
Abstract
Background
Glycogen storage disease type Ia (GSDIa) is an inborn metabolic disorder caused by the deficiency of glucose-6-phospatase-α (G6Pase-α) leading to mitochondrial dysfunction. It remains unclear whether mitochondrial dysfunction is present in patients’ peripheral blood mononuclear cells (PBMC) and whether dietary treatment can play a role. The aim of this study was to investigate mitochondrial function in PBMC of GSDIa patients.
Methods
Ten GSDIa patients and 10 age-, sex- and fasting-time matched controls were enrolled. Expression of genes involved in mitochondrial function and activity of key fatty acid oxidation (FAO) and Krebs cycle proteins were assessed in PBMC. Targeted metabolomics and assessment of metabolic control markers were also performed.
Results
Adult GSDIa patients showed increased CPT1A, SDHB, TFAM, mTOR expression (p < 0.05) and increased VLCAD, CPT2 and citrate synthase activity in PBMC (p < 0.05). VLCAD activity directly correlated with WC (p < 0.01), BMI (p < 0.05), serum malonycarnitine levels (p < 0.05). CPT2 activity directly correlated with BMI (p < 0.05).
Conclusion
Mitochondrial reprogramming is detectable in PBMC of GSDIa patients. This feature may develop as an adaptation to the liver enzyme defect and may be triggered by dietary (over)treatment in the frame of G6Pase-α deficiency. PBMC can represent an adequate mean to assess (diet-induced) metabolic disturbances in GSDIa.
Similar content being viewed by others


Introduction
Glycogen storage disease type Ia (GSDIa) (MIM# 232200) is an inborn disorder of carbohydrate metabolism due to G6PC gene variants. G6PC gene is expressed in the liver, kidney and intestine and encodes for the enzyme glucose 6-phosphatase-α (G6Pase-α). G6Pase-α deficiency leads to impairment of both glycogenolysis and gluconeogenesis and is associated with accumulation of glycogen and fat in the liver and kidney [1]. Hepatomegaly, fasting hypoglycemia, elevated lactate, metabolic acidosis, hyperlipidemia, hyperuricemia and elevated transaminases are common manifestations of GSDIa [2]. Dietary management is the cornerstone of treatment for GSDIa patients. It is based on frequent feedings, including uncooked cornstarch (UCCS)/Glycosade and/or continuous gastric drip-feeding (CNGDF). Despite dietary treatment, GSDIa patients may develop various (long-term) complications, including liver adenomas and chronic kidney disease (CKD) [3]. Medical treatment can be employed to correct secondary metabolic disturbances or delay disease complications [4].
Reduced mitochondria number in GSDIa patients’ liver was first reported in 1980 [5]. More recently, reduced mitochondrial content, abnormal mitochondrial morphology, impaired mitochondrial respiration, and disturbed TCA cycle function have been demonstrated in the liver of G6pc−/− mice [6]. Lower concentration of malate has been found in fibroblasts from one GSDIa patient [7]. Decreased succinate dehydrogenase and NADH dehydrogenase activity have been demonstrated in peripheral lymphocytes from GSDI patients [8]. We have previously reported on indirect markers of tricarboxylic acid (TCA) cycle and fatty acid oxidation (FAO) overload in GSDIa patients’ urine and plasma, respectively [9]. Yet, it remains unclear whether changes in mitochondrial function can be systematically detected in a minimally invasive manner in GSDIa patients’ peripheral blood mononuclear cells (PBMC). It also remains unknown whether dietary treatment can concur to mitochondrial dysfunction in GSDIa.
The aim of the present study was to investigate mitochondrial function in PBMC of GSDIa patients.
Methods
Study design
This was a cross-sectional pilot study. The study protocol was in accordance with the Italian regulations on privacy protection and with the Helsinki Doctrine for Human Experimentation of 1975 as revised in 2013. All participants (or participants’ parents) provided written informed consent prior to inclusion in the study. 10 GSDIa patients were enrolled and compared to 10 age and sex matched controls (Supplemental figure 1). The study was designed to assess in all study participants: 1) the expression of a subset of genes implicated in mitochondrial activity in PBMC, 2) the activity of a subset of proteins involved in mitochondrial FAO and TCA cycle in PBMC and 3) the levels of metabolites associated with FAO or TCA cycle in serum and urine, respectively. Additionally, biochemical markers of metabolic control were assessed in GSDIa patients. All samples were collected in a pre-prandial/fasted state. To minimize the possible effect of diet and fasting time, each adult control (n = 6, control group 1 (C1)) was asked to follow a “GSDIa- like” dietary regimen for 7 days and subsequently have blood collection at the same fasting time as his/her matched patient. “GSDIa- like” dietary regimen followed by each control included same meal schedule, same Kcal/day and same % of daily macronutrients (carbohydrates, lipid, protein) as compared to his/her matched patient; UCCS was substituted with an equivalent amount of natural complex carbohydrates (e.g., pasta, bread, potatoes). As GSDIa patients are dependent on frequent feedings (including overnight) due to G6Pase-α deficiency, to further assess the effect of (short) fasting time, analysis were also performed on one additional blood sample collected after 8-h fasting in adult controls (n = 6, control group 2 (C2)). Due to the difficulty of following a “GSDIa-like” dietary regimen in children, each pediatric control (n = 4) had blood sampling under standard dietary regimen after the same fasting time of his/her age and sex matched patient. Similar to adult controls, in whom fasting time was matched between each patient and his/her matched control, this group is also indicated as C1 (n = 4). Whether C1 refers to adult or pediatric controls is clarified in the text and/or figures and/or tables at each occurrence. Blood and urine samples were collected between 8–11 am depending on each patient’s meal schedule, in order to have the same fasting time for GSDIa patients, C1 and pediatric controls. Blood samples were collected at 8 am for C2.
