
Abstract
Serological diagnostics is generally defined as the detection of specific human immunoglobulins developed against viral, bacterial, or parasitic diseases. Serological tests facilitate the detection of past infections, evaluate immune status, and provide prognostic information. Serological assays were traditionally implemented as indirect immunoassays, and their design has not changed for decades. The advantages of straightforward setup and manufacturing, analytical sensitivity and specificity, affordability, and high-throughput measurements were accompanied by limitations such as semi-quantitative measurements, lack of universal reference standards, potential cross-reactivity, and challenges with multiplexing the complete panel of human immunoglobulin isotypes and subclasses. Redesign of conventional serological tests to include multiplex quantification of immunoglobulin isotypes and subclasses, utilize universal reference standards, and minimize cross-reactivity and non-specific binding will facilitate the development of assays with higher diagnostic specificity. Improved serological assays with higher diagnostic specificity will enable screenings of asymptomatic populations and may provide earlier detection of infectious diseases, autoimmune disorders, and cancer. In this review, we present the major clinical needs for serological diagnostics, overview conventional immunoassay detection techniques, present the emerging immunoassay detection technologies, and discuss in detail the advantages and limitations of mass spectrometry and immunoaffinity proteomics for serological diagnostics. Finally, we explore the design of novel immunoaffinity-proteomic assays to evaluate cell-mediated immunity and advance the sequencing of clinically relevant immunoglobulins.
Similar content being viewed by others
Background
Serological diagnostics is generally defined as the detection of the specific immunoglobulins developed against viral, bacterial, or parasitic diseases and circulating in patient blood or proximal fluids. Serological tests present simple and informative tools for clinical diagnostics and facilitate the detection of previous and current infections, evaluation of immune status, and disease prognosis. Serological tests date back to 1906 and the development of the Wassermann reaction, a complement fixation test to detect anti-cardiolipin antibodies and diagnose syphilis [1]. Currently, serological tests provide complementary clinical information and allow for earlier and more accurate diagnosis of a variety of non-infectious diseases including autoimmune disorders, cancer, celiac disease, rheumatoid arthritis, and others [2,3,4].
The conventional serological tests are exclusively based on the concept of indirect Enzyme-Linked Immunosorbent Assays (ELISA) or their modifications, such as Lateral Flow Immunoassays (LFI) or Fluorophore Linked Immunosorbent Assays (FLISA). Indirect immunoassays, however, may suffer from non-specific binding and cross-reactivity, which result in higher false positive rates and lower diagnostic specificity. Lower diagnostic specificity prohibits the use of serological tests to screen populations with low disease prevalence (early stages of a pandemic, rare diseases, asymptomatic general populations, etc.) [5]. Furthermore, the complete panel of human immunoglobulin isotypes (IgG, IgM, IgA, IgE, and IgD) and subclasses (IgG1, IgG2, IgG3, IgG4, IgA1, and IgA2) is rarely evaluated; the majority of serological studies traditionally evaluate only the antigen-specific total IgG, total IgA, IgM, or their combinations [6]. Redesign of conventional serological tests to evaluate and minimize cross-reactivity, utilize universal reference standards, and measure disease-relevant subclasses and isotypes would facilitate the development of assays with higher diagnostic specificity, reliable implementation, and correct interpretation of serological tests for diagnosis of lower-prevalence diseases, screening of asymptomatic populations, and earlier detection [7,8,9,10].
Innovative and comprehensive immunoassay detection technologies such as mass spectrometry and protein microarrays promise to resolve limitations of conventional indirect immunoassays and provide multiplex measurements of immunoglobulin isotypes and subclasses, allowing for a more detailed analysis of disease states and improving disease prognosis [10]. In this review, we discuss the current clinical needs for serological diagnostics, overview the conventional and emerging immunoassay detection technologies, discuss in detail proteomics and mass spectrometry as a comprehensive immunoassay detection technology, and explore the design of novel immunoaffinity-proteomic assays to evaluate cell-mediated immunity and advance sequencing of clinically-relevant immunoglobulins.
Main text
Clinical needs addressed with serological diagnostics
Diagnostics of infectious diseases
Serological testing remains an essential tool to aid PCR and RT-PCR diagnostics of infectious diseases and provide complimentary clinical information (Table 1). For example, prenatal screening for infectious diseases is aimed at preventing the transfer of infections from mother to child before birth thus identifying and decreasing the risk of pregnancy complications [11]. Around 40% of pregnancies with syphilis infections result in the death of the fetus while surviving newborns could develop a wide array of physiological abnormalities [11]. Likewise, hepatitis B (HBV) infections can lead to serious complications, and 90% of infected infants develop chronic HBV infections [12]. Serological testing, such as the detection of anti-HBc IgM, can assist in the prognosis of infections, allowing for the appropriate preventative measures [12].
