising estimation of genomic relationships. For example, [23] found that parameter estimates may be biased if the genomic relationship coefficients are scaled differently from the pedigree-based estimates. They found that a reasonable scaling was possible by drawing on observed allele frequencies and scaling the relationship matrix such that the diagonal elements averaged 1. Scaling the diagonal elements to average 1 is the same logic that we applied when constructing CRM2 from the NCD values. This helps addressing the potential for biases in parameter estimation. In another example of new methods for genomic prediction, [9] published a method to derive relationships based on runs of homozygosity (ROH). They found that the ROH method produced more accurate GEBV than two alternative methods based on population-wide linkage disequilibrium and linkage. We hypothesize that this ROH method is related to our compression method. Shared ROH will obviously be clustered by compression, and thus, will contribute to the construction of the CRM. However, NCD also detects complex haplotypes beyond ROH. Understanding the biological meaning of ROH length and their implications for inbreeding and population bottlenecks is an active area of research [24]. However, it is currently not clear what inference can be drawn from the shared complex haplotypes that we are able to detect by NCD. The GRM is computed one SNP at a time, so it is not formally connected to haplotype structure.
Detection of sweeps
Previously, we reported that by sliding a CE-based population-level window along genomes, we could identify haplotypes that are likely the result of recent positive selection [8]. Obvious examples are genomic regions with a high level of homozygosity that arise in response to selection for a single beneficial haplotype (i.e. a ‘hard’ selective sweep). The consequence is an increase in CE and, applied to human data, this approach identified classic signatures of selection such as European eye and skin colour, Asian hair texture and European and Masaai Kenyan lactase persistence [8].
In this study, we applied the compression-based sliding window analysis to two bovine populations. Analysis of genomic windows with the largest difference in CE between the two populations successfully identified regions with major effects on production traits in tropically adapted cattle [12]. The top ranked region contains two inhibin genes (INHBE and INHBC), which have been previously identified as associated with fertility traits in tropically adapted cattle [25]. The precise mechanism that drives the outlier behaviour of CE is not clear.
Inspection of the genome-wide profile revealed that nearly all extreme genomic regions were characterised by a greater CE in BB cattle, well above the genome-wide average CE (Fig. 5). This suggests that selection specific to the BB population may have caused this difference in CE between these two populations. However, the reduced CE observed in the TC cattle also appeared to have an effect on the identification of differences between populations. The top ranked region on Bos taurus chromosome 5 (BTA5) (between 56.2 and 56.5 Mb; Fig. 5; Table 9) co-located with an association signal with large effects on yearling weight, body condition score and coat colour, which was reported for tropically adapted cattle [12]. While the analytical approaches between these two results are very different (genome-wide association analysis (GWAS) versus CE), the two studies used the same populations of Australian BB and TC cattle. This prompted a comparison with genes that were identified as putatively under selection in five independent populations of indicine (BB and N’Dama) and taurine cattle (Holstein, Angus and Charolais) [26]. None of the genes that were identified in our study appeared to be under selection in either of the indicine breeds but 59 genes located on BTA5 [between 56.2 and 57.8 Mb; (see Additional file 4: Table S1] were previously identified in the Charolais breed [26]. Our results indicate that CEh recapitulates previous findings based on established metrics for detecting selection sweeps. However, it is interesting to note that many of the top ranked genomic windows based on CEh would not have been classified as outliers using F ST and may constitute novel findings.
Commercial application
In a commercial context, broiler chicken and dairy cattle breeding populations have small effective population sizes, high levels of inbreeding and both use genomic prediction as part of their modern breeding strategies, as discussed in [27]. We anticipate that the NCD method used here also has value in those two industries because it can sensitively discriminate the closely related individuals they make use of when developing their breeding programs. According to [28], for a given effective population size, the major drivers of genomic EBV accuracy are (1) LD between markers and QTL, (2) training set size, (3) heritability and (4) distribution of QTL effects. Therefore, in order to generalise our findings of the possible utility of our method in genomic prediction, we propose that future work should explore phenotypes with a lower heritability and different genetic architectures.