Subjects
Participants were recruited over a 12-month period. Ten GSDIa patients (6 males and 4 females, median age 20.50 ± 9.44 years, age range: 5.20–34.25 years) were enrolled. Based on their age, patients were classified as children (age < 16 years, n = 4) or adults (age ≥ 16 years, n = 6). The diagnosis of GSDIa was based on mutation analysis of the G6PC gene. Mean age at diagnosis was 0.79 ± 0.21 years. 1 GSDIa patient had GH deficiency and was treated with rhGH. 2 GSDIa patients were treated with ACE-inhibitors. 2 GSDIa patients were treated with both ACE-inhibitors and fenofibrate.
All patients were on dietary treatment (Supplemental Table 1). Each patient received uncooked cornstarch (UCCS), CNGDF or a combination of the two. Dietary regimens varied among different patients according to their families’ requests and attitudes. Carbohydrate intake was 8.82 ± 3.90 g/kg/day. The dose of UCCS was 1.0–1.5 g/kg/meal. Nocturnal carbohydrate intake was 2.21 ± 0.55 g/kg in patients receiving UCCS and 3.59 ± 1.41 g/kg in patients receiving CNGDF. In patients receiving CNGDF, the rate of glucose administration varied according to the patients’ age, ranging from 4 to 10 mg/kg/min.
Ten age- and sex-matched subjects with normal random blood glucose and no history of hypoglycemia were included as healthy control participants.
Clinical and biochemical parameters
The following clinical parameters were recorded: body mass index (BMI), waist circumference (WC). Biochemical markers of metabolic control included: serum glucose, triglycerides (TG), cholesterol, lactate, uric acid, aspartate aminotransferase (AST), alanine aminotransferase (ALT).
Gene expression analysis
Relative expression of a subset of genes implicated in mitochondrial activity was assessed using predeveloped TaqMan assay primers and probes (Thermo Fisher Scientific, Waltham, MA, USA). mRNA isolation was carried out as previously described [10]. One µg of RNA was reverse transcribed in a 20 μl reaction mixture using the High Capacity cDNA Reverse Transcription kit (Applied Biosystems, Foster City, CA, USA) and the resulting cDNA was diluted tenfold in 180 μl nuclease-free water. To detect the expression levels of all investigated genes, real-time quantitative PCR (RT-qPCR) was performed with TaqMan Gene Expression PCR Master Mix (Applied Biosystems, Foster City, CA, USA) on ABI 7900 Real-Time PCR instrument (Thermo Fisher Scientific), according to manufacturer’s instructions. Beta-2-microglobulin (B2M) and hypoxanthine phosphoribosyltransferase 1 (HPRT1) housekeeping genes were used as internal controls. The data were analyzed with the SDS relative quantification software version 1.2.1 (Thermo Fisher Scientific). Relative quantification was performed using the Pfaffl method [10]. Selected genes included:
-
1) genes involved in mitochondrial function: carnitine palmitoyltransferase 1A (CPT1A), succinate dehydrogenase complex flavoprotein subunit A (SDHA), succinate dehydrogenase complex flavoprotein subunit B (SDHB), succinate dehydrogenase complex flavoprotein subunit C (SDHC), ATP-citrate lyase (ACLY), nicotinamide phosphoribosyltransferase (NAMPT);
-
2) genes involved in mitochondrial biogenesis: nuclear respiratory factor 1 (NRF1), transcription factor A mitochondrial (TFAM), uncoupling protein 3 (UCP3), peroxisome proliferative activated receptor, gamma, coactivator 1, Alpha (PCG1α), sirtuin 1 (SIRT1), sirtuin 2 (SIRT2);
-
3) genes involved in intracellular sensing: mechanistic target of rapamycin (mTOR), AKT serine/threonine kinase 1 (AKT1), AKT serine/threonine kinase 2 (AKT2), AKT serine/threonine kinase 3 (AKT3).
A full list of genes and TaqMan assays used in this study is reported in Supplemental Table 2.
Enzyme assays
Proteins involved in mitochondrial FAO and TCA cycle included medium-chain acyl-CoA dehydrogenase (MCAD), very long-chain acyl-CoA dehydrogenase (VLCAD), citrate synthase (CS) and carnitine palmitoyltransferase II (CPT2). Functional testing was performed as previously reported [11,12,13,14].
Metabolomics studies
Blood and urine samples were collected to measure the levels of 26 Acylcarnitines (ACs) and 76 urine organic acids (UOA). Whole blood and serum were collected in serum tubes and stored between 2 and 8 °C. Fresh urine samples were collected in a test tube and stored frozen. All reagents and internal standards used for LC–MS/MS and for GC–MS were as previously described [15].
The ACs, extracted and derivatized to butyl esters as previously described [15, 16], were analyzed on an API 4000 triple quadrupole mass spectrometer (Applied Biosystems-Sciex, Toronto, Canada) coupled with the high performance liquid chromatograph Agilent 1100 series (Agilent Technologies, Waldbronn, Germany).
