
Abstract
Background
Folic acid and its derivates, known as folates, are chemoprotective micronutrients of great interest because of their essential role in the maintenance of health and genomic integrity. The supplementation of folic acid during pregnancy has long been known to reduce the risk of neural tube defects (NTDs) in the foetus. Folate metabolism can be altered by many factors, including adequate intake through diet. Folate deficiency can compromise the synthesis, repair and methylation of DNA, with deleterious consequences on genomic stability and gene expression. These processes are known to be altered in chronic diseases, including cancer and cardiovascular diseases.
Main body
This review focuses on the association between folate intake and the risk of childhood leukaemia. Having compiled and analysed studies from the literature, we show the documented effects of folates on the genome and their role in cancer prevention and progression with particular emphasis on DNA methylation modifications. These changes are of crucial importance during pregnancy, as maternal diet has a profound impact on the metabolic and physiological functions of the foetus and the susceptibility to disease in later life. Folate deficiency is capable of modifying the methylation status of certain genes at birth in both animals and humans, with potential pathogenic and tumorigenic effects on the progeny. Pre-existing genetic polymorphisms can modify the metabolic network of folates and influence the risk of cancer, including childhood leukaemias. The protective effects of folic acid might be dose dependent, as excessive folic acid could have the adverse effect of nourishing certain types of tumours.
Conclusion
Overall, maternal folic acid supplementation before and during pregnancy seems to confer protection against the risk of childhood leukaemia in the offspring. The optimal folic acid requirements and supplementation doses need to be established, especially in conjunction with other vitamins in order to determine the most successful combinations of nutrients to maintain genomic health and wellbeing. Further research is therefore needed to uncover the role of maternal diet as a whole, as it represents a main factor capable of inducing permanent changes in the foetus.
Similar content being viewed by others


Background
Nutrition represents one of the leading preventable risk factors in the development of cancer, accounting for nearly 10% of total cases in the UK [1]. The concept of chemoprevention in the insurgence of cancer was first introduced by Sporn [2, 3] and has since been employed in the attempt to arrest, retard or reverse tumorigenic processes by the use of biological and nutritional compounds such as phytochemicals (e.g. carotenoids, allyl sulphur compounds, glucosinolates, isothiocyanates and polyphenols) and vitamins. Diet and the assimilation of micronutrients, therefore, have a substantial impact on health and disease.
Folic acid (or vitamin B9) and its derivatives, collectively known as folates, are chemoprotective micronutrients of great interest belonging to the B vitamin group. They are water-soluble vitamins that function as co-factors in a variety of enzymatic reactions within the cell. Folates are naturally found in leafy vegetables, eggs, legumes, bran and dry fruit, whereas the synthetic form, which has a higher bioavailability, is added as a food fortifier in cereal grain products or used as a dietary supplementation [4, 5].
Folate is essential for the correct functioning of the human body and the maintenance of genomic integrity. Within the cell, it participates in two types of reactions, biosynthesis of nucleotides and methylation reactions, which are required in the fundamental biological processes of DNA synthesis, DNA repair and DNA methylation [6,7,8]. Folic acid is also needed for correct functioning of mitochondria and maintenance of mitochondrial DNA (mtDNA) [9] (Fig. 1).

The relationship between folic acid deficiency and the genomic instability. Low levels of folic acid interfere with the normal biological functions of the cell, compromising the genomic stability for both nuclear and mitochondrial DNA. Folic acid deficiency affects the synthesis of nucleotides, causing DNA damage that cannot be repaired efficiently because of an overall decrease in the nucleotide availability. As methyl donors, the unavailability of folates can alter DNA methylation, causing changes in gene expression and compromising the integrity of chromosomes. Deprivation of folic acid also causes oxidative stress in the cell with consequences affecting the integrity of mitochondrial DNA
Folate deficiency is associated with several disorders such as neural tube defects (NTDs) and malformations in the developing foetus, as well as cardiovascular diseases, depression and Alzheimer’s disease in adults [10,11,12]. Given its role in the maintenance of genomic stability, insufficient folic acid also appears to be involved in the insurgence of cancer [13]. In addition to the above, there is evidence that folic acid deficiency during pregnancy could represent a risk factor for the development of childhood leukaemia in the offspring.
The chemoprotective properties of folic acid with respect to cancer initiation and progression will be explained. In particular, the effects of folate deficiency on genomic health and its potential impact on the insurgence of childhood leukaemia will be discussed. Several studies and findings have been compiled to provide a comprehensive view of current research covering these aspects:
-
1.
The association between folate intake and the risk of childhood leukaemia, by considering studies on the efficacy of folic acid supplementation before or during pregnancy in preventing the disease in the offspring
-
2.
The role of folic acid in influencing the methylation patterns of DNA, an inheritable epigenetic regulatory mechanism capable of altering gene expression in the progeny
The biological role of folates in the cell
Folates are involved in the cellular one-carbon metabolism, acting as one-carbon carriers for the transfer of methyl groups. The metabolically active form of folic acid is tetrahydrofolate (THF), which can be converted into other structurally related molecules, each having a specific function and forming a complex network of enzymatic reactions (Fig. 2).

Intracellular network of reactions in the metabolism of folates. A variety of related compounds derived from folic acid have specific biochemical functions. Folic acid is first converted into dihydrofolate (DHF) and subsequently into tetrahydrofolate (THF) by the enzyme DHF reductase (DHFR). 5,10-methyleneTHF and 10-formylTHF are responsible for the synthesis of purines; 5,10-methyleneTHF also mediates the conversion of dUMP to dTMP for the synthesis of thymidine, catalysed by the enzyme thymidine synthase (TS). 5-methylTHF is involved for methylation reactions, particularly in the conversion of homocysteine to methionine for the formation of SAM, a major methyl donor for DNA (figure taken from [6])
Nucleotide biosynthesis
Folates are responsible for the synthesis of purines and pyrimidines for the correct assembly of DNA and RNA. In particular, they mediate the only known reaction for the de novo synthesis of thymidine, which consists in the conversion of deoxyuridine monophosphate (dUMP) to deoxythymidine monophosphate (dTMP) by the transfer of a methyl group [14, 15].
Methylation reactions
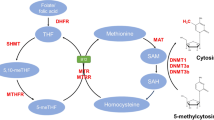
As methyl group donors, folates are involved in methylation reactions including DNA methylation, a major epigenetic process capable of influencing gene expression. Specific loci known as CpG islands are methylated through the modification of cytosine to form 5-methylcytosine by an enzymatic reaction involving the transfer of a methyl group from S-adenosylmethionine (SAM) [16]. Folates are involved in the conversion of homocysteine to methionine required for the formation of SAM. This is achieved by a one-carbon transfer to vitamin B12, which will then be used for the methylation of homocysteine to methionine. By the addition of adenine to methionine, SAM is formed, which is the main methyl donor for DNA [17].
Folate deficiency and genomic damage
Abnormalities in nucleotide biosynthesis and methylation reactions are capable of affecting DNA synthesis, DNA repair and DNA methylation, potentially leading to genomic instability of the cell (Fig. 3).

Processes and consequences contributing to the genomic instability of the cell. Altered DNA synthesis, DNA repair and DNA methylation compromise the genomic stability of the cell. DNA damage and chromosomal abnormalities result from an incorrect assembly of DNA and the inability to repair errors efficiently, potentially affecting the next generation of cells or leading to cell death. If DNA methylation is disrupted, epigenetic changes affecting gene expression can occur, including incorrect methylation patterns or changes in chromatin structure
In conditions of folic acid depletion, the conversion of dUMP cannot proceed, leading to its abnormal intracellular accumulation and misincorporation in the DNA instead of thymine [18]. Excessive uracil content causes point mutations, single- and double-strand DNA breaks, chromosome breaks and formation of micronuclei [19, 20]. The inability to provide nucleotides adequately renders DNA synthesis inefficient, compromising the regenerative power of tissues [21] and the ability to repair DNA efficiently [22].
Disruption of methylation reactions is not limited to altered gene expression. Demethylation of centromeres causes structural and functional aberrations within the chromosome, notably during mitosis, leading to abnormal chromosome segregation and aneuploidy [23]. Low folate has also been shown to alter the expression of microRNAs (miRNAs) due to abnormal DNA methylation [24]. miRNAs are non-coding oligonucleotide RNAs detaining an important role in gene regulation. When abnormally expressed, they can gain oncogenic properties and initiate tumorigenesis [25] and NTDs [26]. Studies on the expression profile of miRNAs have been conducted to uncover a possible role in leukaemogenesis [27] because of their regulatory function in haematopoiesis [28]. The data collected from in vitro experiments on DNA damage and aberrant DNA methylation are consistent with results obtained in vivo in mice subjected to a diet with extreme folic acid deficiency [29,30,31].
Nuclear abnormalities and aneuploidy
The discovery of Howell-Jolly bodies in erythrocytes with megaloblastic anaemia was the first evidence of chromosome damage caused by folic acid deficiency [32,33,34]. Howell-Jolly bodies are chromosomal fragments that lag behind during anaphase, a form of micronuclei present solely in erythrocytes. Studies on sufferers of Chron disease found that their frequency was linked with low serum and intracellular folate [35].
Similar results have been obtained for human lymphocytes cultured in folic acid-depleted media. The formation of micronuclei and other nuclear abnormalities such as nucleoplasmic bridges and nuclear buds were observed, which are indicators of genomic damage [36, 37]. Under similar conditions, particularly in the absence of thymidine, fragile sites and chromosome breakage also occur, which are attributable to the misincorporation of uracil in the DNA [19,39,, 20, 38–40]. Increased chromosomal damage and the inability to efficiently repair aberrant hypoxanthine bases was observed in deprived lymphocyte cultures when compared to folate-replete controls [22]. Accumulation of S phases and subsequent induction of apoptosis has also been described [41].