Conclusions
NCD clusters a genome in a manner that is broadly similar to correlation-based approaches. Unlike GRM, NCD exploits patterns that are present in haplotypes and unlike ROH-based methods, NCD can identify and exploit haplotypes that have a more complex genotype composition. In this study, CRM2 has been validated by the fact that we show that it has a tendency to reduce the missing heritability and increase the phenotype accuracy for a moderately heritable complex trait in two bovine populations. The fine-grained resolution that NCD appears to possess may lend itself to situations for which the capacity to discriminate very closely related individuals is of particular value. A sliding window version of the analysis aimed at detecting sweeps identified regions caused by introgression of taurine haplotypes.
Notes
The oblique reference to George Orwell’s famous quote in Animal Farm follows the introduction of Cilibrasi and Vitanyi’s Clustering by Compression article.
References
Wright S. Coefficients of inbreeding and relationship. Am Nat. 1922;56:330–8.
Eu-Ahsunthornwattana J, Miller EN, Fakiola M, Jeronimo SMB, Blackwell JM, Cordell HJ. Comparison of methods to account for relatedness in genome-wide association studies with family-based data. PLoS Genet. 2014;10:e1004445.
Henderson CR. Rapid method for computing the inverse of a relationship matrix. J Dairy Sci. 1975;58:1727–30.
VanRaden PM. Efficient methods to compute genomic predictions. J Dairy Sci. 2008;91:4414–23.
Wolc A, Arango J, Settar P, Fulton JE, O’Sullivan NP, Preisinger R, et al. Persistence of accuracy of genomic estimated breeding values over generations in layer chickens. Genet Sel Evol. 2011;43:23.
Cilibrasi R, Vitanyi PMB. Clustering by compression. IEEE Trans Inf Theory. 2005;51:1523–45.
Hudson N, Kijas J, Porto Neto L, Reverter A. Compression efficiency relationship matrix: developing new methods to determine genomic relationships for improved breeding. In: 10th World Congress on Genetics Applied to Livestock Production: 17–22 August 2015, Vancouver; 2014.
Hudson NJ, Porto-Neto LR, Kijas J, McWilliam S, Taft RJ, Reverter A. Information compression exploits patterns of genome composition to discriminate populations and highlight regions of evolutionary interest. BMC Bioinformatics. 2014;15:66.
Luan T, Yu X, Dolezal M, Bagnato A, Meuwissen T. Genomic prediction based on runs of homozygosity. Genet Sel Evol. 2014;46:64.
McTavish EJ, Hillis DM. A genomic approach for distinguishing between recent and ancient admixture as applied to cattle. J Hered. 2014;105:445–56.
Roman-Ponce SI, Samore AB, Dolezal MA, Bagnato A, Meuwissen THE. Estimates of missing heritability for complex traits in Brown Swiss cattle. Genet Sel Evol. 2014;46:36.
Porto-Neto LR, Reverter A, Prayaga KC, Chan EK, Johnston DJ, Hawken RJ, et al. The genetic architecture of climatic adaptation of tropical cattle. PLoS One. 2014;9:e113284.
Matukumalli LK, Lawley CT, Schnabel RD, Taylor JF, Allan MF, Heaton MP, et al. Development and characterization of a high density SNP genotyping assay for cattle. PLoS One. 2009;4:e5350.
Bolormaa S, Pryce JE, Kemper K, Savin K, Hayes BJ, Barendse W, et al. Accuracy of prediction of genomic breeding values for residual feed intake and carcass and meat quality traits in Bos taurus, Bos indicus, and composite beef cattle. J Anim Sci. 2013;91:3088–104.
Browning SR, Browning BL. High-resolution detection of identity by descent in unrelated individuals. Am J Hum Genet. 2010;86:526–39.
Ziv J, Lempel A. A universal algorithm for sequential data compression. IEEE Trans Inf Theory. 1977;23:337–43.
Shepard RN. Toward a universal law of generalization for psychological science. Science. 1987;237:1317–23.
Perez-Enciso M, Misztal I. Qxpak. 5: old mixed model solutions for new genomics problems. BMC Bioinformatics. 2011;2:202.
Nicholson G, Smith AV, Jonsson F, Gustafsson O, Stefansson K, Donnelly P. Assessing population differentiation and isolation from single-nucleotide polymorphism data. J R Stat Soc Ser B Stat Methodol. 2002;64:695–715.