Creatinine concentration of urine samples was determined by using automatic analysis system BM/Hitachi 904. Organic acids were extracted from urine and analysed on a GC–MS system including an Agilent 7890A (Agilent Technologies, Santa Clara, CA, USA) gas chromatograph and an Agilent 5975C mass spectrometer. Extraction and analysis were performed as previously described [17]; the concentration of organic acids was normalized to the creatinine concentration of the urine sample and expressed as mmol organic acid/mol creatinine.
Statistical analysis
All data in the text or shown in the figures are expressed as mean ± SE. Statistical analysis was performed using Statistical Package for Social Science (SPSS 10 for Windows Update; SPSS Inc., Chicago, Illinois, USA). Since this was a pilot study, no formal sample size calculation was performed. The comparisons between numerical variables were performed by Student’s t-test corrected for Fisher’s exact test. The normality of the distribution was checked by the Shapiro–Wilk test. Due to small sample size (n = 4), the normality of the distribution could not be checked in children; thus, it was assumed that these data points were non normally distributed. Correlation study was performed by Spearman's rank correlation. Statistical significance was set at p < 0.05.
Results
Clinical and biochemical parameters
Clinical and biochemical parameters are summarized in Table 1. As expected, GSDIa patients showed higher BMI, WC, serum cholesterol, TG, lactate, uric acid, AST, ALT levels compared to controls [9, 18, 19].
Abnormalities in mitochondrial metabolism are present in serum and urine of GSDIa patients
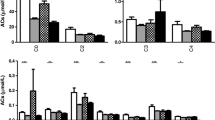
In line with previous report [9], higher serum free carnitine levels were found both in adult GSDIa patients and children compared to age-, sex- and fasting time-matched controls (C1) (Fig. 1A). Adult GSDIa patients also showed higher serum palmitoylcarnitine and stearoylcarnitine concentrations compared to C1. GSDIa children showed higher miristoylcarnitine and palmitoylcarnitine concentrations compared to C1 (Fig. 1B-D). Higher urine pyruvate, ethylmalonate and 2-ketoglutarate excretion were found both in adult GSDIa patients and children compared to C1. Adult GSDIa patients also showed increased urine pyruvate, 2-ketoglutarate, fumarate, ethylmalonate excretion compared to C1 (Fig. 1E-H). These data confirmed that abnormalities in mitochondrial metabolism are present in extra-hepatic tissues in GSDIa.

Serum acylcarnitines (A-D) and urine organic acids (E–H) in GSDIa patients and control groups. For all metabolites mean value is shown for each group of participants. C1: controls who had blood collection after the same fasting time as their matched patients (adults: n = 6, children: n = 4). C14: miristoylcarnitine; C16: palmitoylcarnitine; C18: stearoylcarnitine * p < 0.05; ** p < 0.01; ***p < 0.001
Expression of genes involved in mitochondrial biogenesis, function and intracellular sensing is increased in PBMC of adult GSDIa patients
To ascertain whether mitochondrial dysfunction was also detectable in PBMC of GSDIa patients’, the expression of a subset of key genes involved in mitochondrial activity was assessed. Adult GSDIa patients showed significantly higher CPT1A, SDHB, SDHC, TFAM, mTOR and AKT1 expression compared to age-, sex-, fasting time- and diet-matched controls (C1) and significantly increased CPT1A, SDHB, TFAM, and mTOR expression compared to age-, sex- and not fasting-time/diet-matched controls (C2). CPT1A, SDHC and AKT1 expression was significantly higher in C2 compared to C1 (Fig. 2). Despite not statistically significant, a trend towards increased CPT1A expression in GSDIa children compared to C1 was observed (p = 0.08, supplemental figure 2).

Gene expression analysis in adult GSDIa patients (n = 6) and control groups. For all genes mean value is shown for each group of participants. C1: adult controls who had blood collection after the same fasting time as their matched patient (n = 6). C2: adult controls who had blood collection after 8-h fasting (n = 6).* p < 0.05; ** p < 0.01; ***p < 0.001
Fatty acid oxidation is upregulated in PBMC of GSDIa patients
Since fatty acid oxidation (FAO) and TCA cycle are major mitochondrial functions, we investigated the activity of a subset of proteins involved in these pathways in PBMC. Increased MCAD activity was found in all adult GSDIa patients (range 122.7–426.9%) (Fig. 3A) and 2/4 GSDIa children (range 62.4–147.1%) (Supplemental Fig. 3). Similarly, palmitoyl-CoA oxidation rate as indicator of VLCAD activity was increased in all adult GSDIa patients (range 136.8–214%) (Fig. 3A) and 2/4 children (range 64.9–156%) (Supplemental figure 3). Increased CPT2 activity was found in all adult GSDIa patients (range 144.2–171.1%) (Fig. 3A) and 3/4 children (range 88.8–143.4%) (Supplemental Figure 3). Adult GSDIa patients showed increased VLCAD and CPT2 activity compared to age-, sex-, fasting time- and diet-matched controls (C1) (Fig. 3A). CS activity was assessed in 8/10 GSDIa patients and 8/10 controls (10 adults and 6 children). Adult GSDIa patients showed higher CS activity compared to C1, suggesting mitochondrial proliferation (Fig. 3B). No significant difference in CS activity was observed between children with GSDIa and C1 (Supplemental Figure 3).