Blood transfusion is another area where serological testing for infectious diseases is critical. Blood transfusions have revolutionized hematologic treatments, and there have been increases in safety measures over the last few decades to eliminate infections transmitted during transfusion [13]. Transfusion complications are a relatively common phenomenon and remain a serious concern. Notably, arboviruses, bacteria, and parasites present the most common sources of transfusion-transmitted infections [13]. For example, malaria infections through blood transfusion remain a concern in malaria-endemic and non-endemic countries due to travel and lack of screening [13]. Babesiosis, a zoonotic disease caused by tick-borne piroplasmids, presents another risk to the recipient population [13].
Viral hepatitis and the associated inflammation of the liver is one of the leading causes of mortality worldwide [14]. Five different hepatitis viruses A to E, each of a distinct viral family and with numerous genotypes, lead to viral hepatitis [14]. Hepatitis A (HAV), B (HBV), and C (HCV) viruses include 7, 10, and 7 genotypes, respectively [12, 14, 15]. Hepatitis D virus (HDV) has 8 genotypes and relies on coinfection with HBV and its lipid envelope (HBsAg) for replication [14]. Hepatitis E virus (HEV) has 4 genotypes but a single serotype [14, 16]. Upon hepatitis infection, specific IgM immunoglobulins are produced and manifest an acute infection [14, 16]. HBV testing is a prominent example of detailed serological diagnostics which evaluates the presence of viral protein antigens and the corresponding IgG and IgM; different combinations of positive and negative outcomes for protein antigens and antibodies provide a detailed interpretation of HBV status, such as acute, chronic, past infections, etc. [17]. Elimination of inconsistencies with diagnostics sensitivity and specificity of serological testing for hepatitis viruses and their distinct genotypes is a recognized need [16].
Travel-related infectious diseases represent another area where diagnosis and vaccination status rely on accurate and timely serological testing. Yellow fever, an arbovirus transmitted by mosquitos [18, 19], is currently one of the biggest concerns for travelers [18]. Yellow fever vaccinations provide long-term protection but are not required in non-endemic countries. Typhoid fever mediated by the gram-negative Salmonella bacteria also remains a large concern [20]. While vaccinations result in sustained serum IgG antibodies [20], typhoid fever serological tests require further validation to be used in clinics [21]. Influenza virus remains a concern due to its high mutation rates and adaptability, but its epidemics could be prevented through vaccinations and assessment of antibody protection in regions susceptible to the rapid evolution and adaptability of influenza [22].
Emerging infections in populations lacking prior immunity present global health concerns but also opportunities for the rapid implementation of innovative serological assays. Dengue fever, a re-emerging disease caused by the dengue virus, has 4 different serotypes of IgM and IgG [23]. Pre-existing antibodies often result in dengue hemorrhagic fever in patients with secondary dengue infection [24]. West Nile Virus, a mosquito-transmitted disease, triggers the IgM and IgG response [25] and raises concerns due to high morbidity rates [25].
The novel SARS-CoV-2 coronavirus and the COVID-19 pandemic have recently impacted the entire world. At the early stages of the COVID-19 pandemic novel serological tests were not thoroughly validated and reported diagnostic specificity as low as 95%, preventing the correct interpretation of test results [5]. To achieve positive predictive values > 90%, serological tests with 95% diagnostic specificity could only be informative in populations with COVID-19 prevalence > 33%. At the early stages of the pandemic (~ 0.1% prevalence estimated by RT-PCR), the poorly validated serological assays resulted in highly over-estimated and vastly incorrect rates of asymptomatic disease and “herd immunity” [26, 27], potentially undermining the public trust in the evidence-based medicine. The lessons learned demonstrated the importance of the rational design and development of serological tests and the need for thorough and independent validation of their diagnostic performance [10]. It should also be mentioned that the humoral immune response to SARS-CoV-2 has been thoroughly evaluated mostly for IgM, IgG, and IgA isotypes [28, 29], while the evaluation of the dynamics and cooperation of IgG1-4 and IgA1-2 subclasses could provide additional knowledge on the complexity of humoral and cellular immune responses [10].
Autoimmune diseases
Rheumatoid arthritis is the most common inflammatory autoimmune disease of the joints with a prevalence of 0.5-1% [30]. Serological testing for anti-rheumatoid factor IgM and anti-citrullinated protein IgG antibodies is useful to assist with the diagnosis and provide early interventions, to mitigate symptoms [2]. However, 30–50% of individuals with confirmed rheumatoid arthritis test negative [2]. Improved serological testing for a variety of antigens and distinct IgG subclasses could improve early diagnosis of asymptomatic rheumatoid arthritis [7].
The development of specific serological tests will greatly benefit the diagnosis and treatment of inflammatory bowel diseases, such as Crohn’s disease and ulcerative colitis, which significantly affect the quality of life, increase the risk of death, and are currently incurable [31, 32]. Multiple antibodies have been tested for diagnosis of the specific types of inflammatory bowel diseases, primarily anti-Saccharomyces cerevisiae IgG and IgA antibodies and anti-neutrophil cytoplasmic antibodies [33, 34]. The serological testing could distinguish between Crohn’s disease and ulcerative colitis but suffered from low diagnostic sensitivity [33, 34]. Detailed investigation of the levels of antibody isotypes and subclasses against a variety of antigens could aid in earlier and more specific diagnosis of inflammatory bowel diseases [35].
