
Abstract
Background
Male sex reversal syndrome is a rare genetic cause of male infertility with an overall incidence of 1/20,000–1/100,000 males. There is mismatching between the genetic make-up and the apparent clinical features. The clinical presentation of such cases is variable ranging from ambiguous genitalia at birth, failed puberty, up to normal male phenotype with infertility and hypogonadism. The exact molecular and genetic bases of this syndrome are still unclear. Most of the recorded cases were SRY positive (i.e. representing 80–90% of all cases), and they showed translocated SRY gene on the Y chromosome. Moreover, fewer cases of male sex reversal (46, XX) were SRY negative.
Case presentation
Herby, we report a rare case of a 35-year-old infertile male patient who presented with azoospermia, hypergonadotropic hypogonadism, and abnormal classical (46, XX) karyotype, as well as negative FISH for SRY gene. He had a previous negative biopsy and was asking for redoing micro-TESE, whoever he was discouraged as chances to find sperm is eventually nil, and instead, he was prescribed testosterone replacement therapy to correct hypogonadism.
Conclusion
Therefore, any case of non-obstructive azoospermia should be offered genetic testing trying to exclude non-treatable cases and for genetic counseling.
Similar content being viewed by others


1 Background
Male sex reversal syndrome, also known as “46, XX male, 46, XX testicular DSD or de la Chapelle syndrome “ is a disorder of sexual differentiation (DSD) in which the gonadal sex (i.e. male phenotype) is not matching with the chromosomal sex (i.e. 46, XX). It’s over overall incidence about 1:20,000–100,000 of the male population, being a rare cause of male infertility [1]. The first case to be recorded in literature returns back to 1964, where it was first described by Albert de la Chapelle [2], and up till now few cases had been reported.
Interestingly, the exact pathogenesis of this rare syndrome is still unknown, whereas in SRY positive cases (represent 80–90% of cases); it was found that the (SRY) gene was translocated into a sex chromosome or an autosome. On the other hand, in SRY negative cases (represent 10–20% of cases); two mechanisms had been suggested either overexpression of pro-testis genes (i.e. SOX-3, SOX-9, and SOX-10), or decreased expression of anti-testis genes, such as DAX-1, WNT4, and RSPO1 [3,4,5,6].
The clinical presentations of (46, XX) males are variable, whereas patients may present in early life with variable degrees of ambiguous genitalia. Undescended testes, micropenis, and hypospadias, as well as the presence of residual embryonic remnants of the Müllerian duct structures, could be encountered at this stage. Later on, patients may present with failed puberty or infertility with preserved male internal and external genitalia [7].
Due to the rarity and underdiagnosis of such cases, we report this case of SRY negative (46, XX) infertile male aiming at adding to the previously reported cases in the literature hoping to find alternative modalities for managing such cases.
2 Case presentation
A 35‐years‐old male presented with primary infertility for 3 years. His wife was 29-year-old, and all her investigations concerning her fertility potential were found to be completely normal. He was a non-smoker with no special habits of medical importance. His past medical history was irrelevant. He underwent a diagnostic testicular biopsy one year ago elsewhere and unfortunately was negative for sperm or spermatogenic cells. Histopathological revision of the slides showed extensive tubular fibrosis (sclerosis) and hyalinization along with Leydig cells hyperplasia as shown in Fig. 1.

Testicular histopathology showing tubular fibrosis and hyalinization with leydig cell hyperplasia
General examination showed short stature, hypogonadal features, and mild obesity, with Grade (II) gynecomastia. Moreover, local genital examination showed stretched penile length of 9 cm with no hypospadias. In addition, there was a hypoplastic scrotum, bilateral small firm testes, with no varicocele or hydrocele.
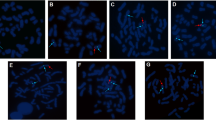
Semen analysis showed repeated low volume azoospermia. Hormone profile showed hypergonadotropic hypogonadism pattern with high FSH: 68.21 mIU/mL, high LH: 59.03 mIU/mL, low total testosterone (T): 0.63 ng/mL and elevated Estradiol (E2): 58 pg/mL along with disturbed T/E2 ratio. Genetic testing showed abnormal karyotyping (46, XX) in all examined cells as shown in Fig. 2, besides that FISH analysis for the SRY gene was found to be negative.

Karyotyping showing (46, XX) genotype
Radiological imaging in the form of transrectal ultrasound (TRUS) examination showed hypoplastic seminal vesicles and prostate along with visualization of both vasa differentia. In addition, scrotal duplex assessment of both testes showed bilateral small-sized testes (Right testis: 2.5 ml, and left testis: 2.1 ml) with atrophic changes with diminished intratesticular vascularity, and no focal lesions could be detected.