FAO enzymes (A) and citrate synthase (B) activity in adult GSDIa patients (n = 6) and controls. FAO enzyme activity is presented as % activity compared to the mean calculated on a pool of healthy subjects. C1: adult controls who had blood collection after the same fasting time as their matched patients (n = 6). For all proteins mean value is shown for each group of participants. * p < 0.05; ** p < 0.01; ***p < 0.001
Mitochondrial markers correlate with dietary data in GSDIa patients
In GSDIa patients a direct correlation was found between: a) VLCAD activity and WC (p < 0.01), BMI (p < 0.05) (Fig. 4A), serum malonycarnitine levels (p < 0.05) and ACLY mRNA levels (p < 0.05) (Fig. 4B) and b) CPT2 activity and BMI (p < 0.05) (Fig. 4C). A direct correlation between VLCAD activity and WC (p < 0.05), BMI (p < 0.05), and ACLY mRNA levels (p < 0.05) as well as between CS activity and ACLY expression (p < 0.05) was found when analyzing adult GSDIa patients separately (Supplemental Figure 4). None of the above-mentioned correlation was found in GSDIa children, in whom a direct correlation between ACLY expression level and serum AST (p < 0.01) and ALT (p < 0.05) was observed (Supplemental Figure 5).

Correlation analysis in GSDIa patients. A Correlation between VLCAD activity and waist circumference (WC, black circles, ρ = 0.89, n = 7 subjects) and BMI (grey triangles, ρ = 0.77, n = 10 subjects). B Correlation between VLCAD activity and serum malonylcarnitine (C3DC, black circles, ρ = 0.67, n = 9 subjects) and ACLY mRNA levels (grey triangles, ρ = 0.66, n = 10 subjects). C Correlation between CPT2 activity and BMI (black circles, ρ = 0.64, n = 10 subjects). * p < 0.05

Correlation analysis in GSDIa patients not receiving continuous nocturnal gastric drip feeding (CNGDF). A Correlation between VLCAD activity and daily carbohydrate (CH) intake (black circles, ρ = -0.79, n = 7 subjects). B Correlation between citrate synthase activity and daily carbohydrate (CH) intake (black circles, ρ = -0.89, n = 6 subjects) and daily caloric intake (grey triangles, ρ =—0.89, n = 6 subjects). * p < 0.05
In GSDIa patients who did not receive CNGDF an inverse correlation was found between: a) VLCAD activity and daily carbohydrate intake (ρ = -0.79, p < 0.05) (Fig. 5A) and b) CS activity and daily carbohydrate intake (ρ = -0.89, p < 0.05) and daily kcal intake (ρ =-0.89, p < 0.05) (Fig. 5B).
Discussion
To the best of our knowledge this is the first study to systematically assess mitochondrial activity in PBMC of GSDIa patients. The expression of a subset of genes implicated in mitochondrial function and biogenesis and intracellular sensing as well as the activity of a subset of proteins involved in mitochondrial FAO and TCA cycle were evaluated in GSDIa patients and age-, and gender- matched healthy controls (C2). To gather information on the possible relationship between mitochondrial (dys)function and dietary treatment, genomic and enzyme activity analysis were also performed in adult age-, gender-, fasting time- and diet-matched controls (C1). Targeted metabolomics was also performed on participants' serum and urine. Adult GSDIa patients showed significantly higher CPT1A, SDHB, SDHC, TFAM, mTOR and AKT1 expression compared to age-, sex-, fasting time- and diet-matched controls (C1) and significantly increased CPT1A, SDHB, TFAM, and mTOR expression compared to age-, sex- and not fasting-time/diet-matched controls (C2). CPT1A, SDHC and AKT1 expression was significantly higher in C2 compared to C1. Increased VLCAD, CPT2 an citrate synthase activity were found in PBMC of adult GSDIa patients. A direct correlation was found between VLCAD and WC, BMI and serum malonylcarnitine levels in adult GSDIa patients. A direct correlation between CPT2 activity and BMI was also found. Accumulation of free carnitine and long-chain acylcarnitines as well as increased urine excretion of various TCA cycle metabolites were found in GSDIa patients.
Metabolomics findings were consistent with previous research reporting accumulation of various serum acylcarnitines and increased urine excretion of various TCA cycle metabolites in GSDIa patients [8, 20]. Notably, the degree of increased urinary excretion was higher for downstream mitochondrial metabolites (i.e. 2-ketoglutarate, fumarate, ethylmalonate) than pyruvate. This likely reflects the multiple pathways towards which excess pyruvate is hijacked (i.e. reduction to lactate, acetyl-CoA synthesis, pyruvate/malate shuttle). Adult GSDIa patients also showed increased CPT1A, SDHB, TFAM, mTOR expression in PBMC. Because the activation of mTOR mediates the fatty acid metabolic reprogramming with upregulation of mitochondrial degradation via PPARα [21, 22] we were not surprised to observe a parallel increase of the corresponding enzymes VLCAD, CPT2 and CS activity in PBMC of GSDIa patients. These data strongly suggest that mitochondrial reprogramming associated with increased oxidative processes for energy production are detectable in PBMC of GSDIa patients.