Autoimmune gastritis, an inflammatory autoimmune disorder, may lead to chronic atrophic gastritis followed by pernicious anemia [36, 37]. Autoimmune gastritis is typically asymptomatic until pernicious anemia develops and is then diagnosed by endoscopic biopsy [36]. Serological tests for antibodies targeting parietal cells and intrinsic Castle’s factor are informative for initial assessment but their diagnostic specificity and sensitivity are not sufficient to replace diagnostic biopsies [36, 37].
Celiac disease is another chronic autoimmune disorder characterized by inflammation of the small intestine in response to gluten [38]. It is associated with an increased risk of death and decreased quality of life due to a variety of complications including gastrointestinal distress, malabsorption, and anemia [38]. Diagnosis is based on the detection of anti-tissue transglutaminase and anti-endomysial IgA antibodies followed by an endoscopic duodenal biopsy. In some populations, such as symptomatic children, a diagnosis can be made based on serology alone [3]. However, nearly 10% of patients with confirmed celiac disease on biopsy tested negative for the established serological markers [39]. Testing the levels of specific antibody subclasses in celiac disease may facilitate more accurate diagnosis and potentially reduce the need for biopsies.
Multiple sclerosis is a major contributor to neurological-derived disability in young adults [40]. Diagnosis of multiple sclerosis is based on symptoms, imaging, and analysis of cerebrospinal fluid for IgG and IgM [41]. Early diagnosis and treatment help improve outcomes of multiple sclerosis [42].
Systemic lupus erythematosus (SLE), a chronic autoimmune disorder, could be manifested either through only minor symptoms, such as skin rashes, or a variety of severe symptoms such as organ failures [43]. SLE diagnosis is complex and is based on several criteria or biopsy confirmations [44]. The presence of antinuclear IgG or IgM autoantibodies targeting DNA, phospholipids, and nuclear antigens is included in the diagnostic criteria [44, 45].
Infertility
Globally, nearly 15% of couples are infertile, and around half of those cases are due to male factor infertility [46,47,48,49,50]. Antisperm antibodies (ASA), primarily of IgA and IgG isotypes, were detected in nearly 16% of infertile men and were associated with male immune infertility [51]. ASA were detected either directly, as bound to sperm cell surface, or indirectly, as soluble antibodies [51]. While the identity of the ASA antigens was not studied in detail, the cell-surface testis-specific proteins could be potential candidates [52]. It should be noted that about 2% of fertile men test positive for ASA, so the presence of ASA alone is not sufficient to diagnose immune infertility [51]. Analysis of data on ASA prevalence and their impact on fertility was complicated by the variability of tests, sample types, and thresholds used to report a positive result [51, 53].
Cancer
Detection of autoantibodies generated against specific tumor-associated antigens (TAAs), such as cancer/testis antigens, may facilitate early cancer diagnosis and prognosis [8, 9, 54]. Autoantibodies could potentially be detected in serum before cancer becomes clinically significant [8, 55]. Serological diagnostics of cancers, however, have not been well established. Common challenges of cancer autoantibody testing include insufficient understanding of the identity of cancer-specific antigens, high analytical sensitivity of assays to detect extremely low levels of antigens secreted by small tumors and transient levels of the corresponding autoantibodies, lack of tools for the independent evaluation of diagnostic specificity of serological tests, and lack of standards [9].
Over the past decades, autoantibodies were identified in serum of patients with breast, lung, ovarian, and prostate cancers [8, 55]. For example, CA125 in combination with the human epididymis protein 4 antigen-autoantibody complexes increased sensitivity from 63 to 81% to detect early-stage ovarian cancer [4]. Several autoantibody panels including Videssa Breast [56], EarlyCDT-Lung [57], and MitogenDx [58] are now available as Laboratory Developed Tests and justify the use of autoantibodies as cancer biomarkers. MitogenDx cancer test to diagnose the paraneoplastic syndrome, often the first manifestation of neoplasms [59], measures autoantibodies against several testis- and brain-specific proteins in lung, breast, ovarian, and other major cancers [60]. There was also a strong association found between paraneoplastic syndrome, IgG4-related disease, and cancer [61, 62]. Previous studies detected IgG, IgA, and IgE autoantibodies [8, 55, 63] but rarely evaluated autoantibody subclasses. Few studies revealed IgG1 and IgG3 as the most abundant cancer autoantibodies [64, 65]. Interestingly, IgG4, a “blocking” antibody subclass often generated after long-term exposure to antigens in non-infectious settings [66], was found associated with immune evasion, immunotherapy inefficiency, and poor survival in cancer [67,68,69]. Innovative high-specificity assays for serological testing may revolutionize cancer autoantibody studies, validate cancer/testis antigen hypothesis, discover novel immunotherapy targets, and enable precision approaches to immunotherapy.