Wang et al. [42] and Beetstra et al. [43] revealed an association between folic acid deficiency and the incidence of aneuploidies of chromosomes 17 and 21, often observed in breast cancer and leukaemia. This was also observed for chromosome 8 [44], found abnormal in number in prostate, skin and breast cancers, cholesteatoma and leukaemia [45,46,47,48]. In particular, trisomy 8 has been reported as a recurrent chromosomal abnormality in acute myeloid leukaemia (AML) and hence considered a cytogenetic marker of AML [49, 50]. Ni et al. [44] demonstrated that aneuploidy of chromosome 8 is influenced by folic acid deficiency similarly to chromosome 17. In addition, riboflavin deficiency seemed not to aggravate the risk of aneuploidy, which is coherent with other studies in which riboflavin had no influence on the formation of micronuclei [51].
Telomere abnormalities
The consumption of vital micronutrients through diet, most notably folic acid, represents an important determinant for the maintenance of telomere length and health [52,53,54]. Telomeres consist of repeated hexameric sequences (TTAGGG) found at the end of chromosomes together with other accessory proteins. This complex is called “telosome” and protects chromosome ends from degradation and chemical damage, which could result in chromosomal instability and breakage [55]. Because of their chemical composition rich in thymidine, chromosome ends are thought to be susceptible to uracil misincorporation and impaired repair.
Telomeric abnormalities and dysfunction have been associated with ageing, cancer and degenerative diseases [55,56,57]. Telomeric shortening is commonly observed in initial stages of tumorigenesis, but in some cancers, excessive telomerase activity can cause abnormally elongated telomeres [56]. Epigenetic changes, especially DNA methylation, can also disrupt the normal maintenance of the telomere length by interfering with the expression of the machinery involved [58,59,60]. These effects are particularly critical during foetal life, as early programming events in utero can have a permanent impact on health and susceptibility to disease.
Low maternal folic acid levels have been shown to cause shorter telomeres in the newborn, although the clinical implications in later life are not known [61, 62]. In population studies, low folate status was associated with telomere abnormalities in a non-linear manner. Considering that the normal concentration in population ranges between 13.5 and 45.3 nmol/L [63], with a plasma concentration below 11.6 nmol/L, telomere length increased, whereas shortening was observed above the median [64]. In vitro, human lymphoblasts cultured in folic acid-deficient medium showed that chromosomes undergo an initial telomeric elongation followed by a rapid shortening, both being indicators of genomic instability [54].
Damage to mitochondrial DNA
Genomic instability is not limited to nuclear DNA damage, as also mitochondrial DNA (mtDNA) appears to be affected by lack of folic acid. Folates possess anti-oxidant properties against reactive oxygen species (ROS) and lipid peroxidation and are able to process harmful metabolites, preventing mitochondrial toxicity [9, 65, 66]. Folic acid deficiency can cause oxidative stress and initiate apoptosis [67] with deleterious consequences on the integrity of mtDNA. Most studies, conducted on rodents, found an increased number of mtDNA deletions when folic acid was deficient [68,69,70,71,72]. Studies on different tissue types in rats demonstrated that mtDNA deletions in the liver induced by ageing are associated to folate levels, indicating that folic acid supplementation reduces the occurrence of these deletions by two- to threefolds when compared to depleted rats [72]. This has also been observed in rat lymphocytes, where folic acid deficiency causes increased deletions by nearly fourfolds [71].
Accumulation of somatic deletions and mutations in the mtDNA may play a role in tumorigenesis, as mtDNA is subjected to detrimental factors originating from the environment, including dietary deficits [73]. Damage to mtDNA is capable of inducing reactions that can damage the nuclear DNA as well, by both genetic and epigenetic mechanisms, including methylation, chromatin remodelling and signalling pathways [74]. This is particularly relevant in cancer because damaged mitochondria can induce changes in the genome and in the surrounding microenvironment, both capable of creating an advantageous setting for tumorigenesis [75].
In haematological malignancies, somatic mutations and changes in mitochondrial gene expression have mainly been observed in myelodysplastic syndromes [76, 77]. Acquired mitochondrial mutations have also been found in the bone marrow of nearly 40% of patients with adult leukaemia, when compared to normal tissue with no mutations [78]. These results are similar to other studies on different cancers, where only a fraction of patients with the same malignancy showed mtDNA mutations [79,80,81,82,83].
Acquired mtDNA deletions and low folate status have been associated with incidence of hepatocellular carcinoma, suggesting that carcinogenesis is attributable to the deleterious effects of folate deficiency on the stability of both nuclear and mitochondrial DNA [84]. Studies on the impact of nutrients and correlated mitochondrial damage in the offspring are limited, but it is known that mtDNA in the placenta responds to environmental factors. For instance, airborne pollution is capable of damaging the mitochondria and altering methylation patterns, potentially leading to adverse health outcomes for both mother and foetus [85, 86].
The mitochondrial genome is highly variable among different populations, which poses a limitation when searching possible pathogenic mutations [87, 88]. Also, the consequences depend on the locus and the extent of the mutations, as certain mutations seem to have no effect on the function, metabolism or phenotype of the mitochondrion or the cell as a whole [78]. In fact, potentially pathogenic mutations are found in the population, although the vast majority is not clinically expressing the disease [89].
Genomic damage caused by folate deficiency and ionising radiations
To better comprehend the extent of the DNA damage inflicted by folic acid deficiency, comparative studies have been conducted on the similarities with ionising radiation damage. Courtemanche et al. [90] showed that cultured lymphocytes depleted of folic acid or irradiated with high-dose radiation presented similar DNA double-strand breaks and were both subjected to decreased proliferation, cell cycle arrest and apoptosis. However, differences in gene expression analysis indicated that, although similar, the damage seems to arise following different pathways. The exact mechanism that the cell employs in response to nutritional deficits has been studied in Caenorhabditis elegans [91]. Folate deficiency also seems to be an enhancing factor of DNA damage resulting from radiation in vivo [92], which led to the investigation of folic acid acting as a radioprotective agent in vitro [93].
Folates as modulators of DNA methylation
Methylation of CpG islands at the promoter or regulatory regions of a given gene represses its expression, whereas unmethylation allows the transcription to proceed (Fig. 4). The exact mechanism is not completely understood. The positioning of the methyl group is thought to physically impede the binding of the transcription machinery and block the activation of that gene. However, exceptions to this mechanism exist. For instance, methylation seems to activate the transcription of the gene for telomerase [94]. Hypo- and hypermethylation of CpG islands is of particular interest in carcinogenesis, as tumour-suppressor genes and proto-oncogenes become erroneously inactivated and activated, respectively, causing uncontrolled growth [95, 96].

Methylation can modify the expression of genes. Methylated regions upstream the start site of transcription are a signal of gene repression. Conversely, the absence of methylation promotes the transcription of the gene
DNA methylation forms distinctive patterns in different tissue types that can be transmitted to the next generation of cells to perpetuate the expression profile for the appropriate differentiation of tissues. However, it appears that the maintenance of the methylation per se is not sufficient to guarantee the stable expression pattern throughout the entire genome [97]. The poor availability of folates as a source of methyl groups can influence the ability to maintain the correct methylation patterns, causing mainly hypomethylation of genomic DNA [98, 99], reversible upon re-supplementation [17,101,, 100–102]. The disruption of methylation patterns by lack of folates can also occur by hypermethylation of certain loci, although this might seem counterintuitive. Different cell types and specific loci in the genome respond to folate status in different ways, thus affecting gene expression by various mechanisms [16, 103, 104].
Folates and DNA methylation patterns in foetal life
Folate deficiency has been shown to influence the methylation status of certain genes at birth in both animals and humans. Recent findings in rats clarified that gestational folic acid intake can influence the progeny’s gene expression and this occurs in an organ-specific manner, with the brain being the most susceptible to these changes [105].
This is particularly relevant in conception, which occurs before the DNA methylation pattern re-programming. The critical window period to determine the DNA methylation pattern is right after fertilisation, when the zygote is in between the morula and blastocyst phases [106]. The periconceptional period is also crucial for the establishment of correct genetic, epigenetic and metabolic settings for successful reproduction, and nutrients of the one-carbon metabolism play a pivotal role in these processes [107].
Gonseth et al. [108] demonstrated that exposure to folates 12 months before conception is capable of modifying DNA methylation in healthy newborns. Genes involved in neural crest development (TFAP2A), acute myeloid leukaemia (STX11), cystic kidney disease (CYS1) and other genes involved in foetal facial (OTX2) and neural development were sensitive to the modifying effects of folate deficiency on methylation patterns. Deficiency in periconceptional folate consumption was associated with methylation of promoter regions of these genes resulting in downregulation, with potential tumorigenic effects on genes with tumour-suppressor properties. Steegers-Theunissen et al. [109] examined the use of folic acid during pregnancy and the methylation status of the offspring. Maternal folic acid supplementation resulted in a 4.5% increase in the methylation of insulin-like growth factor 2 (IGF-2) in the progeny, which is positively associated with maternal SAM levels, inversely proportional to weight at birth, and it has been linked with several chronic disturbances. Chang et al. [110] compared DNA methylation levels in different tissues from aborted foetuses affected by NTDs with normal healthy controls. The brain tissue was hypomethylated in foetuses with NTDs. The folate in serum was also lower in mothers whose foetuses presented NTDs, further confirming the association between folic acid levels and methylation status.
However, the hypothesis that folate deficiency acts as limiting factors during the embryonic development is controversial. In fact, studies on the association between the exposure to folates during foetal life and the global DNA methylation status at birth in folate-replete populations have yielded contrasting results. It is plausible that the relationship between folic acid consumption and its effects on DNA methylation is dose dependent, meaning that populations with severe folic acid deficiency are more prone to the modifying effects of folate on methylation than folate-replete populations [108, 111]. This is coherent with studies from Heijmans et al. [112] and Tobi et al. [113] who investigated the epigenetic alterations in individuals who suffered hunger during war with a severe deficiency in folic acid. Heijmans et al. found that IGF-2 was hypomethylated, and Tobi et al. identified hypomethylation in INSIGF and hypermethylation in IL-10, LEP, ABCA1, GNASAS and MEG3, which are associated with growth and metabolic disorders.
Contribution of diet in the epigenetic control of gene expression
Maternal diet during pregnancy can influence many physiological and metabolic functions in the foetus and determine susceptibility in the development of diseases later in life [114]. The intrauterine process of foetal programming has been associated with physical, psychological, metabolic and pharmacological stress factors. These exogenous events are capable of inducing permanent changes in the foetus, with potential post-natal consequences [115]. The developing foetus is subjected to an extensive programme of cell division, growth and differentiation. Differentiation requires a precise organisation of gene expression, which is regulated by DNA methylation, chromatin structure and other genetic and epigenetic determinants [116]. Any interference with these events of epigenetic modification can permanently compromise gene expression and have serious repercussions on the development of the organism [115, 117].