CPT1A, CPT2 and VLCAD play a key role in the mitochondrial FAO. The ubiquitously expressed CPT1A gene encodes for the CPT1, which is located in the mitochondrial outer membrane and catalyzes the rate-limiting step of the mitochondrial FAO [23, 24]. As CPT1 is inhibited by the cytosolic accumulation of malonyl-CoA [25, 26], a reduction of liver mitochondrial FAO has been postulated in in GSDIa [27]. Consistently, studies in G6pc−/− mice demonstrated reduced cpt1a and acadvl (i.e. the gene encoding for the VLCAD) expression in the liver (and kidney), yet associated with normal cpt2 expression [28,29,30,31]. CPT1 protein over-representation was found in the liver of older G6pc−/− mice [32]. These data may suggest that, although the transport of acyl-residues across the membrane seems to be inhibited as indicated by the enhanced concentration of CPT1 protein, the intramitochondrial activation of fatty acids for FAO appears to be maintained. The upregulated activity of mitochondrial FAO in PBMC from GSDIa patients reported in this work corroborates this hypothesis. We hypothesize that enhanced expression of CPT1A accompanied by the increased VLCAD and CPT2 activity in PBMC may likely represents a mechanism to counter-act malonyl-CoA-induced inhibition of hepatic CPT1 activity in response to high circulating fatty acids [25]. A possible correlation between (increased) expression of genes involved in FAO and residual G6Pase-α activity has been recently reported in G6pc−/− mice [33], supporting this hypothesis. Since 8/10 GSDIa patients carried G6PC mutations which completely abolished G6Pase-α activity [1], a correlation between FAO upregulation and estimated G6Pase-α residual activity could not be assessed in the present study.
Besides the enzyme defect per se, mitochondrial FAO can be affected by the diet. Plasma fatty acids levels from obese individuals are usually elevated [34]. Increased CPT1A expression was reported in PBMC from obese individuals [35, 36] and lymphocytes from patients with type 2 diabetes [37]. Significantly different CPT1A expression was found between the two control groups in the present study (Fig. 2), suggesting a role of dietary treatment in FAO upregulation in GSDIa patients. We hypothesize that an increase in mitochondrial FAO in response to dietary (over) treatment can occur up to a certain threshold in GSDIa patients. Beyond this limit mitochondrial compensation is exceeded and FAO becomes overloaded. This hypothesis is line with experimental data from a computational model of mitochondrial FAO, showing increased FAO flux occurring up to 50 mM palmitoyl-CoA followed by a subsequent decreased flux (together with acyl-CoA accumulation) when the concentration of palmitoyl-CoA was further increased [38]. We speculate that enhanced mitochondrial FAO in PBMC may represent an adaptive endogenous response to counteract lipid overload and lipotoxicity (secondary to defective liver mitochondrial FAO and increased lipogenesis) in GSDIa patients otherwise concurring to the development of metabolic syndrome and insulin-resistance [8, 17]. Similarly to type 2 diabetes, increased lipid flux through the CPT1 may lead to FAO overload over time in GSDIa with the accumulation of long-chain acylcarnitines in PBMC [37]. In this small sample size study, adaptations to the various dietary regimens (i.e. UCCS/Glycosade and CNGDF) could not be compared. As decreased mitochondrial FAO contributes to CKD progression [39] we cannot rule out that such adaptive response may also occur in other tissues possibly playing a role in the development of long-term complications in GSDIa.
Consistent with our hypothesis it is not surprising that increased mTOR, TFAM and SDHB expression as well as increased CS activity were found in PBMC of adult GSDIa patients. mTOR activation concurs to the detrimental effects of fatty acids in various cell types [40, 41]. Particularly, obesity and type 2 diabetes results in mTOR hyperexpression in various tissues [42], including PBMC [43]. TFAM is a key regulator of mitochondrial DNA replication and transcription. Its expression is modulated by a number of factors, including sirtuins and PCG1α [44]. Previous studies showed that the role of TFAM in regulating mitochondrial fuel metabolism and ROS emission is tissue specific [45, 46]. Specifically, muscle-specific TFAM overexpression was associated with increased FAO, CS and TCA cycle activity in mice fed a high-fat diet [47]. Notably, decreased TFAM expression and unchanged mTOR mRNA levels were found in the liver G6pc−/− mice [5, 48]. Data from the present study support the hypothesis that extra-hepatic tissue may deploy an adaptive (i.e. opposite) mitochondrial reprogramming in response to the metabolic abnormalities secondary to the liver G6Pase-α defect. m-TOR modulator AMPK signaling has been shown to be downregulated in the liver of GSDIa mice [48]. Further studies assessing AMPK signaling in GSDIa patients are worthy.
Although no significant difference in mean values was found, higher CPT1A mRNA levels as well as increased VLCAD and CPT2 activity were found in 2/4, 3/4 and 2/4 GSDIa children compared to their matched controls, respectively. These data support the hypothesis that FAO upregulation in GSDIa patients’ PBMC arises from the combination of G6Pase-α deficiency and (long-term) dietary treatment. Additional studies with larger children’s groups are warranted.
The study has some potential limitations. First, a small number of patients with GSDIa was included. Since this was a pilot study, no formal sample size calculation was performed. The limited sample size could have contributed to the lack of statistical significance, especially in children. Second, it is unclear whether the study population adequately reflects the large clinical and biochemical heterogeneity displayed by patients with GSDIa. Particularly, 8/10 GSDIa patients carried the p.Arg83Cys variant (which completely abolish G6Pase-α function). Therefore, effect of genotype on mitochondrial activity could not be assessed. Third, only 3/10 GSDIa patients received CNGDF with the remainders receiving UCCS/Glycosade in the present study. Thus, contribution of mitochondrial adaptation to the various dietary regimens (i.e. CNGDF and UCCS/Glycosade) to mitochondrial reprogramming could not be assessed. Fourth, downstream and side mitochondrial pathways (e.g. AMPK, autophagy) were not investigated in the present study.