Serological diagnostics utilizing conventional immunoassay detection techniques
Since measurements of pathogen-specific antibodies circulating in serum rely on highly specific antigen-antibody interactions and affinity enrichments, serological assays could be generalized as immunoassays. The only exception of serological testing implemented without the requirement for antigen-antibody affinity interactions would be a detection of ‘M-proteins’ in monoclonal gammopathy or multiple myeloma [70]. ‘M-proteins’ can be directly measured by LC-MS or other techniques due to the extremely high levels in patient serum (up to 30 mg/mL) and monoclonal sequences [71, 72]. In this review, we discuss immunoassays with either conventional detection techniques, such as enzyme-linked absorbance measurements, or the emerging detection technologies, such as mass spectrometry. Other differences between serological assays include direct or indirect detection of antibody constant heavy chains, label-based or label-free approaches, multiplexing capabilities, affordability, analytical sensitivity and specificity, reproducibility, and throughput. In this section, we will overview immunoassays with conventional detection techniques.
Indirect ELISA
Enzyme-linked immunosorbent assay (ELISA) is a well-established technique in research and clinical laboratories [6]. Indirect ELISA (Fig. 1A) relies on affinity enrichment of antigen-specific polyclonal antibodies followed by their detection using the secondary anti-human antibodies conjugated to an enzyme (horseradish peroxidase, alkaline phosphatase, or beta-galactosidase). In the case of horseradish peroxidase, the signal amplification is provided through the enzyme-catalyzed conversion of 3,3′,5,5′-tetramethylbenzidine substrate into its oxidized form which has a specific absorbance at 450 nm [73, 74]. In a fluorophore-linked immunosorbent assay (FLISA), secondary antibodies are conjugated to a fluorescent molecule. While indirect ELISA is a highly sensitive assay, it suffers from non-specific binding (such as non-specific adsorption of analytes and reagents to the microplate surface), cross-reactivity (such as cross-reactivity of secondary antibodies), and challenges with assay standardization [6]. Due to the lack of established standards of antigen-specific human polyclonal antibodies, indirect ELISA measurements typically report relative units, such as signal intensities, binding antibody units (BAU)/mL, or antibody ‘titers’ (the highest sample dilution factors which result in positive signals), but not absolute concentrations (µg/mL). Relative measurements are considered one of the major limitations which restrict inter-hospital and international standardization of serological testing [75]. The conventional immunoassays could hardly be multiplexed for the complete panel of human antibody isotypes and subclasses due to the cross-reactivity of the secondary antibodies, lack of the multiple spectrally-resolved fluorescent labels, and the limited dynamic range resulting in the need to measure multiple dilutions of the same sample [76, 77].

Serological immunoassays with conventional detection techniques. (A) Colorimetric indirect ELISA: antigens of interest are immobilized on the microplate surface and incubated with diluted blood serum samples. Specific human antibodies (blue) are captured, and non-specific antibodies and proteins are removed with microplate washing. Enzyme-conjugated secondary anti-human antibodies (green) oxidize the substrate (yellow), and the absorbance of the product is measured by a spectrophotometer. Relative antibody titers are determined by the highest dilution of a positive blood serum sample that provided a positive result. (B) Lateral flow immunoassay: Specific antibodies in patient samples bind to an antigen immobilized on colloidal gold nanoparticles on a sample pad (S). Capillary flow transfers complexes to the conjugation pad, where the complexes interact with the immobilized anti-human secondary antibodies, aggregate, and precipitate at the test line (T). Precipitation of gold nanoparticles results in color change (red stripe). As a control for test completion, rabbit antibodies conjugated to gold nanoparticles travel along through the T region, interact with the goat-anti-rabbit antibodies at the control line C, precipitate, and result in color change. (C) Multiplex particle-based flow cytometry: Each antigen is conjugated to a bead of a unique “color” which is predetermined by a unique combination of ten infrared dyes at different concentrations. Beads are mixed and incubated with serum, and the antigens capture corresponding human antibodies. Secondary anti-human IgG antibodies are conjugated to a fluorescent dye (green) used for quantification. Particle-based flow cytometry utilizes two different lasers to detect the bead identity (red laser) and signal intensity (green) of a single bead passing through the detection region. To map antibody isotypes and subclasses across multiple antigens, the analysis is repeated with the secondary antibodies specific for human IgM, IgA, or IgG1-4
Lateral Flow Immunoassay
Lateral Flow Immunoassay (LFI; Fig. 1B) is a variant of indirect immunoassay intended for rapid, straightforward, instrument-free, and point-of-care detection of pathogen-specific antibodies in a variety of clinical samples [78]. A paper-based LFI device is composed of several compartments, including a sample pad, conjugation pad, test line (immobilized anti-human IgG antibody), control line (immobilized anti-rabbit IgG antibody), and an absorbent pad [78]. LFI limitations include those of indirect ELISA (cross-reactivity and challenges with standardization), as well as semi-quantitative measurements, lower sensitivity and reproducibility, and batch-to-batch variability [79].
Multiplexed particle-based flow cytometry
Particle-based flow cytometry immunoassays were developed to enable multiplexed measurements. The most common platforms, such as Luminex assays (Fig. 1C), comprised highly multiplexed bead-based immunoassays in which dozens of different analytes were measured in a single sample [80]. Particle-based flow cytometry immunoassays are robust, reproducible, require minimal expertise, and are widely used in clinical and research laboratories.