Experiments on agouti mice proved that nutrition can induce epigenetic changes in the offspring by interfering with normal DNA methylation, influencing the susceptibility to disease [118,119,120]. The coat colour of agouti mice is determined by the methylation of the agouti gene, which is strongly dependent on the maternal diet [120]. This has led to the hypothesis that the risk of developing diseases might partially be attributable to parental nutrition [121,122,123]. An intracisternal A particle (IAP) is present in the upstream region of the agouti gene. The gene is regulated by the promoter activity of IAP by methylation. The availability of methyl groups from the maternal diet during pregnancy increases the methylation of DNA in the IAP gene, inducing phenotypic changes that affect the gene expression of the progeny (i.e. change in coat colour) [118, 119]. In a separate study, Waterland et al. [121] identified similar loci in the human genome with distinct methylation patterns depending on the season of birth.
In mice, a paternal diet low in protein induced epigenetic changes in the progeny, when compared to controls with an equilibrated protein intake. The changes affected the methylation of DNA in the liver in the genes involved in the biosynthesis of lipids and cholesterol [124]. Paternal fat-rich diets reduce the methylation status of the Il13ra2 gene in pancreatic cells in female progeny [59]. A protein-depleted diet in the mother during pregnancy has also been shown to dysregulate DNA methylation and gene expression but was reversible if folic acid was supplemented [114].
Folates and cancer
The capability of folates of modulating DNA methylation, repair and synthesis suggests a role in tumorigenesis. Folate deficiency has been proposed as a contributing factor in the development of cervical, lung, breast, brain, colorectal and pancreatic cancers [6,126,, 14, 125–127]. In particular, a large number of studies on humans have shown that a higher intake of folates through diet and higher plasma levels of folate are associated with lower risk of developing polyps and tumour in the colon [21,129,130,, 128–131].
Interaction of folates with cancer-related genes
It is now well established that mutations in the BRCA1 and BRCA2 genes can result in defective DNA repair and lead to the development of breast cancer. As folate deprivation is linked with chromosomal abnormalities, it has been proposed that carriers of germinal mutations to these genes are more susceptible to the genomic damage caused by low levels of folic acid, when compared to individuals without the mutation. The study revealed that the mutation of BRCA1 and BRCA2 did not increase the magnitude of damage caused by folic acid deficiency. Moderate folic acid deficiency showed a greater chromosomal instability than the damage observed in mutation carriers [132].
Lack of folic acid can also affect the genetic and epigenetic integrity of p53, which is a well-characterised tumour suppressor [29, 133]. Inactivating mutations of p53 have been described in cancer [134]. However, it appears that structural damage to the gene caused by folate deficiency has no effect on gene transcription, expression and function and has not prevented folic acid-deprived lymphocytes from undergoing apoptosis via p53 activation [41, 135]. In rats, p53 mRNA transcript levels in the colon were decreased by folic acid deprivation, but no changes were observed in its methylation status, suggesting that colon cancer tumorigenesis is initiated by other mechanisms than p53 inactivation [136].
Excessive folic acid intake and cancer progression
It is generally agreed that high levels of folic acid confer protective action lowering the risk of malignant disease [137, 138]. According to the World Health Organization (WHO) guidelines regarding folic acid intake for pregnant women, over-supplementation has no negative outcomes on health [139], which has however been contested in a number of studies. Selhub and Rosenberg [140] discussed various issues in discordance with the claims from the WHO, while Smith et al. [98] highlighted the possibility of folic acid supplementation not being beneficial for the population as a whole. For instance, an excessive intake of folic acid may nourish tumours that have already initiated. The stage of the malignancy and the time period of folic acid supplementation could make a substantial difference in whether folic acid acts as suppressor of malignant transformation or promoter of growth for established tumours [21, 141]. This has been particularly evident for colorectal cancer [21, 142] and prostate cancer [143]. In vitro studies on colon cancer cell lines showed that folic acid supplementation in medium is capable of changing DNA methylation patterns, as well as altering the proliferative capacity and phenotype of these cells in culture. This is of particular importance in understanding the role of folic acid supplementation on the behaviour of tumours in terms of phenotypic changes, motility and invasion [144]. Other studies have reported an increased proliferation due to altered DNA methylation in response to excessive folic acid supplementation [6, 145].
Folate intake, haematopoiesis and leukaemia
Folate in haematopoiesis
Folic acid and vitamin B12 are involved in the correct production and maturation of blood cells from haematopoietic stem cells (HSCs), particularly for the production of red blood cells [146]. Bills et al. [147] proved that folic acid-depleted mice showed an ineffective haematopoiesis, with changes affecting the maturation of progenitor cells.
This is evident in megaloblastic anaemia, a disorder of erythropoiesis that arises from folic acid or vitamin B12 deficiency. This haematological disorder is characterised by the accumulations of enlarged, immature erythroblasts in the bone marrow due to an impaired DNA synthesis and capacity to divide [148], highlighting the role of these vitamins in the maintenance of genomic health.
The prenatal origin of leukaemia
Leukaemia is characterised by the presence of acquired chromosomal rearrangements that are confined to the diseased bone marrow cells. Retrospective studies carried out on Guthrie cards of individuals that developed leukaemia during childhood showed the presence of genetic rearrangements in those archival samples. This constitutes the proof that the initiating genetic events leading to leukaemia were already present in utero [149, 150]. Furthermore, secondary events must occur after birth to promote the cancer through clonal expansion [151]. Notwithstanding, the exact aetiology of childhood leukaemia is largely unknown. The causes for the arising of chromosomal translocations and the methylation patterns that accompany different leukaemia phenotypes are still not fully understood. Leukaemogenesis appears to be a result of genetic and environmental factors, occurring prior and during pregnancy, but also post-partum and later in life (Fig. 5). Several environmental factors have been identified, including exposure to radiation, certain chemicals or infections [152, 153]. The role of nutrition is also gaining importance, as micronutrients are capable of interfering with the genomic stability, and studies are already being undertaken to assess the extent of this influence [154, 155].

Multi-hit hypothesis for the insurgence of childhood leukaemia. The initiation of childhood leukaemia requires multiple oncogenic events to occur, with the first event occurring in utero and a second hit occurring after birth or later in life. The first event may be called the predisposing condition at the genomic level that is necessary but not sufficient for the insurgence of cancer. Leukaemia is initiated when a second event promotes abnormal cell proliferation
Maternal intake of folic acid and childhood leukaemia
The current recommended consumption and supplementation of folic acid intake is summarised in Fig. 6. The recommended daily assumption (RDA) of folic acid is 400 μg/day in adults, 600 μg/day in pregnant women and 500 μg/day during lactation. The Estimated Average Requirements (EARs) are 320, 520 and 450 μg/day, respectively [5]. Folic acid supplementation is required during pregnancy, first and foremost for the prevention of NTDs [156]. It appears that consumption of folic acid and multivitamin supplements reduces the risk of leukaemia in the offspring, one of the most prevalent cancers in children under 15 years of age [157]. Although research on this topic has been contradictory [158,159,160,161,162], recent publications suggest that folic acid does play a protective role against these malignancies.

Current recommendations for folic acid nutritional requirements per age. The Institute of Medicine (Food and Nutrition Board) determined the recommended daily assumption (RDA) and Estimated Average Requirements (EARs) for folate according to age and status. For infants (0–12 months), adequate intake (AI) is presented instead, as the influence of maternal nutrients can interfere with experimental evidence for RDA and EAR. During pregnancy and lactation, the requirements are higher, as the developing foetus needs high levels of folic acid. At doses above 200 μg, unmetabolised folate is detectable in plasma
Metayer et al. [163] considered 12 studies conducted in ten countries from 1980 to 2012 to extract data on the intake of folic acid and other vitamins in women during and before pregnancy. According to this epidemiological study, the risk of acute lymphoblastic leukaemia (ALL) is lowered when folic acid plus multivitamins were taken during the year before and during pregnancy. Similarly, the risk for AML decreased with folic acid intake before and during pregnancy, but other vitamin intake seemed not to have the same protective effect. In particular, folic acid supplementation seemed to have a more evident protective effect towards AML than ALL, with a diminished incidence of 32% for AML versus 21% for ALL. Vitamin intake was associated with a decreased risk for ALL of 15%, whereas no correlation was found with the risk of developing AML. Despite the lack of information on dosage, it is clear that folic acid and other vitamins are required during the entire course of pregnancy, from the very early stages, as has long been known in regard to NTDs [156]. At doses above 200 μg/day, unmetabolised folic acid is observed in circulation [164]. Since the long-term effects of this phenomenon have not been investigated, the recommended dosage (Fig. 6) remains questionable, as well as the potential risks of the metabolite in plasma [140, 165, 166].
In a recent matched case-control study from Singer et al. [167], maternal intake of folate, vitamins B12 and B6, riboflavin and methionine 1 year before pregnancy were examined, confirming their protective action against ALL and AML. Similar findings are shown in a more comprehensive study focusing on the maternal dietary quality as a whole, rather than individual nutrients [168]. Other case-control studies [169] have investigated the risk for ALL only, the most common form of childhood leukaemia, finding reduced risk for the disease linked to maternal supplementation of folic acid.
Role of genetic polymorphisms in the development of childhood leukaemia
Polymorphisms in genes participating in folate metabolism are capable of modifying the pathways through which folic acid is processed but also influence the susceptibility to cancer (leukaemia, lung, breast, brain, colorectal, gastric, head and neck malignancies) and other diseases. Mutations have been identified in the MTHFR, TS, MTR and MTRR genes (Fig. 2) [170]. The most common variants of the MTHFR gene are C677T, characterised by a C → T transition, and A1298C, where an A → C transversion occurs; both result in reduced enzymatic activity [171, 172].