After proper counseling, the patient was discouraged from undergoing testicular sperm extraction (TESE) as both testes were atrophic, and chances to find spermatogenic focus are eventually nil due to the absence of genes regulating spermatogenesis. He was prescribed hormone replacement therapy for hypogonadism to correct low testosterone levels for physical and sexual wellbeing in the form of testosterone enanthate 250 mg IM every 3 weeks with regular follow-up every 3 months. Finally, regular self-examination of both testes and scrotal ultrasound assessment was recommended.
3 Discussion
Male sex reversal syndrome (46, X male) is a rare genetic cause of male infertility. The first reported case of 46, XX male was described by Albert de la Chapelle in 1964. Since that time few cases have been reported and most of them were SRY positive (accounting for 80–90% of all cases) [8], whereas the remaining cases were SRY negative [5]. Despite being SRY negative, some cases exhibited normal male phenotype, however up till now the exact molecular and genetic mechanism are still questionable and only hypotheses are postulated [4]. It is characterized by a (46, XX) karyotype with variability in clinical presentation; ambiguous genitalia, failed puberty, and infertility [9].
Most of the reported cases were accidentally discovered in clinical and laboratory evaluation of infertile azoospermic male patients with abnormal karyotyping (46, XX) and hypergonadotropic hypogonadism (i.e. due to primary testicular failure) [10]. Only a single report by Yalcin et al. had described a (46, XX) male with hypogonadotropic hypogonadism that was diagnosed retrospectively by inadequate response to gonadotropin therapy due to associated pituitary macroadenoma [11].
Up till now, none of the reported classical (46, XX) cases had a chance for parenthood due to total testicular atrophy and complete absence of spermatogenic cells due to the absence of genes regulating spermatogenesis located on the Y chromosome especially the “azoospermia factor” (AZF) in the Yq locus. Only chimeric cases with mosaicism (46, XX/46, XY) could have sperm either in the ejaculate or intratesticular and had the chance to father children through assisted reproductive techniques (ARTs) [12,13,14]. Therefore, classical (46, XX) patients seeking fertility should be only offered either adoption or using donor sperm to have a child. Otherwise, HRT should not be missed to correct the clinical manifestation of hypogonadism for physical and sexual wellbeing.
Finally, patients with (46, XX) DSD are at an increased risk of gonadoblastoma on top of the dysgenetic gonads. Therefore, regular self‑examinations should be encouraged along with regular testicular ultrasound examinations for screening as well as early detection of any focal lesions. In addition, testicular biopsy ± orchidectomy is only suggested in cases of suspicious testicular lesions [1, 15].
4 Conclusion
Male sex reversal syndrome is a DSD and a rare genetic cause of male factor infertility with a discrepancy between genotypic and phenotypic sex. Every physician either uro/andrologist or endocrinologist dealing with such cases should be oriented with clinical management and prognosis. Classical (46, XX) male patients seeking fertility should be always discouraged from undergoing TESE and should be offered either adoption or donor sperm for fathering. HRT with testosterone supplementation should be always in mind to correct hypogonadism for physical and sexual wellbeing.
Availability of data and material
Authors can confirm that all relevant data are included in the article and/or its supplementary information files.