Serological diagnostics utilizing the emerging immunoassay detection technologies
Recent infectious disease epidemics and pandemics have accelerated the development of innovative approaches for serological testing (Fig. 2).

Emerging immunoassay detection technologies for serological diagnostics. (A) Surface plasmon resonance assays detect antibodies through their binding to antigens immobilized on gold surfaces. Specific binding results in plasmon resonance and changes in the refractive index. (B) A biolayer interferometry immunosorbent assay detects the shift in the wavelength of the reflected light upon antigen-specific antibody binding to the surface of a fiber optic sensor. (C) Electrochemical biosensors are transistors that detect antibody-antigen binding onto a gate electrode and measure changes in voltage across the source and drain electrodes. (D) Protein microarrays consist of numerous antigens that are printed onto the surface, enrich specific antibodies, and facilitate their relative quantification using fluorophore-labeled secondary antibodies. (E) Following affinity enrichment and trypsin digestion, MS facilitates highly specific quantification of peptides representing human immunoglobulin isotypes and subclasses
Surface plasmon resonance (SPR) biosensors
Surface plasmon resonance (SPR) biosensors present an emerging approach for serological diagnostics with direct label-free quantification [81]. Its major limitations include relatively poor analytical sensitivity, low spatial resolution, expensive equipment, and extensive training of personnel [82, 83]. Rapid detection of protein biomarkers, human antibodies, and monoclonal antibody therapeutics is a promising application of SPR biosensors [84, 85].
Biolayer interferometry
Similar to surface plasmon resonance, biolayer interferometry (BLI) provides fast, label-free, and real-time detection of antigen-antibody interactions and provides information about interaction affinity and kinetics [86,87,88]. Some examples of BLI assays include and label-free measurements of monoclonal antibody therapeutics (LOQs 2–10 µg/mL in serum) and detection of anti-COVID-19 antibodies [89,90,91,92].
Electrochemical biosensors
Electrochemical biosensors are becoming an increasingly useful diagnostic tool and can rapidly detect proteins in biological fluids [93, 94]. Some recent examples include the detection of tau protein and its interactions [95] and measurements of COVID-19 mRNA, proteins, and serological antibodies [96].
Protein microarrays
Protein microarrays facilitate simultaneous analysis of thousands of human proteins and their functional interactions [97,98,99]. Ultra-high-density ‘human proteome’ microarrays (HuProt, ProtoArray, NAPPA, etc.) enable serological profiling of hundreds of patient samples across > 20,000 full-length human protein antigens [100,101,102]. Limitations of protein microarrays include challenges with standardization, high costs, and lack of clinical translation.
Immunoaffinity – mass spectrometry for serological diagnostics
Overview of mass spectrometry technologies for proteomics
Mass spectrometry (MS) technologies, with their numerous approaches to measuring the variety of analytes ranging from small molecules to large proteins and intact viral particles, are rapidly reshaping clinical laboratories. Protein analysis is currently dominated by the bottom-up proteomic approaches, in which unique enzyme-derived peptides are used as proxies for the identification and quantification of the corresponding proteins [103, 104]. While trypsin is still the most common enzyme, alternative proteases (Lys-C, Glu-C, chymotrypsin, etc.) have been actively investigated to complement trypsin [105]. Common LC-MS proteomic workflows include LC separation of peptides, peptide ionization, separation of ions by their mass-over-charge (m/z) ratios, and measurements of the intensity of each molecular ion [106]. Ionization of thermally labile biological molecules, such as proteins, is typically achieved with soft ionization techniques including electrospray ionization (ESI) and matrix-assisted laser desorption/ionization (MALDI). A variety of mass analyzers are utilized to separate molecular ions in the gas phase based on separation in time (time-of-flight mass analyzers, TOF), filtering (quadrupole analyzers), trapping (ion trap mass analyzers), as well as trapping with comprehensive signal processing (Fourier-transform ion cyclotron resonance (FT-ICR) and Orbitrap mass analyzers) [107, 108]. To facilitate deep analysis of complex biological samples, common proteomic approaches often utilize hybrid mass analyzers (quadrupole-TOF, ion trap-Orbitrap, etc.) and tandem mass spectrometry (MS/MS) approaches: separation of the molecular ions by their m/z in the first mass analyzer, fragmentation of ions with a variety of mechanisms, separation of the fragment ions by their m/z in another analyzer, and measurement of ion intensity of the molecular ions and their fragments [109]. The variety of peptide fragmentation mechanisms (collision-induced dissociation, CID; electron-transfer dissociation, ETD; electron capture dissociation, ECD; ultraviolet photodissociation, UVPD, and others) provide complimentary structural information on peptide sequences and post-translational modifications [110].