A number of studies have been undertaken to understand the role of polymorphisms in genes involved in folic acid metabolism in the development of childhood leukaemia, yielding discordant results. While a reduced susceptibility to ALL has been disproven by some [173,174,175,176,177,178], others showed a significant decrease in its insurgence linked to the presence of polymorphisms [179,180,181,182]. Five studies on the MTHFR 677CT polymorphism did not find any significant difference in the susceptibility to ALL [173,174,175,176,177], whereas four studies proved that the polymorphism conferred a diminished risk in the development of the malignancy in the Brazilian and Western European populations [179,180,181,182]. Similar results come from the MTHFR 677TT variant. While most studies agree on an insignificant difference in risk for ALL, the Korean population showed to have an increased susceptibility due to the polymorphism [173, 176, 182].
The A1298C variant shows similar conflicting results. Some data suggest that it plays a role in increasing the risk for childhood leukaemia [182, 183], although this has been disproven by others [173, 176, 180]. The MTRR A66G polymorphism seemed to confer a reduced risk of developing ALL in most populations apart from the Korean population, where the variant did not show to influence the susceptibility to the disease [184]. Polymorphisms of the TSER, the promoter enhancer region of the TS gene, did not show to be a significant factor in determining the risk for ALL [185,186,187,188].
Milne et al. [161] investigated the risk of ALL in a population-based case-control study, in which 392 individuals with polymorphisms in the MTHFR, MTR, MTRR and CBS genes and 535 controls were analysed, investigating both parents and progeny. The risk of developing ALL seemed to be diminished in offspring of fathers with the genotype MTRR 66GG. The authors concluded that the risk of ALL in the progeny due to polymorphisms in genes of the folate metabolism can be influenced by maternal intake of folic acid, although more research is needed.
Limited information is available about the susceptibility to AML. Most studies conclude that there is no association between polymorphisms of MTHFR and the risk of AML [189,190,191]. Certain studies, however, have proven that the polymorphic variant MTHFR C677T is a risk factor for the development of AML in the Romanian and Asian populations [192,193,194,195].
The differences in these results could be attributable to genetic and environmental variants in geographic areas and populations [178, 196].
Most studies on the risk of leukaemia in children have focused on the role of single nutrients, rather than a wider understanding of the maternal diet as a whole. It is likely that several factors and interactions of nutrients are accountable for the development of the disease. Further research is needed to uncover the multiple aspects of the diet and their effects on the health of the mother and progeny. Paternal periconceptional folic acid supplementation has also been considered in some studies, but results are inconclusive [197, 198].
Conclusions
Correct nutrition represents one of the most crucial protective factors for many pathological conditions including cancer. Certain nutrients have proven to detain a protective and preventive role for the maintenance of human health. However, the interaction between specific nutrients and development of disease is complex and might be influenced by additional dietary and environmental factors, indicating that the association between maternal folic acid intake and methylation status of the progeny is non-linear and non-definitive.
Folates are required within the cell for synthesis, repair and methylation of nuclear and mitochondrial DNA. The studies examined in this review suggest that folic acid deficiency is capable of interfering with these processes, which are extremely important during foetal development and, if altered, are capable of promoting carcinogenesis and the development of other diseases. In particular, DNA damage due to lack of folates can lead to the formation of chromosomal abnormalities, which are considered a hallmark in cancer and leukaemia. Overall, folic acid intake during pregnancy seems to provide protection against the risk of leukaemia in the offspring.
The exact optimal dosage is still unclear, considering that excessive intake of folic acid might have serious drawbacks, including the nourishment of pre-existing cancers or pre-cancerous conditions. However, the overall dietary plan in pregnancy, disease and non-disease conditions should consider the interaction of multiple nutrients, rather than the accurate dosage of single compounds. Genomic damage and cancer growth can be potentially controlled through a combination of different micronutrients and correct dosage. In this respect, personalised nutrition could be implemented not only to provide a better diet plan during pregnancy but also as an adjuvant to anti-cancer therapies for specific tumours.
Abbreviations
- AI:
-
Adequate intake
- ALL:
-
Acute lymphoblastic leukaemia
- AML:
-
Acute myeloid leukaemia
- dTMP:
-
Deoxythymidine monophosphate
- dUMP:
-
Deoxyuridine monophosphate
- EARs:
-
Estimated Average Requirements
- HSCs:
-
Haematopoietic stem cells
- IAP:
-
Intracisternal insertion A particle
- mtDNA:
-
Mitochondrial DNA
- NTDs:
-
Neural tube defects
- RDA:
-
Recommended daily assumption
- ROS:
-
Reactive oxygen species
- SAM:
-
S-Adenosylmethionine
- THF:
-
Tetrahydrofolate
- WHO:
-
World Health Organization
References
Parkin DM. The fraction of cancer attributable to lifestyle and environmental factors in the UK in 2010. Br J Cancer. 2011;105:S2–5.
Sporn MB. Approaches to prevention of epithelial cancer during the preneoplastic period. Cancer Res. 1976;36(7 Part 2):2699–702.
Sporn MB, Dunlop NM, Newton DL, Smith JM. Prevention of chemical carcinogenesis by vitamin A and its synthetic analogs (retinoids). InFederation Proc. 1976;35(6):1332–8.
Berry RJ, Bailey L, Mulinare J, Bower C, Dary O. Fortification of flour with folic acid. Food Nutr Bull. 2010;31(1_suppl1):S22–35.
Institute of Medicine (US) Standing Committee on the Scientific Evaluation of Dietary Reference Intakes. Dietary reference intakes for thiamin, riboflavin, niacin, vitamin B6, folate, vitamin B12, pantothenic acid, biotin, and choline. Washington (DC): National Academies Press (US); 2000.
Liu J, Lynne WR. Folate and one-carbon metabolism and its impact on aberrant DNA methylation in cancer. Adv Genet. 2010;71:79.
Nijhout HF, Reed MC, Ulrich CM. Mathematical models of folate-mediated one-carbon metabolism. Vitam Hormones. 2008;79:45–82.
Wagner C. Biochemical role of folate in cellular metabolism. Clin Res Regul Aff. 2001;18(3):161–80.
Depeint F, Bruce WR, Shangari N, Mehta R, O’Brien PJ. Mitochondrial function and toxicity: role of B vitamins on the one-carbon transfer pathways. Chem Biol Interact. 2006;163(1):113–32.
Erickson JD. Folic acid and prevention of spina bifida and anencephaly. 10 years after the US Public Health Service recommendation. MMWR. Recommendations and reports: morbidity and mortality weekly report. MMWR Recomm Rep. 2002;51(RR-13):1–3.
Lamprecht SA, Lipkin M. Chemoprevention of colon cancer by calcium, vitamin D and folate: molecular mechanisms. Nat Rev Cancer. 2003;3(8):601–14.
Holmquist C, Larsson S, Wolk A, de Faire U. Multivitamin supplements are inversely associated with risk of myocardial infarction in men and women—Stockholm Heart Epidemiology Program (SHEEP). J Nutr. 2003;133(8):2650–4.
Duthie SJ. Folate and cancer: how DNA damage, repair and methylation impact on colon carcinogenesis. J Inherit Metab Dis. 2011;34(1):101–9.
Choi SW, Mason JB. Folate status: effects on pathways of colorectal carcinogenesis. J Nutr. 2002;132(8):2413S–8S.
Bailey LB, Gregory JF. Polymorphisms of methylenetetrahydrofolate reductase and other enzymes: metabolic significance, risks and impact on folate requirement. J Nutr. 1999;129(5):919–22.
Crider KS, Yang TP, Berry RJ, Bailey LB. Folate and DNA methylation: a review of molecular mechanisms and the evidence for folate’s role. Adv Nutr. 2012;3(1):21–38.
Choi SW, Friso S, Keyes MK, Mason JB. Folate supplementation increases genomic DNA methylation in the liver of elder rats. Br J Nutr. 2005;93(01):31–5.
Eto I, Krumdieck CL. Role of vitamin B12 and folate deficiencies in carcinogenesis. Essential nutrients in carcinogenesis. New York: Plenum Press. 1986;313–330.
Blount BC, Mack MM, Wehr CM, MacGregor JT, Hiatt RA, Wang G, Wickramasinghe SN, Everson RB, Ames BN. Folate deficiency causes uracil misincorporation into human DNA and chromosome breakage: implications for cancer and neuronal damage. Proc Natl Acad Sci. 1997;94(7):3290–5.
Blount BC, Ames BN. DNA damage in folate deficiency. Baillière’s Clin Haematol. 1995;8(3):461–78.
Kim YI. Folate and colorectal cancer: an evidence-based critical review. Mol Nutr Food Res. 2007;51(3):267–92.
Libbus BL, Borman LS, Ventrone CH, Branda RF. Nutritional folate-deficiency in Chinese hamster ovary cells: chromosomal abnormalities associated with perturbations in nucleic acid precursors. Cancer Genet Cytogenet. 1990;46(2):231–42.
Zingg JM, Jones PA. Genetic and epigenetic aspects of DNA methylation on genome expression, evolution, mutation and carcinogenesis. Carcinogenesis. 1997;18(5):869–82.
Marsit CJ, Eddy K, Kelsey KT. MicroRNA responses to cellular stress. Cancer Res. 2006;66(22):10843–8.
He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, Powers S, Cordon-Cardo C, Lowe SW, Hannon GJ, Hammond SM. A microRNA polycistron as a potential human oncogene. Nature. 2005;435(7043):828–33.
Shookhoff JM, Gallicano GI. A new perspective on neural tube defects: folic acid and microRNA misexpression. Genesis. 2010;48(5):282–94.
Zanette DL, Rivadavia F, Molfetta GA, Barbuzano FG, Proto-Siqueira R, Falcão RP, Zago MA, Silva-Jr WA. miRNA expression profiles in chronic lymphocytic and acute lymphocytic leukemia. Braz J Med Biol Res. 2007;40(11):1435–40.
Chen CZ, Lodish HF. MicroRNAs as regulators of mammalian hematopoiesis. Seminars in immunology. Acad Press. 2005;17(2):155–65.
Kim YI, Pogribny IP, Basnakian AG, Miller JW, Selhub J, James SJ, Mason JB. Folate deficiency in rats induces DNA strand breaks and hypomethylation within the p53 tumor suppressor gene. Am J Clin Nutr. 1997;65(1):46–52.