In conclusion, we showed that mitochondrial reprogramming (i.e., FAO and TCA cycle hyperactivation) is detectable in PBMC from GSDIa patients as opposed to previously reported liver findings. We hypothesize that mitochondrial reprogramming in PBMC occurs as an adaptation to metabolic abnormalities secondary to the G6Pase-α defect in the liver. Likely, dietary (over)treatment (i.e. frequent feedings, excess UCCS/Glycosade) may trigger mitochondrial reprogramming in the frame of G6Pase-α deficiency-induced pathologic environment. PBMC can represent an adequate mean to assess secondary (diet-induced) disturbances in GSDIa in a minimally invasive way [49].
References
Chou JY, Mansfield BC. Mutations in the glucose-6-phosphatase-alpha (G6PC) gene that cause type Ia glycogen storage disease. Hum Mutat. 2008;29(7):921–30.
Kishnani PS, Austin SL, Abdenur JE, Arn P, Bali DS, Boney A, et al. Diagnosis and management of glycogen storage disease type I: a practice guideline of the American College of Medical Genetics and Genomics. Genet Med. 2014;16(11):e1.
Rake JP, Visser G, Labrune P, Leonard JV, Ullrich K, Smit GP, European Study on Glycogen Storage Disease Type I (ESGSD I). Guidelines for management of glycogen storage disease type I - European Study on Glycogen Storage Disease Type I (ESGSD I). Eur J Pediatr. 2002;161(Suppl 1):S112-9.
Derks TGJ, Rodriguez-Buritica DF, Ahmad A, de Boer F, Couce ML, Grünert SC, et al. Glycogen Storage Disease Type Ia: Current Management Options, Burden and Unmet Needs. Nutrients. 2021;13(11):3828.
Riede UN, Spycher MA, Gitzelmann R. Glycogenosis type I (glucose 6-phosphatase deficiency): I. Ultrastructural morphometric analysis of juvenile liver cells. Pathol Res Pract. 1980;167(1):136–50.
Farah BL, Sinha RA, Wu Y, Singh BK, Lim A, Hirayama M, et al. Hepatic mitochondrial dysfunction is a feature of Glycogen Storage Disease Type Ia (GSDIa). Sci Rep. 2017;20(7):44408.
Hannibal L, Theimer J, Wingert V, et al. Metabolic Profiling in Human Fibroblasts Enables Subtype Clustering in Glycogen Storage Disease. Front Endocrinol (Lausanne). 2020;11:579981 Published 2020 Nov 23.
Kurbatova OV, Izmailova TD, Surkov AN, Namazova-Baranova LS, Poliakova SI, Miroshkina LV, et al. Mitochondrial dysfunction in children with hepatic forms of glycogen storage disease. Vestn Ross Akad Med Nauk. 2014;69(7–8):78–84.
Rossi A, Ruoppolo M, Formisano P, Villani G, Albano L, Gallo G, Crisci D, Moccia A, Parenti G, Strisciuglio P, Melis D. Insulin-resistance in glycogen storage disease type Ia: linking carbohydrates and mitochondria? J Inherit Metab Dis. 2018;41(6):985–95.
Assunto A, Ferrara U, De Luca A, Pivonello C, Lombardo L, Piscitelli A, et al. Isoform-specific NF1 mRNA levels correlate with disease severity in Neurofibromatosis type 1. Orphanet J Rare Dis. 2019;14(1):261.
Tucci S, Herebian D, Sturm M, Seibt A, Spiekerkoetter U. Tissue-specific strategies of the very-long chain acyl-CoA dehydrogenase-deficient (VLCAD-/-) mouse to compensate a defective fatty acid β-oxidation. PLoS ONE. 2012;7(9):e45429.
Hesse J, Braun C, Behringer S, Matysiak U, Spiekerkoetter U, Tucci S. The diagnostic challenge in very-long chain acyl-CoA dehydrogenase deficiency (VLCADD). J Inherit Metab Dis. 2018;41(6):1169–78.
Tucci S, Behringer S, Sturm M, Grünert SC, Spiekerkoetter U. Implementation of a fast method for the measurement of carnitine palmitoyltransferase 2 activity in lymphocytes by tandem mass spectrometry as confirmation for newborn screening. J Inherit Metab Dis. 2019;42(5):850–6.
Tucci S, Wagner C, Grünert SC, Matysiak U, Weinhold N, Klein J, Porta F, Spada M, Bordugo A, Rodella G, Furlan F, Sajeva A, Menni F, Spiekerkoetter U. Genotype and residual enzyme activity in medium-chain acyl-CoA dehydrogenase (MCAD) deficiency: Are predictions possible? J Inherit Metab Dis. 2021;44(4):916–25.
Scolamiero E, Cozzolino C, Albano L, Ansalone A, Caterino M, Corbo G, et al. Targeted metabolomics in the expanded newborn screening for inborn errors of metabolism. Mol BioSyst. 2015;11(6):1525–35.
De Pasquale V, Caterino M, Costanzo M, Fedele R, Ruoppolo M, Pavone LM. Targeted Metabolomic Analysis of a Mucopolysaccharidosis IIIB Mouse Model Reveals an Imbalance of Branched-Chain Amino Acid and Fatty Acid Metabolism. Int J Mol Sci. 2020;21(12):E4211.
Villani GR, Gallo G, Scolamiero E, Salvatore F, Ruoppolo M. “Classical organic acidurias”: diagnosis and pathogenesis. Clin Exp Med. 2017;17(3):305–23.