Commonly used analytical modes of MS include global proteome-wide approaches for protein identification (‘shotgun’ MS) and targeted approaches for protein quantification (parallel reaction monitoring, PRM; multiple reaction monitoring, MRM; and selected reaction monitoring, SRM, with the terms MRM and SRM often used interchangeably). Combinations of liquid chromatography (LC) separations with targeted MS measurements allow for the multiplexing of several hundred analytes and their measurements in dozens of biological and clinical samples [111,112,113,114,115,116,117,118,119,120,121,122]. MS has been an invaluable tool to identify the human proteome and reveal its complexity, post-translational modifications, and protein-protein interactions [123]. Targeted proteomics empowered with high-quality standards facilitated quantitative analysis of multiple proteins across disease and healthy states [124,125,126,127,128,129,130,131,132]. The high analytical specificity of MS facilitated measurements of monoclonal antibodies for their structural information, post-translational modifications, purity, and therapeutic levels in blood plasma [133]. Likewise, LC-MS has previously been used to quantify the levels of total IgG subclasses in serum [134]. However, the relatively slow progress of MS for serological diagnostics could be explained by insufficient MS sensitivity, the complexity of polyclonal antibodies (numerous isotypes, subclasses, allotypes, and variable regions), the lack of standards, and inability to predict sequences of hypervariable regions from the germline genomic sequences. While mass analyzers, analytical modes, sample preparation, and data analysis are continuously improving, conventional sandwich immunoassays are still 2–3 orders more sensitive than state-of-the-art LC-MS assays [135,136,137].
Mass spectrometry as a comprehensive detection technology for serological immunoassays
MS can be viewed as an emerging immunoassay detection technology, with the potential for proteome-wide identification and quantification of human proteins. Combinations of immunoaffinity enrichments with MS detection of proteins and peptides are known by a variety of terms including Mass Spectrometry Immunoassays [138], Stable Isotope Standards and Capture by Anti-Peptide Antibodies (SISCAPA) [139], Immuno-Precipitation Mass Spectrometry (IP-MS) [140], Immuno-MALDI [141], and others. Immunoaffinity-targeted proteomic assays (Fig. 3) provide a sensitivity of ~ 100 pg/mL in serum, approaching the sensitivity of common sandwich immunoassays (~ 10 pg/mL) [142, 143]. It should be emphasized that the readout of immunoassays with some conventional detection techniques (e.g. absorbance at 450 nm) is relatively simple and two-dimensional (wavelength and signal intensity). On the contrary, MS output is complex and multi-dimensional (LC retention time, precursor m/z, precursor intensity, fragment m/z, fragment intensity) and thus results in very high analytical specificity. Thus, the suggested IA-MS assays may well resolve the potential false positives and facilitate independent evaluation of diagnostic specificity of serological tests.

Serological diagnostics with immunoaffinity-targeted proteomics. (A) Specific antibodies are affinity enriched from the patient serum, denatured, and digested with trypsin, and the peptides are analyzed by targeted MS assays. (B) Following electrospray ionization (ESI), targeted MS assays, such as Selected Reaction Monitoring, enable the isolation of peptide ions of interest with the first quadrupole (Q1), their fragmentation in Q2, isolation of fragments of interest in Q3, and measurement of fragment ion intensities. (C) Peak areas of the “light” endogenous peptides and the corresponding spiked-in “heavy” isotope-labeled internal standards are used to calculate peptide ratios and absolute concentrations of the complete panel of the human immunoglobulin isotypes and subclasses
Recently, immunoaffinity proteomics was demonstrated as a conceptually novel platform for serological diagnostics [10, 144, 145]. Simple assay design and targeted proteomics measurements provided high sensitivity (1 ng/mL), high reproducibility (CV < 10%), and relatively high throughput (~ 100 samples/day) [10]. IA-SRM assays for testing new antigens can be rapidly developed and enable rapid response to epidemics and pandemics [10]. Advantages of IA-SRM assays in comparison to the conventional indirect immunoassays include (i) multiplex quantification of the complete panel of human immunoglobulin isotypes (IgG, IgM, IgA, IgE, IgD) and subclasses (IgG1-4, IgA1-2) in clinical samples; (ii) absolute quantification (ng/mL); (iii) high analytical specificity of MS which provided high diagnostic specificity (fewer false-positives); (iv) standardization using short synthetic stable isotope-labeled peptide reference standards which can be synthesized in large amounts and distributed across numerous clinical laboratories; (v) ~ 10-fold higher dynamic range (70–70,000 ng/mL for IgG1 in serum) which enabled measurements without multiple dilutions of clinical samples [10]. IA-SRM analysis of negative or positive convalescent COVID-19 plasma confirmed the true positive immune response with IgG1/IgG3/IgA1 pairing and provided a 385 ng/mL cut-off for anti-RBD IgG1 to detect COVID-19 convalescent plasma with nearly 100% sensitivity and specificity [10].