Pogribny IP, Basnakian AG, Miller BJ, Lopatina NG, Poirier LA, James SJ. Breaks in genomic DNA and within the p53 gene are associated with hypomethylation in livers of folate/methyl-deficient rats. Cancer Res. 1995;55(9):1894–901.
Pogribny IP, Muskhelishvili L, Miller BJ, James SJ. Presence and consequence of uracil in preneoplastic DNA from folate/methyl-deficient rats. Carcinogenesis. 1997;18(11):2071–6.
Discombe G. L’origine des corps de Howell-Jolly et des anneaux de Cabot. Sangre. 1948;29(262):270.
Koyama S. Studies on Howell-Jolly body. Acta Haematol Japan. 1960;23:20–5.
Bessis M, Lessin LS, Beutler E. Morphology of the erythron, Hematology. 3rd ed. New York: McGraw-Hill; 1983. p. 257–79.
Everson RB, Wehr CM, Erexson GL, MacGregor JT. Association of marginal folate depletion with increased human chromosomal damage in vivo: demonstration by analysis of micronucleated erythrocytes. J Natl Cancer Inst. 1988;80(7):525–9.
Lindberg HK, Wang X, Järventaus H, Falck GC, Norppa H, Fenech M. Origin of nuclear buds and micronuclei in normal and folate-deprived human lymphocytes. Mutat Res. 2007;617(1):33–45.
Fenech M, Crott JW. Micronuclei, nucleoplasmic bridges and nuclear buds induced in folic acid deficient human lymphocytes—evidence for breakage–fusion-bridge cycles in the cytokinesis-block micronucleus assay. Mutat Res. 2002;504(1):131–6.
Sutherland GR. Heritable fragile sites on human chromosomes I. Factors affecting expression in lymphocyte culture. Am J Hum Genet. 1979;31(2):125.
Jacky PB, Beek B, Sutherland GR. Fragile sites in chromosomes: possible model for the study of spontaneous chromosome breakage. Science. 1983;220(4592):69–70.
Reidy JA, Zhou X, Chen AT. Folic acid and chromosome breakage I. Implications for genotoxicity studies. Mutat Res Lett. 1983;122(2):217–21.
Lin HL, Chen CJ, Tsai WC, Yen JH, Liu HW. In vitro folate deficiency induces apoptosis by a p53, Fas (Apo-1, CD95) independent, bcl-2 related mechanism in phytohaemagglutinin-stimulated human peripheral blood lymphocytes. Br J Nutr. 2006;95(05):870–8.
Wang XU, Thomas P, Xue J, Fenech M. Folate deficiency induces aneuploidy in human lymphocytes in vitro—evidence using cytokinesis-blocked cells and probes specific for chromosomes 17 and 21. Mutat Res. 2004;551(1):167–80.
Beetstra S, Thomas P, Salisbury C, Turner J, Fenech M. Folic acid deficiency increases chromosomal instability, chromosome 21 aneuploidy and sensitivity to radiation-induced micronuclei. Mutat Res. 2005;578(1):317–26.
Ni J, Lu L, Fenech M, Wang X. Folate deficiency in human peripheral blood lymphocytes induces chromosome 8 aneuploidy but this effect is not modified by riboflavin. Environ Mol Mutagen. 2010;51(1):15–22.
Mark HF, Afify AM, Werness BA, Das S, Mark S, Samy M. Trisomy 8 in stage I and stage III ovarian cancer detected by fluorescence in situ hybridization. Exp Mol Pathol. 1999;66(1):76–81.
Rummukainen J, Kytölä S, Karhu R, Farnebo F, Larsson C, Isola JJ. Aberrations of chromosome 8 in 16 breast cancer cell lines by comparative genomic hybridization, fluorescence in situ hybridization, and spectral karyotyping. Cancer Genet Cytogenet. 2001;126(1):1–7.
Bofin AM, Ytterhus B, Fjøsne HE, Hagmar BM. Abnormal chromosome 8 copy number in cytological smears from breast carcinomas detected by means of fluorescence in situ hybridization (FISH). Cytopathology. 2003;14(1):5–11.
Yildirim MS, Ozturk K, Acar H, Arbag H, Ulku CH. Chromosome 8 aneuploidy in acquired cholesteatoma. Acta Otolaryngol. 2003;123(3):372–6.
Maserati E, Aprili F, Vinante F, Locatelli F, Amendola G, Zatterale A, Milone G, Minelli A, Bernardi F, Lo Curto F, Pasquali F. Trisomy 8 in myelodysplasia and acute leukemia is constitutional in 15–20% of cases. Genes Chromosom Cancer. 2002;33(1):93–7.
Paulsson K, Johansson B. Trisomy 8 as the sole chromosomal aberration in acute myeloid leukemia and myelodysplastic syndromes. Pathol Biol. 2007;55(1):37–48.
Kimura M, Umegaki K, Higuchi M, Thomas P, Fenech M. Methylenetetrahydrofolate reductase C677T polymorphism, folic acid and riboflavin are important determinants of genome stability in cultured human lymphocytes. J Nutr. 2004;134(1):48–56.
Paul L. Diet, nutrition and telomere length. J Nutr Biochem. 2011;22(10):895–901.
Moores CJ, Fenech M, O’Callaghan NJ. Telomere dynamics: the influence of folate and DNA methylation. Ann N Y Acad Sci. 2011;1229(1):76–88.
Bull CF, Mayrhofer G, O’Callaghan NJ, Au AY, Pickett HA, Low GK, Zeegers D, Hande MP, Fenech MF. Folate deficiency induces dysfunctional long and short telomeres; both states are associated with hypomethylation and DNA damage in human WIL2-NS cells. Cancer Prev Res. 2014;7(1):128–38.
Fenech M. Nutriomes and personalised nutrition for DNA damage prevention, telomere integrity maintenance and cancer growth control. In Advances in Nutrition and Cancer. Springer Berlin Heidelberg; 2014:427-441.
Svenson U, Roos G. Telomere length as a biological marker in malignancy. Biochim Biophys Acta. 2009;1792(4):317–23.
Blackburn EH. Telomeres: no end in sight. Cell. 1994;77(5):621–3.
Blasco MA. The epigenetic regulation of mammalian telomeres. Nat Rev Genet. 2007;8(4):299–309.
Ng SF, Lin RC, Laybutt DR, Barres R, Owens JA, Morris MJ. Chronic high-fat diet in fathers programs [bgr]-cell dysfunction in female rat offspring. Nature. 2010;467(7318):963–6.
Gonzalo S, Jaco I, Fraga MF, Chen T, Li E, Esteller M, Blasco MA. DNA methyltransferases control telomere length and telomere recombination in mammalian cells. Nat Cell Biol. 2006;8(4):416–24.
Louis-Jacques AF, Salihu HM, King LM, Paothong A, Sinkey RG, Pradhan A, Riggs BM, Siegel EM, Salemi JL, Whiteman VE. A positive association between umbilical cord RBC folate and fetal TL at birth supports a potential for fetal reprogramming. Nutr Res. 2016;36(7):703–9.
Entringer S, Epel ES, Lin J, Blackburn EH, Buss C, Shahbaba B, Gillen DL, Venkataramanan R, Simhan HN, Wadhwa PD. Maternal folate concentration in early pregnancy and newborn telomere length. Ann Nutr Metab. 2015;66(4):202–8.
WHO. Serum and red blood cell folate concentrations for assessing folate status in populations. Vitamin and Mineral Nutrition Information System. Geneva: World Health Organization; 2015. p. 1–5.
Paul L, Cattaneo M, D’angelo A, Sampietro F, Fermo I, Razzari C, Fontana G, Eugene N, Jacques PF, Selhub J. Telomere length in peripheral blood mononuclear cells is associated with folate status in men. J Nutr. 2009;139(7):1273–8.
Rezk BM, Haenen GR, van der Vijgh WJ, Bast A. Tetrahydrofolate and 5-methyltetrahydrofolate are folates with high antioxidant activity. Identification of the antioxidant pharmacophore. FEBS Lett. 2003;555(3):601–5.
Joshi R, Adhikari S, Patro BS, Chattopadhyay S, Mukherjee T. Free radical scavenging behavior of folic acid: evidence for possible antioxidant activity. Free Radic Biol Med. 2001;30(12):1390–9.
Huang RF, Yaong HC, Chen SC, Lu YF. In vitro folate supplementation alleviates oxidative stress, mitochondria-associated death signalling and apoptosis induced by 7-ketocholesterol. Br J Nutr. 2004;92(06):887–94.
Fenech M. Folate (vitamin B9) and vitamin B12 and their function in the maintenance of nuclear and mitochondrial genome integrity. Mutat Res. 2012;733(1):21–33.
Branda RF, Brooks EM, Chen Z, Naud SJ, Nicklas JA. Dietary modulation of mitochondrial DNA deletions and copy number after chemotherapy in rats. Mutat Res. 2002;501(1):29–36.
Chou YF, Yu CC, Huang RF. Changes in mitochondrial DNA deletion, content, and biogenesis in folate-deficient tissues of young rats depend on mitochondrial folate and oxidative DNA injuries. J Nutr. 2007;137(9):2036–42.
Chou YF, Huang RF. Mitochondrial DNA deletions of blood lymphocytes as genetic markers of low folate-related mitochondrial genotoxicity in peripheral tissues. Eur J Nutr. 2009;48(7):429–36.
Crott JW, Choi SW, Branda RF, Mason JB. Accumulation of mitochondrial DNA deletions is age, tissue and folate-dependent in rats. Mutat Res. 2005;570(1):63–70.
Ormazabal A, Casado M, Molero-Luis M, Montoya J, Rahman S, Aylett SB, Hargreaves I, Heales S, Artuch R. Can folic acid have a role in mitochondrial disorders? Drug Discov Today. 2015;20(11):1349–54.
Chinnery PF, Elliott HR, Hudson G, Samuels DC, Relton CL. Epigenetics, epidemiology and mitochondrial DNA diseases. Int J Epidemiol. 41(1):177–187.
Wallace DC. Mitochondria and cancer. Nat Rev Cancer. 2012;12(10):685–98.
Schildgen V, Wulfert M, Gattermann N. Impaired mitochondrial gene transcription in myelodysplastic syndromes and acute myeloid leukemia with myelodysplasia-related changes. Exp Hematol. 2011;39(6):666–75.