Melis D, Rossi A, Pivonello R, Salerno M, Balivo F, Spadarella S, et al. Glycogen storage disease type Ia (GSDIa) but not Glycogen storage disease type Ib (GSDIb) is associated to an increased risk of metabolic syndrome: possible role of microsomal glucose 6-phosphate accumulation. Orphanet J Rare Dis. 2015;29(10):91.
Rossi A, Simeoli C, Salerno M, Ferrigno R, Della Casa R, Colao A, Strisciuglio P, Parenti G, Pivonello R, Melis D. Imbalanced cortisol concentrations in glycogen storage disease type I: evidence for a possible link between endocrine regulation and metabolic derangement. Orphanet J Rare Dis. 2020;15(1):99.
Saavedra H, Yu A, Rodriguez-Buritica DF. The use of alanine, free carnitine and IGFBP-1 as potential biomarkers for glycogen storage disease type I. Mol Genet Metab. 2022;136(Supplement 1):S18 ISSN 1096-7192.
Soliman GA. The integral role of mTOR in lipid metabolism. Cell Cycle. 2011;10(6):861–2.
Ricoult SJ, Manning BD. The multifaceted role of mTORC1 in the control of lipid metabolism. EMBO Rep. 2013;14(3):242–51.
Gobin S, Thuillier L, Jogl G, Faye A, Tong L, Chi M, et al. Functional and structural basis of carnitine palmitoyltransferase 1A deficiency. J Biol Chem. 2003;278(50):50428–34.
Houten SM, Violante S, Ventura FV, Wanders RJ. The Biochemistry and Physiology of Mitochondrial Fatty Acid β-Oxidation and Its Genetic Disorders. Annu Rev Physiol. 2016;78:23–44.
Bandsma RH, Smit GP, Kuipers F. Disturbed lipid metabolism in glycogen storage disease type 1. Eur J Pediatr. 2002;161(Suppl 1):S65–9.
Das AM, Lücke T, Meyer U, Hartmann H, Illsinger S. Glycogen storage disease type 1: impact of medium-chain triglycerides on metabolic control and growth. Ann Nutr Metab. 2010;56(3):225–32.
Gjorgjieva M, Oosterveer MH, Mithieux G, Rajas F. Mechanisms by Which Metabolic Reprogramming in GSD1 Liver Generates a Favorable Tumorigenic Environment. JIEMS. 2016.
Monteillet L, Gjorgjieva M, Silva M, Verzieux V, Imikirene L, Duchampt A, et al. Intracellular lipids are an independent cause of liver injury and chronic kidney disease in non alcoholic fatty liver disease-like context. Mol Metab. 2018;16:100–15.
Waskowicz LR, Zhou J, Landau DJ, Brooks ED, Lim A, Yavarow ZA, Kudo T, Zhang H, Wu Y, Grant S, Young SP, Huat BB, Yen PM, Koeberl DD. Bezafibrate induces autophagy and improves hepatic lipid metabolism in glycogen storage disease type Ia. Hum Mol Genet. 2019;28(1):143–54.
Yavarow ZA, Kang HR, Waskowicz LR, Bay BH, Young SP, Yen PM, Koeberl DD. Fenofibrate rapidly decreases hepatic lipid and glycogen storage in neonatal mice with glycogen storage disease type Ia. Hum Mol Genet. 2020;29(2):286–94.
Monteillet L, Labrune P, Hochuli M, Do Cao J, Tortereau A, Miliano AC, et al. Cellular and metabolic effects of renin-angiotensin system blockade on glycogen storage disease type I nephropathy. Hum Mol Genet. 2022;31(6):914–28.
Cangelosi D, Resaz R, Petretto A, Segalerba D, Ognibene M, Raggi F, et al. A Proteomic Analysis of GSD-1a in Mouse Livers: Evidence for Metabolic Reprogramming, Inflammation, and Macrophage Polarization. J Proteome Res. 2019;18(7):2965–78.
Rutten MGS, Derks TGJ, Huijkman NCA, Bos T, Kloosterhuis NJ, van de Kolk KCWA, et al. Modeling Phenotypic Heterogeneity of Glycogen Storage Disease Type 1a Liver Disease in Mice by Somatic CRISPR/CRISPR-associated protein 9-Mediated Gene Editing. Hepatology. 2021;74(5):2491–507.
Mihalik SJ, Goodpaster BH, Kelley DE, Chace DH, Vockley J, Toledo FG, DeLany JP. Increased levels of plasma acylcarnitines in obesity and type 2 diabetes and identification of a marker of glucolipotoxicity. Obesity (Silver Spring). 2010;18(9):1695–700.
Díaz-Rúa R, Palou A, Oliver P. Cpt1a gene expression in peripheral blood mononuclear cells as an early biomarker of diet-related metabolic alterations. Food Nutr Res. 2016;23(60):33554.
Costa A, Reynés B, Konieczna J, Martín M, Fiol M, Palou A, Romaguera D, Oliver P. Use of human PBMC to analyse the impact of obesity on lipid metabolism and metabolic status: a proof-of-concept pilot study. Sci Rep. 2021;11(1):18329.
Nicholas DA, Proctor EA, Agrawal M, Belkina AC, Van Nostrand SC, Panneerseelan-Bharath L, et al. Fatty Acid Metabolites Combine with Reduced β Oxidation to Activate Th17 Inflammation in Human Type 2 Diabetes. Cell Metab. 2019;30(3):447-461.e5.
van Eunen K, Simons SM, Gerding A, Bleeker A, den Besten G, Touw CM, Houten SM, Groen BK, Krab K, Reijngoud DJ, Bakker BM. Biochemical competition makes fatty-acid β-oxidation vulnerable to substrate overload. PLoS Comput Biol. 2013;9(8):e1003186.