In the future, serological testing by IA-SRM may find its unique niche in clinical laboratories. There are ongoing initiatives to standardize protein measurements by mass spectrometry, such as the three-tier system using a fit-for-purpose approach for the discovery of protein biomarkers [146], and the Clinical and Laboratory Standards Institute (CLSI) C64 guideline “Quantitative Measurement of Proteins and Peptides by Mass Spectrometry” which provides a framework for the design, development, and validation of quantitative clinical protein and peptide assays [147]. While the throughput of IA-SRM is currently insufficient for large-scale population testing, IA-SRM may emerge as “gold standard” assays for independent validation and standardization of serological tests using stable, quantifiable, and affordable reference standards [10]. The relatively low throughput of IA-SRM assays could be improved through automation of IA and proteomic sample preparation, rapid digestion, fast microflow separations using short LC columns and sub-2 μm particles, multichannel turbulent flow LC, and scheduling for parallel analysis [10]. Rapid LC-independent approaches may include MALDI-TOF [148] and paper spray ion mobility MS [149] and may revolutionize serological diagnostics to the same extent as MALDI-TOF revolutionized clinical microbiology [150].
Advancing immunoaffinity-proteomics for antibody sequencing
The challenge of antibody sequencing
Monoclonal antibodies are a rapidly growing class of innovative therapies with a recognized impact in oncology, autoimmune disorders, and chronic inflammation [151]. Sequencing of pathogen-specific antibodies enriched directly from patient samples may facilitate the rapid development of therapeutic antibodies. However, the immense diversity of antibody clones makes de novo sequencing a formidable challenge [152].
Variable heavy domains (VH) of human antibodies are assembled through the rearrangement of IGHV, IGHD, and IGHJ genes (V-D-J recombination) and subsequent affinity maturation, while the set of IGHC genes encodes the constant heavy (CH) domains and defines immunoglobulin isotypes and subclasses. Likewise, variable light domains (VL) are assembled through the rearrangement of IGKV, IGLV, IGKJ, and IGLJ genes (V-J recombination), while IGKC and IGLC genes define the constant light (CL) domains. VH and VL chains provide unique paratope conformations for the antigen binding with high affinity (Kd 0.01-10 nM), while the CH domains mediate protein-protein interactions with numerous Fc receptors and complement component 1 (C1) complexes to enable diverse immunological functions, such as complement-dependent cytotoxicity and antibody-dependent cellular cytotoxicity.
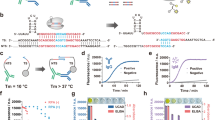
An antibody clonotype is defined as a group of sequences derived from a common progenitor B cell and sharing one of the variable (V), diversity (D), joining (J), and constant (C) genes [153, 154]. Recent studies on B cell receptor repertoire sequencing reported that the clonotype sharing between patients was much higher than expected by chance [155,156,157,158]. Inspection of immunoglobulin sequence diversity reveals semi-variable framework regions (FR1-4) and hypervariable CDRs, with CDR-H3 being the most diverse domain (Fig. 4). The framework regions are encoded by the germline V- and J-genes, are minimally affected by affinity maturation, and include some well-conserved sequences (YYCAR, etc.) suitable as sequence tags for the hybrid de novo sequencing approaches.
Numerous molecular biology, mass spectrometry, and bioinformatic approaches were developed to attempt antibody de novo sequencing [159]. MS sequencing of circulating antibodies was demonstrated using patient-specific proteomic databases generated through the single B cell receptor mRNA sequencing [159, 160]. Neural networks and deep learning approaches were developed to resolve complex and ambiguous antibody mass spectra and enable MS de novo sequencing without the patient-specific B cell receptor databases [161, 162].

Challenges of characterization and sequencing of polyclonal antibodies by MS. Variable heavy (VH) and light (VL) chains of human immunoglobulins include semi-variable framework regions (FR1-4) and hypervariable CDRs, with CDR-H3 being the most diverse domain. Sequence logos present the experimental diversity of the matched heavy and light variable chains of 199 B-cell clones secreting anti-spike SARS-CoV-2 antibodies, based on reanalysis of data for a single convalescent donor [158]. The framework regions are encoded by the germline V- and J-genes, are minimally affected by affinity maturation, and include some well-conserved sequences (YYCAR, etc.) suitable as sequence tags for the hybrid de novo sequencing approaches
Antibody sequencing with immunoaffinity-proteomics
IA-MS has the potential to advance de novo sequencing of high-affinity antibodies enriched from patient samples. Epitope-directed enrichments using short linear epitope antigens, increasing stringency (salts, pH, or temperature), and high-efficiency separation approaches may facilitate the enrichment of high-affinity, high-abundance, and low-diversity clonotype pools, as we previously demonstrated for aptamers and small molecules [163,164,165,166,167,168,169]. Our recent MS sequencing results for SARS-CoV-2 antibodies (unpublished), as well as other studies [157, 170], revealed that some pools of anti-RBD polyclonal antibodies included low numbers of clonotypes. While some of those high-abundance IGHV/IGKV clonotypes matched published B cell RNAseq data [171], additional high-abundance clonotypes potentially missed by the B cell RNAseq were identified. Intriguingly, even a single patient sample with a major and high-abundance antibody clonotype could be sufficient to sequence the most abundant clone, as demonstrated for a single patient with circulating sepsis-specific IgG1 [156]. Mass spectrometry sequencing of the mature high-affinity and high-abundance antibodies enriched directly from the patient’s samples will enable novel approaches to generate or refine therapeutic antibodies, extend the use of monoclonal antibody therapies, and advance precision immunology [172].