Wulfert M, Küpper AC, Tapprich C, Bottomley SS, Bowen D, Germing U, Haas R, Gattermann N. Analysis of mitochondrial DNA in 104 patients with myelodysplastic syndromes. Exp Hematol. 2008;36(5):577–86.
He L, Luo L, Proctor SJ, Middleton PG, Blakely EL, Taylor RW, Turnbull DM. Somatic mitochondrial DNA mutations in adult-onset leukaemia. Leukemia. 2003;17(12):2487–91.
Penta JS, Johnson FM, Wachsman JT, Copeland WC. Mitochondrial DNA in human malignancy. Mutat Res. 2001;488(2):119–33.
Fliss MS, Usadel H, Caballero OL, Wu L, Buta MR, Eleff SM, Jen J, Sidransky D. Facile detection of mitochondrial DNA mutations in tumors and bodily fluids. Science. 2000;287(5460):2017–9.
Jeronimo C, Nomoto S, Caballero OL, Usadel H, Henrique R, Varzim G, Oliveira J, Lopes C, Fliss MS, Sidransky D. Mitochondrial mutations in early stage prostate cancer and bodily fluids. Oncogene. 2001;20(37):5195.
Jones JB, Song JJ, Hempen PM, Parmigiani G, Hruban RH, Kern SE. Detection of mitochondrial DNA mutations in pancreatic cancer offers a “mass”-ive advantage over detection of nuclear DNA mutations. Cancer Res. 2001;61(4):1299–304.
Kirches E, Krause G, Warich‐Kirches M, Weis S, Schneider T, Meyer‐Puttlitz B, Mawrin C, Dietzmann K. High frequency of mitochondrial DNA mutations in glioblastoma multiforme identified by direct sequence comparison to blood samples. Int J Cancer. 2001;93(4):534–8.
Wu MY, Kuo CS, Lin CY, Lu CL, Huang RF. Lymphocytic mitochondrial DNA deletions, biochemical folate status and hepatocellular carcinoma susceptibility in a case-control study. Br J Nutr. 2009;102(5):715.
Janssen BG, Munters E, Pieters N, Smeets K, Cox B, Cuypers A, Fierens F, Penders J, Vangronsveld J, Gyselaers W, Nawrot TS. Placental mitochondrial DNA content and particulate air pollution during in utero life. Environ Health Perspect. 2012;120(9):1346.
Janssen BG, Byun HM, Gyselaers W, Lefebvre W, Baccarelli AA, Nawrot TS. Placental mitochondrial methylation and exposure to airborne particulate matter in the early life environment: an ENVIR ON AGE birth cohort study. Epigenetics. 2015;10(6):536–44.
Van Oven M, Kayser M. Updated comprehensive phylogenetic tree of global human mitochondrial DNA variation. Hum Mutat. 2009;30(2):E386–94.
Herrnstadt C, Elson JL, Fahy E, Preston G, Turnbull DM, Anderson C, Ghosh SS, Olefsky JM, Beal MF, Davis RE, Howell N. Reduced-median-network analysis of complete mitochondrial DNA coding-region sequences for the major African, Asian, and European haplogroups. Am J Hum Genet. 2002;70(5):1152–71.
Elliott HR, Samuels DC, Eden JA, Relton CL, Chinnery PF. Pathogenic mitochondrial DNA mutations are common in the general population. Am J Hum Genet. 2008;83(2):254–60.
Courtemanche C, Huang AC, Elson-Schwab I, Kerry N, Ng BY, Ames BN. Folate deficiency and ionizing radiation cause DNA breaks in primary human lymphocytes: a comparison. FASEB J. 2004;18(1):209–11.
Ortbauer M, Ripper D, Fuhrmann T, Lassi M, Auernigg‐Haselmaier S, Stiegler C, König J. Folate deficiency and over-supplementation causes impaired folate metabolism: regulation and adaptation mechanisms in Caenorhabditis elegans. Mol Nutr Food Res. 2016;60:949–56.
Branda RF, Blickensderfer DB. Folate deficiency increases genetic damage caused by alkylating agents and γ-irradiation in Chinese hamster ovary cells. Cancer Res. 1993;53(22):5401–8.
Padula G, Ponzinibbio MV, Seoane AI. Possible radioprotective effect of folic acid supplementation on low dose ionizing radiation-induced genomic instability in vitro. 2016.
Guilleret I, Yan P, Grange F, Braunschweig R, Bosman FT, Benhattar J. Hypermethylation of the human telomerase catalytic subunit (hTERT) gene correlates with telomerase activity. Int J Cancer. 2002;101(4):335–41.
Taberlay PC, Jones PA. DNA methylation and cancer. Epigenetics and Disease. Springer Basel; 2011:1-23.
Gaudet F, Hodgson JG, Eden A, Jackson-Grusby L, Dausman J, Gray JW, Leonhardt H, Jaenisch R. Induction of tumors in mice by genomic hypomethylation. Science. 2003;300(5618):489–92.
Jones PA, Liang G. Rethinking how DNA methylation patterns are maintained. Nat Rev Genet. 2009;10(11):805–11.
Smith AD, Kim YI, Refsum H. Is folic acid good for everyone? Am J Clin Nutr. 2008;87(3):517–33.
Cravo M, Fidalgo P, Pereira AD, Gouveia-Oliveira A, Chaves P, Selhub J, Mason JB, Mira FC, Leitao CN. DNA methylation as an intermediate biomarker in colorectal cancer: modulation by folic acid supplementation. Eur J Cancer Prev. 1994;3(6):473–80.
Bohnsack BL, Hirschi KK. Nutrient regulation of cell cycle progression. Annu Rev Nutr. 2004;24:433–53.
Pufulete M, Al-Ghnaniem R, Khushal A, Appleby P, Harris N, Gout S, Emery PW, Sanders TA. Effect of folic acid supplementation on genomic DNA methylation in patients with colorectal adenoma. Gut. 2005;54(5):648–53.
Rampersaud GC, Kauwell GP, Hutson AD, Cerda JJ, Bailey LB. Genomic DNA methylation decreases in response to moderate folate depletion in elderly women. Am J Clin Nutr. 2000;72(4):998–1003.
Hayashi I, Sohn KJ, Stempak JM, Croxford R, Kim YI. Folate deficiency induces cell-specific changes in the steady-state transcript levels of genes involved in folate metabolism and 1-carbon transfer reactions in human colonic epithelial cells. J Nutr. 2007;137(3):607–13.
Jhaveri MS, Wagner C, Trepel JB. Impact of extracellular folate levels on global gene expression. Mol Pharmacol. 2001;60(6):1288–95.
Ly A, Ishiguro L, Kim D, Im D, Kim SE, Sohn KJ, Croxford R, Kim YI. Maternal folic acid supplementation modulates DNA methylation and gene expression in the rat offspring in a gestation period-dependent and organ-specific manner. J Nutr Biochem. 2016;33:103–10.
Kelsey G. Epigenome changes during development. Epigenetic Epidemiology. Springer Netherlands; 2012:77–103.
Steegers-Theunissen RP, Twigt J, Pestinger V, Sinclair KD. The periconceptional period, reproduction and long-term health of offspring: the importance of one-carbon metabolism. Hum Reprod Update. 19(6):640–655.
Gonseth S, Roy R, Houseman EA, de Smith AJ, Zhou M, Lee ST, Nusslé S, Singer AW, Wrensch MR, Metayer C, Wiemels JL. Periconceptional folate consumption is associated with neonatal DNA methylation modifications in neural crest regulatory and cancer development genes. Epigenetics. 2015;10(12):1166–76.
Steegers-Theunissen RP, Obermann-Borst SA, Kremer D, Lindemans J, Siebel C, Steegers EA, Slagboom PE, Heijmans BT. Periconceptional maternal folic acid use of 400 μg per day is related to increased methylation of the IGF2 gene in the very young child. PLoS One. 2009;4(11):e7845.
Chang H, Zhang T, Zhang Z, Bao R, Fu C, Wang Z, Bao Y, Li Y, Wu L, Zheng X, Wu J. Tissue-specific distribution of aberrant DNA methylation associated with maternal low-folate status in human neural tube defects. J Nutr Biochem. 2011;22(12):1172–7.
Boeke CE, Baccarelli A, Kleinman KP, Burris HH, Litonjua AA, Rifas-Shiman SL, Tarantini L, Gillman M. Gestational intake of methyl donors and global LINE-1 DNA methylation in maternal and cord blood: prospective results from a folate-replete population. Epigenetics. 2012;7(3):253–60.
Heijmans BT, Tobi EW, Stein AD, Putter H, Blauw GJ, Susser ES, Slagboom PE, Lumey LH. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci. 2008;105(44):17046–9.
Tobi EW, Lumey LH, Talens RP, Kremer D, Putter H, Stein AD, Slagboom PE, Heijmans BT. DNA methylation differences after exposure to prenatal famine are common and timing- and sex-specific. Hum Mol Genet. 2009;18(21):4046–53.
Altobelli G, Bogdarina IG, Stupka E, Clark AJ, Langley-Evans S. Genome-wide methylation and gene expression changes in newborn rats following maternal protein restriction and reversal by folic acid. PLoS One. 2013;8(12):e82989.
Langley‐Evans SC. Nutritional programming of disease: unravelling the mechanism. J Anat. 2009;215(1):36–51.
Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33:245–54.
Gluckman PD, Lillycrop KA, Vickers MH, Pleasants AB, Phillips ES, Beedle AS, Burdge GC, Hanson MA. Metabolic plasticity during mammalian development is directionally dependent on early nutritional status. Proc Natl Acad Sci. 2007;104(31):12796–800.
Waterland RA. Do maternal methyl supplements in mice affect DNA methylation of offspring? J Nutr. 2003;133(1):238.
Waterland RA, Jirtle RL. Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol Cell Biol. 2003;23(15):5293–300.
Dolinoy DC. The agouti mouse model: an epigenetic biosensor for nutritional and environmental alterations on the fetal epigenome. Nutr Rev. 2008;66 suppl 1:S7–S11.