Abbreviations
- AZF:
-
Azoospermia factor
- DSD:
-
Disorder of sexual differentiation
- E2:
-
Estradiol
- FISH:
-
Fluorescent in situ hybridizations
- FSH:
-
Follicle-stimulating hormone
- HRT:
-
Hormone replacement therapy
- ICSI:
-
Intracytoplasmic sperm injection
- LH:
-
Luteinizing hormone
- SRY:
-
Sex-determining region on the Y chromosome
- T:
-
Testosterone
- TESE:
-
Testicular sperm extraction
References
Akinsal EC, Baydilli N, Demirtas A, Saatci C, Ekmekcioglu O (2017) Ten cases with 46, XX testicular disorder of sex development: single center experience. Int Braz J Urol 43(4):770–775. https://doi.org/10.1590/s1677-5538.ibju.2016.0505
Chen T, Tian L, Wu F, Xuan X, Ma G, Tang R, Lu J (2019) Clinical and genetic analysis in males with 46, XX disorders of sex development: a reproductive centre experience of 144 cases. Andrologia 51(4):e13232. https://doi.org/10.1111/and.13232
Delachapelle A, Hortling H, Niemi M, Wennstroem J (1964) XX Sex Chromosomes in a human male first case. Acta Med Scand 175:25–28. https://doi.org/10.1111/j.0954-6820.1964.tb04630.x
Grinspon RP, Rey RA (2019) Molecular characterization of XX maleness. Int J Mol Sci 20(23):6089. https://doi.org/10.3390/ijms20236089
Laursen RJ, Alsbjerg B, Vogel I, Gravholt CH, Elbaek H, Lildballe DL, Humaidan P, Vestergaard EM (2018) Case of successful IVF treatment of an oligospermic male with 46, XX/46, XY chimerism. J Assist Reprod Genet 35(7):1325–1328. https://doi.org/10.1007/s10815-018-1194-5
Li TF, Wu QY, Zhang C, Li WW, Zhou Q, Jiang WJ, Cui YX, Xia XY, Shi YC (2014) 46, XX testicular disorder of sexual development with SRY-negative caused by some unidentified mechanisms: a case report and review of the literature. BMC Urol 14:104. https://doi.org/10.1186/1471-2490-14-104
Majzoub A, Arafa M, Starks C, Elbardisi H, Al Said S, Sabanegh E (2017) 46 XX karyotype during male fertility evaluation; case series and literature review. Asian J Androl 19(2):168. https://doi.org/10.4103/1008-682x.181224
Rajender S, Rajani V, Gupta NJ, Chakravarty B, Singh L, Thangaraj K (2006) SRY-negative 46, XX male with normal genitals, complete masculinization and infertility. Mol Hum Reprod 12(5):341–346. https://doi.org/10.1093/molehr/gal030
Ryan NA, Akbar S (2013) A case report of an incidental finding of a 46, XX, SRY-negative male with masculine phenotype during standard fertility workup with review of the literature and proposed immediate and long-term management guidance. Fertil Steril 99(5):1273–1276. https://doi.org/10.1016/j.fertnstert.2012.11.040
Sugawara N, Tokunaga Y, Maeda M, Komaba R, Araki Y (2005) A successful pregnancy outcome using frozen testicular sperm from a chimeric infertile male with a 46, XX/46, XY karyotype: case report. Human Reprod (Oxford, England) 20(1):147–148. https://doi.org/10.1093/humrep/deh587
Sugawara N, Kimura Y, Araki Y (2012) Successful second delivery outcome using refrozen thawed testicular sperm from an infertile male true hermaphrodite with a 46, XX/46 XY karyotype: case report. Hum Cell 25(4):96–99. https://doi.org/10.1007/s13577-012-0054-3
Terribile M, Stizzo M, Manfredi C, Quattrone C, Bottone F, Giordano DR, Bellastella G, Arcaniolo D, De Sio M (2019) 46, XX testicular disorder of sex development (DSD): a case report and systematic review. Medicina (Kaunas) 55(7):371. https://doi.org/10.3390/medicina55070371
Wu QY, Li N, Li WW, Li TF, Zhang C, Cui YX, Xia XY, Zhai JS (2014) Clinical, molecular and cytogenetic analysis of 46, XX testicular disorder of sex development with SRY-positive. BMC Urol 14:70. https://doi.org/10.1186/1471-2490-14-70
Yalcin MM, Ozkan C, Akturk M, Percin FE, Altinova A, Karakoc A, Ayvaz G, Cakir N (2018) 46 XX male syndrome with hypogonadotropic hypogonadism: A case report. Northern Clinics Istanbul 6(3):308–311. https://doi.org/10.14744/nci.2018.57625
Yue F, Zhang H, Xi Q, Jiang Y, Li L, Liu R, Wang R (2019) Molecular cytogenetic analysis and genetic counseling: a case report of eight 46, XX males and a literature review. Mol Cytogenet 12:44. https://doi.org/10.1186/s13039-019-0456-y
Funding
None.
Author information
Authors and Affiliations
Contributions
All authors (M.A., N.I. and N.E.) shared equally in this manuscript including; data collection/interpretation, manuscript writing, and revision. In addition, all authors have approved the final version of the manuscript.
Corresponding author
Ethics declarations
Consent for publication
A written informed consent has been obtained from the patient to collect and publish relevant medical data and figures anonymously.
Competing interests
No competing interest to be declared by all authors.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Abd El Salam, M.A., Ibrahim, N.H. & Eskarous, N.N. A rare case of male sex reversal syndrome (46, XX) with negative SRY gene: a disorder of sexual differentiation (DSD). Afr J Urol 27, 101 (2021). https://doi.org/10.1186/s12301-021-00210-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12301-021-00210-5