Lachance J, Tishkoff SA. SNP ascertainment bias in population genetic analyses: why it is important, and how to correct it. Bioessays. 2013;35:780–6.
Porto-Neto LR, Sonstegard TS, Liu GE, Bickhart DM, Da Silva MV, Machado MA, et al. Genomic divergence of zebu and taurine cattle identified through high-density SNP genotyping. BMC Genomics. 2013;14:876.
VanRaden PM, Cooper TA, Wiggans GR, O’Connell JR, Bacheller LR. Confirmation and discovery of maternal grandsires and great-grandsires in dairy cattle. J Dairy Sci. 2013;96:1874–9.
Forni S, Aguilar I, Misztal I. Different genomic relationship matrices for single-step analysis using phenotypic, pedigree and genomic information. Genet Sel Evol. 2011;43:1.
Pemberton TJ, Absher D, Feldman MW, Myers RM, Rosenberg NA, Li JZ. Genomic patterns of homozygosity in worldwide human populations. Am J Hum Genet. 2012;91:275–92.
Fortes MR, Reverter A, Kelly M, McCulloch R, Lehnert SA. Genome-wide association study for inhibin, luteinizing hormone, insulin-like growth factor 1, testicular size and semen traits in bovine species. Andrology. 2013;1:644–50.
Xu L, Bickhart DM, Cole JB, Schroeder SG, Song J, Tassell CP, et al. Genomic signatures reveal new evidences for selection of important traits in domestic cattle. Mol Biol Evol. 2015;32:711–25.
Gianola D, Wu XL, Manfredi E, Simianer H. A non-parametric mixture model for genome-enabled prediction of genetic value for a quantitative trait. Genetica. 2010;138:959–77.
Hayes BJ, Bowman PJ, Chamberlain AJ, Goddard ME. Invited review: genomic selection in dairy cattle: progress and challenges. J Dairy Sci. 2009;92:433–43.
Zhang Q, Lee HG, Han JA, Kim EB, Kang SK, Yin J, et al. Differentially expressed proteins during fat accumulation in bovine skeletal muscle. Meat Sci. 2010;86:814–20.
de Camargo GM, Costa RB, de Albuquerque LG, Regitano LC, Baldi F, Tonhati H. Polymorphisms in TOX and NCOA2 genes and their associations with reproductive traits in cattle. Reprod Fertil Dev. 2014;27:523–8.
Authors’ contributions
AR conceived the study, performed the mathematical analysis, managed and resourced the project. NJH drafted the manuscript and contributed to the development of the concept. LPN and JK performed some of the genetic analyses. LPN, JK and NJH provided biological interpretation. All authors read and approved the final manuscript.
Acknowledgements
We wish to acknowledge funding from Meat and Livestock Australia under project B.BSC.0344. Two colleagues, Miguel Perez-Enciso and Sonja Dominik kindly provided constructive comments and insights on an earlier draft of this manuscript.
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Additional files
12711_2015_158_MOESM1_ESM.docx
Additional file 1. The UNIX scripts for recreating the toy GRM and CRM that are described in Figure 1. This file contains the UNIX scripts allowing the reader to recreate the example GRM and CRM from Figure 1.
12711_2015_158_MOESM2_ESM.tiff
Additional file 2: Figure S1. Comparison of GRM and NCD across the full parameter space including self–self pairs. This figure illustrates the relationship between GRM and NCD across the full parameter space.
12711_2015_158_MOESM3_ESM.jpeg
Additional file 3: Figure S2. Comparison of GRM and NCD without self–self pairs and highlighting each pair with its average Zebu contribution. The clearest relationship between GRM and NCD is for pure bloodline pairs. This figure illustrates how the relationship between GRM and NCD is influenced by the breed characteristics of the pair of animals in question.
12711_2015_158_MOESM4_ESM.xlsx
Additional file 4: Table S1. Genome regions with extreme differences in CEh between BB and TC cattle. This file identifies the genomic regions that are different between BB and TC cattle as determined by sliding window CEh.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

{kind=link}
Cite this article
Hudson, N.J., Porto-Neto, L., Kijas, J.W. et al. Compression distance can discriminate animals by genetic profile, build relationship matrices and estimate breeding values. Genet Sel Evol 47, 78 (2015). https://doi.org/10.1186/s12711-015-0158-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12711-015-0158-9