Waterland RA, Kellermayer R, Laritsky E, Rayco-Solon P, Harris RA, Travisano M, Zhang W, Torskaya MS, Zhang J, Shen L, Manary MJ. Season of conception in rural Gambia affects DNA methylation at putative human metastable epialleles. PLoS Genet. 2010;6(12):e1001252.
Gluckman PD, Hanson MA. Living with the past: evolution, development, and patterns of disease. Science. 2004;305(5691):1733–6.
Barker DJ. A new model for the origins of chronic disease. Med Health Care Philos. 2001;4(1):31–5.
Carone BR, Fauquier L, Habib N, Shea JM, Hart CE, Li R, Bock C, Li C, Gu H, Zamore PD, Meissner A. Paternally induced transgenerational environmental reprogramming of metabolic gene expression in mammals. Cell. 2010;143(7):1084–96.
James SJ, Melnyk S, Pogribna M, Pogribny IP, Caudill MA. Elevation in S-adenosylhomocysteine and DNA hypomethylation: potential epigenetic mechanism for homocysteine-related pathology. J Nutr. 2002;132(8):2361S–6S.
Shrubsole MJ, Jin F, Dai Q, Shu XO, Potter JD, Hebert JR, Gao YT, Zheng W. Dietary folate intake and breast cancer risk. Cancer Res. 2001;61(19):7136–41.
Prinz-Langenohl R, Fohr I, Pietrzik K. Beneficial role for folate in the prevention of colorectal and breast cancer. Eur J Nutr. 2001;40(3):98–105.
Paspatis GA, Kalafatis E, Oros L, Xourgias V, Koutsioumpa P, Karamanolis DG. Folate status and adenomatous colonic polyps. Dis Colon Rectum. 1995;38(1):64–7.
Kennedy DA, Stern SJ, Moretti M, Matok I, Sarkar M, Nickel C, Koren G. Folate intake and the risk of colorectal cancer: a systematic review and meta-analysis. Cancer Epidemiol. 2011;35(1):2–10.
Kim DH, Smith-Warner SA, Spiegelman D, Yaun SS, Colditz GA, Freudenheim JL, Giovannucci E, Goldbohm RA, Graham S, Harnack L, Jacobs EJ. Pooled analyses of 13 prospective cohort studies on folate intake and colon cancer. Cancer Causes Control. 2010;21(11):1919–30.
Mason JB. Folate status and colorectal cancer risk: a 2016 update. Mol Asp Med. 2016;2(53):73–9.
Beetstra S, Salisbury C, Turner J, Altree M, McKinnon R, Suthers G, Fenech M. Lymphocytes of BRCA1 and BRCA2 germ-line mutation carriers, with or without breast cancer, are not abnormally sensitive to the chromosome damaging effect of moderate folate deficiency. Carcinogenesis. 2006;27(3):517–24.
Liu Z, Choi SW, Crott JW, Smith DE, Mason JB. Multiple B-vitamin inadequacy amplifies alterations induced by folate depletion in p53 expression and its downstream effector MDM2. Int J Cancer. 2008;123(3):519–25.
Surget S, Khoury MP, Bourdon JC. Uncovering the role of p53 splice variants in human malignancy: a clinical perspective. OncoTargets Ther. 2013;7:57–68.
Crott JW, Liu Z, Choi SW, Mason JB. Folate depletion in human lymphocytes up-regulates p53 expression despite marked induction of strand breaks in exons 5–8 of the gene. Mutat Res. 2007;626(1):171–9.
Sohn KJ, Stempak JM, Reid S, Shirwadkar S, Mason JB, Kim YI. The effect of dietary folate on genomic and p53-specific DNA methylation in rat colon. Carcinogenesis. 2003;24(1):81–90.
Hsiung DT, Marsit CJ, Houseman EA, Eddy K, Furniss CS, McClean MD, Kelsey KT. Global DNA methylation level in whole blood as a biomarker in head and neck squamous cell carcinoma. Cancer Epidemiol Prev Biomark. 2007;16(1):108–14.
Piyathilake CJ, Macaluso M, Alvarez RD, Chen M, Badiga S, Siddiqui NR, Edberg JC, Partridge EE, Johanning GL. A higher degree of LINE-1 methylation in peripheral blood mononuclear cells, a one-carbon nutrient related epigenetic alteration, is associated with a lower risk of developing cervical intraepithelial neoplasia. Nutrition. 2011;27(5):513–9.
Cordero AM, Crider KS, Rogers LM, Cannon MJ, Berry RJ. Optimal serum and red blood cell folate concentrations in women of reproductive age for prevention of neural tube defects: World Health Organization guidelines. Morb Mortal Wkly Rep. 2015;64(15):421–3.
Selhub J, Rosenberg IH. Excessive folic acid intake and relation to adverse health outcome. Biochimie. 2016;126:71–8.
Kim YI. Will mandatory folic acid fortification prevent or promote cancer? Am J Clin Nutr. 2004;80(5):1123–8.
Cole BF, Baron JA, Sandler RS, Haile RW, Ahnen DJ, Bresalier RS, McKeown-Eyssen G, Summers RW, Rothstein RI, Burke CA, Snover DC. Folic acid for the prevention of colorectal adenomas: a randomized clinical trial. Jama. 2007;297(21):2351–9.
Collin SM. Folate and B12 in prostate cancer. Adv Clin Chem. 2013;60:1–63.
Farias N, Ho N, Butler S, Delaney L, Morrison J, Shahrzad S, Coomber BL. The effects of folic acid on global DNA methylation and colonosphere formation in colon cancer cell lines. J Nutr Biochem. 2015;26(8):818–26.
Yu M, Li W, Luo S, Zhang Y, Liu H, Gao Y, Wang X, Wilson JX, Huang G. Folic acid stimulation of neural stem cell proliferation is associated with altered methylation profile of PI3K/Akt/CREB. J Nutr Biochem. 2014;25(4):496–502.
Koury MJ, Ponka P. New insights into erythropoiesis: the roles of folate, vitamin B12, and iron. Annu Rev Nutr. 2004;24:105–31.
Bills ND, Koury MJ, Clifford AJ, Dessypris EN. Ineffective hematopoiesis in folate-deficient mice. Blood. 1992;79(9):2273–80.
Aslinia F, Mazza JJ, Yale SH. Megaloblastic anemia and other causes of macrocytosis. Clin Med Res. 2006;4(3):236–41.
Greaves M. Molecular genetics, natural history and the demise of childhood leukaemia. Eur J Cancer. 1999;35(14):1941–53.
Greaves M. Infection, immune responses and the aetiology of childhood leukaemia. Nat Rev Cancer. 2006;6(3):193–203.
Mori H, Colman SM, Xiao Z, Ford AM, Healy LE, Donaldson C, Hows JM, Navarrete C, Greaves M. Chromosome translocations and covert leukemic clones are generated during normal fetal development. Proc Natl Acad Sci. 2002;99(12):8242–7.
Wiemels J. Perspectives on the causes of childhood leukemia. Chem Biol Interact. 2012;196(3):59–67.
Wiemels J. New insights into childhood leukemia etiology. Eur J Epidemiol. 2015;30(12):1225–7.
Ames BN. DNA damage from micronutrient deficiencies is likely to be a major cause of cancer. Mutat Res. 2001;475(1):7–20.
Tower RL, Spector LG. The epidemiology of childhood leukemia with a focus on birth weight and diet. Crit Rev Clin Lab Sci. 2007;44(3):203–42.
Hewitt SM, Crowe CM, Navin AW, Miller ME. Recommendations for the use of folic acid to reduce the number of cases of spina bifida and other neural tube defects. Atlanta GA USA. 1992;41:980–4.
Ward E, DeSantis C, Robbins A, Kohler B, Jemal A. Childhood and adolescent cancer statistics, 2014. CA Cancer J Clin. 2014;64(2):83–103.
Amigou A, Rudant J, Orsi L, Goujon-Bellec S, Leverger G, Baruchel A, Bertrand Y, Nelken B, Plat G, Michel G, Haouy S. Folic acid supplementation, MTHFR and MTRR polymorphisms, and the risk of childhood leukemia: the ESCALE study (SFCE). Cancer Causes Control. 2012;23(8):1265–77.
Goh YI, Koren G. Folic acid in pregnancy and fetal outcomes. J Obstet Gynaecol. 2008;28(1):3–13.
Jensen CD, Block G, Buffler P, Ma X, Selvin S, Month S. Maternal dietary risk factors in childhood acute lymphoblastic leukemia (United States). Cancer Causes Control. 2004;15(6):559–70.
Milne E, Royle JA, Miller M, Bower C, de Klerk NH, Bailey HD, van Bockxmeer F, Attia J, Scott RJ, Norris MD, Haber M. Maternal folate and other vitamin supplementation during pregnancy and risk of acute lymphoblastic leukemia in the offspring. Int J Cancer. 2010;126(11):2690–9.
Ross JA, Blair CK, Olshan AF, Robison LL, Smith FO, Heerema NA, Roesler M. Periconceptional vitamin use and leukemia risk in children with Down syndrome. Cancer. 2005;104(2):405–10.
Metayer C, Milne E, Dockerty JD, Clavel J, Pombo-de-Oliveira MS, Wesseling C, Spector LG, Schüz J, Petridou E, Ezzat S, Armstrong BK. Maternal supplementation with folic acid and other vitamins and risk of leukemia in offspring: a Childhood Leukemia International Consortium study. Epidemiology. 2014;25(6):811–22.
Kelly P, McPartlin J, Goggins M, Weir DG, Scott JM. Unmetabolized folic acid in serum: acute studies in subjects consuming fortified food and supplements. Am J Clin Nutr. 1997;65(6):1790–5.
Troen AM, Mitchell B, Sorensen B, Wener MH, Johnston A, Wood B, Selhub J, McTiernan A, Yasui Y, Oral E, Potter JD. Unmetabolized folic acid in plasma is associated with reduced natural killer cell cytotoxicity among postmenopausal women. J Nutr. 2006;136(1):189–94.
Tam C, O’Connor D, Koren G. Circulating unmetabolized folic acid: relationship to folate status and effect of supplementation. Obstet Gynecol Int. 2012;2012:485179.