Stadler K, Goldberg IJ, Susztak K. The evolving understanding of the contribution of lipid metabolism to diabetic kidney disease. Curr Diab Rep. 2015;15(7):40.
Wang J, Chen Y, Song Q, Griffiths A, Song Z. mTORC1-IRE1α pathway activation contributes to palmitate-elicited triglyceride secretion and cell death in hepatocytes. Exp Biol Med (Maywood). 2020;245(14):1268–79.
Marafie SK, Al-Shawaf EM, Abubaker J, Arefanian H. Palmitic acid-induced lipotoxicity promotes a novel interplay between Akt-mTOR, IRS-1, and FFAR1 signaling in pancreatic β-cells. Biol Res. 2019;52(1):44.
Mao Z, Zhang W. Role of mTOR in Glucose and Lipid Metabolism. Int J Mol Sci. 2018;19(7):2043.
Jung UJ, Seo YR, Ryu R, Choi MS. Differences in metabolic biomarkers in the blood and gene expression profiles of peripheral blood mononuclear cells among normal weight, mildly obese and moderately obese subjects. Br J Nutr. 2016;116(6):1022–32.
Ekstrand MI, Falkenberg M, Rantanen A, Park CB, Gaspari M, Hultenby K, Rustin P, Gustafsson CM, Larsson NG. Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Hum Mol Genet. 2004;13(9):935–44.
Ikeda M, Ide T, Fujino T, Arai S, Saku K, Kakino T, et al. Overexpression of TFAM or twinkle increases mtDNA copy number and facilitates cardioprotection associated with limited mitochondrial oxidative stress. PLoS ONE. 2015;10(3):e0119687.
Vernochet C, Mourier A, Bezy O, Macotela Y, Boucher J, Rardin MJ, et al. Adipose-specific deletion of TFAM increases mitochondrial oxidation and protects mice against obesity and insulin resistance. Cell Metab. 2012;16(6):765–76.
Koh JH, Johnson ML, Dasari S, LeBrasseur NK, Vuckovic I, Henderson GC, et al. TFAM Enhances Fat Oxidation and Attenuates High-Fat Diet-Induced Insulin Resistance in Skeletal Muscle. Diabetes. 2019;68(8):1552–64.
Gautam S, Zhang L, Arnaoutova I, Lee C, Mansfield BC, Chou JY. The signaling pathways implicated in impairment of hepatic autophagy in glycogen storage disease type Ia. Hum Mol Genet. 2020;29(5):834–44.
de Mello VD, Kolehmanien M, Schwab U, Pulkkinen L, Uusitupa M. Gene expression of peripheral blood mononuclear cells as a tool in dietary intervention studies: What do we know so far? Mol Nutr Food Res. 2012;56(7):1160–72.
Funding
Nutricia Metabolics Young Investigator Grant 2019 to A.R.
Author information
Authors and Affiliations
Contributions
AR wrote the first draft of the manuscript and neither an honorarium or grant, or other forms of payment was given to anyone to produce the manuscript. MR, PS, GP, DM critically reviewed the manuscript. All authors made substantial contributions to the conception or design of the work or the acquisition, analysis or interpretation of data. AR, PS, GP and DM were involved in the clinical investigation and follow-up of the patients. AA, CR performed gene expression analysis. ST performed enzyme assays. MR, MC, FP performed metabolomics studies. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the Ethical Committee of the Universita` di Napoli “Federico II, protocol number 151/05.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Supplemental figure 1. Study design and subjects. FAO: fatty acid oxidation; PBMC: peripheral blood mononuclear cells; TCA: tricarboxylic acid * GSDIa patients and C1.
Additional file 2:
Supplemental figure 2. CPT1A expression in GSDIa childrenand pediatric controls. Each control had blood sampling under standard dietary regimen after the same fasting time of his/her age and sex matched patient. Mean value is shown for each group of participants. p=0.08.
Additional file 3:
Supplemental figure 3.FAO enzymes in GSDIa children. FAO enzyme activity is presented as % activity compared to the mean calculated on a pool of healthy subjects.Citrate synthase activity in GSDIa childrenand healthy controls. For all proteins mean value is shown for each group of participants. C1: pediatric controls who had blood collection after the same fasting time as their matched patients.
Additional file 4:
Supplemental figure 4. Correlation analysis in adult GSDIa patients.Correlation between VLCAD activity and waist circumferenceand BMI.Correlation between VLCAD activity and ACLY mRNA levels.Correlation between citrate synthase activity and ACLY mRNA levels. * p< 0.05.
Additional file 5:
Supplemental figure 5. Correlation between ACLY mRNA levels and serum ASTand ALT. *p< 0.05; **p< 0.01.
Additional file 6:
Supplemental table 1. Clinical information of GSDIa patients.
Additional file 7:
Supplemental table 2. The list of TaqMan assays used for gene expression analysis by real-time quantitative PCR.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rossi, A., Assunto, A., Rosano, C. et al. Mitochondrial reprogramming in peripheral blood mononuclear cells of patients with glycogen storage disease type Ia. Genes Nutr 18, 10 (2023). https://doi.org/10.1186/s12263-023-00729-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12263-023-00729-y