Singer AW, Selvin S, Block G, Golden C, Carmichael SL, Metayer C. Maternal prenatal intake of one-carbon metabolism nutrients and risk of childhood leukemia. Cancer Causes Control. 2016;27(7):929–40.
Singer AW, Carmichael SL, Selvin S, Fu C, Block G, Metayer C. Maternal diet quality before pregnancy and risk of childhood leukaemia. Br J Nutr. 2016;116(8):1469–78.
Thompson JR, Gerald PF, Willoughby ML, Armstrong BK. Maternal folate supplementation in pregnancy and protection against acute lymphoblastic leukaemia in childhood: a case-control study. Lancet. 2001;358(9297):1935–40.
Nazki FH, Sameer AS, Ganaie BA. Folate: metabolism, genes, polymorphisms and the associated diseases. Gene. 2014;533(1):11–20.
Frosst P, Blom HJ, Milos R, Goyette P, Sheppard CA, Matthews RG, Boers GJ, den Heijer M, Kluijtmans LA, Van den Heuvel LP, Rozen R. A candidate genetic risk factor for vascular disease: a common mutation in methylenetetrahydrofolate reductase. Nat Genet. 1995;10:111–3.
van der Put NM, Gabreëls F, Stevens EM, Smeitink JA, Trijbels FJ, Eskes TK, van den Heuvel LP, Blom HJ. A second common mutation in the methylenetetrahydrofolate reductase gene: an additional risk factor for neural-tube defects? Am J Hum Genet. 1998;62(5):1044–51.
Chiusolo P, Reddiconto G, Cimino G, Sica S, Fiorini A, Farina G, Vitale A, Sorà F, Laurenti L, Bartolozzi F, Fazi P. Methylenetetrahydrofolate reductase genotypes do not play a role in acute lymphoblastic leukemia pathogenesis in the Italian population. Haematol. 2004;89(2):139–44.
Kim HN, Kim YK, Lee IK, Yang DH, Lee JJ, Shin MH, Park KS, Choi JS, Park MR, Jo DY, Won JH. Association between polymorphisms of folate-metabolizing enzymes and hematological malignancies. Leuk Res. 2009;33(1):82–7.
Skibola CF, Smith MT, Hubbard A, Shane B, Roberts AC, Law GR, Rollinson S, Roman E, Cartwright RA, Morgan GJ. Polymorphisms in the thymidylate synthase and serine hydroxymethyltransferase genes and risk of adult acute lymphocytic leukemia. Blood. 2002;99(10):3786–91.
Thirumaran RK, Gast A, Flohr T, Burwinkel B, Bartram C, Hemminki K, Kumar R. MTHFR genetic polymorphisms and susceptibility to childhood acute lymphoblastic leukemia. Blood. 2005;106(7):2590–1.
Oh D, Kim NK, Jang MJ, Kim HC, Lee JH, Lee JA, Ahn MJ, Kim CS, Kim HS, Park S, Chi HS. Association of the 5, 10-methylenetetrahydrofolate reductase (MTHFR C677T and A1298C) polymorphisms in Korean patients with adult acute lymphoblastic leukemia. Anticancer Res. 2007;27(5A):3419–24.
Schnakenberg E, Mehles A, Cario G, Rehe K, Seidemann K, Schlegelberger B, Elsner HA, Welte KH, Schrappe M, Stanulla M. Polymorphisms of methylenetetrahydrofolate reductase (MTHFR) and susceptibility to pediatric acute lymphoblastic leukemia in a German study population. BMC Med Genet. 2005;6(1):23.
de Jonge R, Tissing WJ, Hooijberg JH, Jansen G, Kaspers GJ, Lindemans J, Peters GJ, Pieters R. Polymorphisms in folate-related genes and risk of pediatric acute lymphoblastic leukemia. Blood. 2009;113(10):2284–9.
Franco RF, Simões BP, Tone LG, Gabellini SM, Zago MA, Falcão RP. The methylenetetrahydrofolate reductase C677T gene polymorphism decreases the risk of childhood acute lymphocytic leukaemia. Br J Haematol. 2001;115(3):616–8.
Gemmati D, Ongaro A, Scapoli GL, Della Porta M, Tognazzo S, Serino ML, Di Bona E, Rodeghiero F, Gilli G, Reverberi R, Caruso A. Common gene polymorphisms in the metabolic folate and methylation pathway and the risk of acute lymphoblastic leukemia and non-Hodgkin’s lymphoma in adults. Cancer Epidemiol Prev Biomark. 2004;13(5):787–94.
Zanrosso CW, Hatagima A, Emerenciano M, Ramos F, Figueiredo A, Félix TM, Segal SL, Giugliani R, Muniz MT, Pombo-de-Oliveira MS. The role of methylenetetrahydrofolate reductase in acute lymphoblastic leukemia in a Brazilian mixed population. Leuk Res. 2006;30(4):477–81.
Alcasabas P, Ravindranath Y, Goyette G, Haller A, Del Rosario L, Lesaca‐Medina MY, Darga L, Ostrea EM, Taub JW, Everson RB. 5,10-methylenetetrahydrofolate reductase (MTHFR) polymorphisms and the risk of acute lymphoblastic leukemia (ALL) in Filipino children. Pediatr Blood Cancer. 2008;51(2):178–82.
Gast A, Bermejo JL, Flohr T, Stanulla M, Burwinkel B, Schrappe M, Bartram CR, Hemminki K, Kumar R. Folate metabolic gene polymorphisms and childhood acute lymphoblastic leukemia: a case–control study. Leukemia. 2007;21(2):320–5.
Lauten M, Cario G, Asgedom G, Welte K, Schrappe M. Protein expression of the glucocorticoid receptor in childhood acute lymphoblastic leukemia. Haematologica. 2003;88(11):1253–8.
Lightfoot TJ, Skibola CF, Willett EV, Skibola DR, Allan JM, Coppede F, Adamson PJ, Morgan GJ, Roman E, Smith MT. Risk of non-Hodgkin lymphoma associated with polymorphisms in folate-metabolizing genes. Cancer Epidemiol Prev Biomark. 2005;14(12):2999–3003.
Nazki FH, Masood A, Banday MA, Bhat A, Ganai BA. Thymidylate synthase enhancer region polymorphism not related to susceptibility to acute lymphoblastic leukemia in the Kashmir population. Genet Mol Res. 2012;11(2):906–17.
Rahimi Z, Ahmadian Z, Akramipour R, Madani H, Mozafari H, Vaisi-Raygani A, Shahriari-Ahmadi A. Thymidilate synthase and methionine synthase polymorphisms in children with acute lymphoblastic leukemia in Western Iran. Int J Hematol-Oncol Stem Cell Res. 2010;4(1):9–12.
Zhu XL, Liu ZZ, Yan SX, Wang W, Chang RX, Zhang CY, Guo Y. Association between the MTHFR A1298C polymorphism and risk of cancer: evidence from 265 case–control studies. Mol Gen Genomics. 2016;291(1):51–63.
Qin YT, Zhang Y, Wu F, Su Y, Lu GN, Wang RS. Association between MTHFR polymorphisms and acute myeloid leukemia risk: a meta-analysis. PLoS One. 2014;9(2):e88823.
Vahid P, Farnaz R, Zaker F, Farzaneh A, Parisa R. Methylenetetrahydrofolate reductase gene polymorphisms and risk of myeloid leukemia. Lab Med. 2010;41(8):490–4.
Bănescu C, Iancu M, Trifa AP, Macarie I, Dima D, Dobreanu M. The methylenetetrahydrofolate reductase (MTHFR) 677 C>T polymorphism increases the risk of developing chronic myeloid leukemia—a case-control study. Tumor Biol. 2015;36(4):3101–7.
Bănescu C, Trifa AP. Methylenetetrahydrofolate reductase 677 C>T polymorphism is associated with acute myeloid leukemia. Leuk Lymphoma. 2015;56(4):1172–4.
He H, He G, Wang T, Cai J, Wang Y, Zheng X, Dong Y, Lu J. Methylenetetrahydrofolate reductase gene polymorphisms contribute to acute myeloid leukemia and chronic myeloid leukemia susceptibilities: evidence from meta-analyses. Cancer Epidemiol. 2014;38(5):471–8.
Dong S, Liu Y, Chen J. MTHFR gene polymorphism and risk of myeloid leukemia: a meta-analysis. Tumor Biol. 2014;35(9):8913–9.
Yang B, Liu Y, Li Y, Fan S, Zhi X, Lu X, Wang D, Zheng Q, Wang Y, Wang Y, Sun G. Geographical distribution of MTHFR C677T, A1298C and MTRR A66G gene polymorphisms in China: findings from 15357 adults of Han nationality. PLoS One. 2013;8(3):e57917.
Bailey HD, Miller M, Greenop KR, Bower C, Attia J, Marshall GM, Armstrong BK, Milne E. Paternal intake of folate and vitamins B6 and B12 before conception and risk of childhood acute lymphoblastic leukemia. Cancer Causes Control. 2014;25(12):1615–25.
Wen W, Shu XO, Potter JD, Severson RK, Buckley JD, Reaman GH, Robison LL. Parental medication use and risk of childhood acute lymphoblastic leukemia. Cancer. 2002;95(8):1786–94.
Funding
This study was not supported by a funding body.
Availability of data and materials
Not applicable
Authors’ contributions
CDC and ST handled the conception and design of the review. CDC, DR and ST wrote the manuscript. MG and ST handled the critical revision of the manuscript. CDC, DR, MG and ST gave approval of the final version of the manuscript.
Authors’ information
CDC is a private Practitioner in Nutrition recently graduated in Nutrition Sciences (postgraduate speciality degree) and already holding a PhD in Genetics. DR is a Biomedical Sciences student in the Leukaemia and Chromosome Laboratory. MG is Professor in Endocrinology, Physiology and Nutrition. ST is Head of the Leukaemia and Chromosome Laboratory with particular interest in childhood leukaemia.
Competing interests
The authors declare that they have no competing interests
Consent for publication
Not applicable
Ethics approval and consent to participate
Not applicable
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Cantarella, C.D., Ragusa, D., Giammanco, M. et al. Folate deficiency as predisposing factor for childhood leukaemia: a review of the literature. Genes Nutr 12, 14 (2017). https://doi.org/10.1186/s12263-017-0560-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12263-017-0560-8