Detecting T cell immunity with immunoaffinity-proteomics
Cell-mediated immunity
In addition to the humoral immunity mediated by B cell-derived antibodies, the adaptive immune system induces cellular immunity mediated by T cells. Briefly, CD8 + killer T cells or CD4 + helper T cells engage the specific T-cell receptors (TCRs) on their surface to search for human cells that display unique peptide antigens bound to the cell-surface Major Histocompatibility Complex (MHC) proteins. Recognition of a specific peptide-MHC complex by TCR triggers T cell activation and proliferation to facilitate either the direct cytotoxic activity and lysis of the infected cells (mediated by CD8 + cytotoxic T lymphocytes), or secretion of cytokines to induce proliferation of antibody-producing B cells (CD4 + helper T lymphocytes). The complexity of immune response emerges from the diversity of MHC proteins (three human MHC class I and three MHC II gene products with two alleles), diversity of T-cell receptor repertoires (VDJ recombination), and the numerous displayed peptides per each antigen (8–10 aa peptides for MHC Class I and 13–25 aa peptides for MHC Class II). Only the specific combination of peptide antigen, MHC allele and TCR results in pMHC-TCR interaction and T cell activation.
T-cell immunity has traditionally been evaluated with Enzyme-linked Immunospot (ELISpot) assays [173]. Such assays identify the specific peptide antigens that can activate patient-derived T cells. A sandwich immunoassay to detect interferon-γ secreted from activated T cells is used as a reporter system. While being very sensitive, ELISpot assays are tedious and low-throughput, require viable T cells, and reveal poor reproducibility across laboratories [174]. More robust and high throughput approaches to evaluate cellular immunity are urgently needed.
The prospects of T cell-mediated immunity evaluation with immunoaffinity proteomics
Recent proteomic approaches enabled the identification and de novo sequencing of MHC class I (8–10 aa) or class II (13–25 aa) peptides, epitope mapping, and identification of protein-protein interactions orchestrating the immune response [175]. Immunoaffinity proteomics may enable direct and in vitro evaluation of T cell-mediated immunity (Fig. 5). For instance, peptide-MHC complexes (pMHC) can be generated using the recombinant MHC I or MHC II proteins and matched synthetic MHC I or MHC II peptides, while the patient-specific soluble TCR pools can be generated with lysis of CD8+ (MHC I) or CD4+ (MHC II) T lymphocytes. Synthetic pMHC complexes will enable affinity enrichment of peptide-specific TCRs (with the optional chemical cross-linking to retain low-affinity interactions). The subsequent trypsin digestion and LC-SRM quantification of the unique peptides of α, β1 and β2 TCR constant chains will measure circulating levels of specific CD8 + or CD4 + T lymphocytes (Fig. 5).

The proposed IA-MS workflow for in vitro measurements of pMHC-specific TCRs. (A) Immunopeptidome workflows facilitate the identification of MHC-specific peptides through the isolation of antigen-presenting cells, affinity enrichment of peptide-MHC (pMHC) complexes, dissociation of class I (9–10 aa) or class II (13–25 aa) MHC peptides, and de novo sequencing of MHC peptides by LC-MS. (B) Recombinant MHC I or MHC II proteins representing the patient-specific Human Leukocyte Antigen (HLA) variants are conjugated to magnetic particle-bound streptavidin tetramers and incubated with the synthetic MHC I or MHC II peptides previously identified with immunopeptidome workflows. Numerous pMHC complexes need to be prepared. (C) CD8 + cytotoxic T lymphocytes (MHC I) and CD4 + helper T lymphocytes (MHC II) are isolated from the patient’s blood and lysed to release soluble TCRs (α-β1 or α-β2). (D) T lymphocyte lysates are incubated with the individual pMHC complexes, and endogenous pMHC-specific TCRs are enriched and covalently cross-linked to pMHCs to retain low-affinity interactions. Following trypsin digestion, unique peptides of α constant (TRAC_HUMAN), β1 constant (TRBC1_HUMAN), and β2 constant (TRBC2_HUMAN) chains are quantified by LC-SRM. Corresponding heavy isotope labeled peptide internal standards (IS) facilitate accurate relative or absolute quantification
The major challenges of evaluating T cell-mediated immunity with immunoaffinity proteomics will include (i) the immense diversity of HLA alleles (numerous polymorphic variants of the recombinant MHC monomers need to be multiplexed on a microplate); (ii) relatively low pMHC-TCR affinities (Kd ~1-100 µM [176]); (iii) inability to differentiate between viable and nonviable T cells. Development of proof-of-concept approaches for TCR identification, quantification, and sequencing may utilize some well-established immunotherapy constructs, such as affinity-matured TCRs binding with high affinity (Kd~13 pM) to the recombinant pMHC complexes (such as melanoma-derived gp100 peptide displayed on HLA-A∗02:01) [177].
TCR sequencing
TCR sequencing, similar to BCR and antibody sequencing, presents a formidable challenge. The TCR repertoire is highly diverse and is generated through recombination, addition, and deletion of the various segments of the alpha and beta chains [178]. TCRs, however, do not undergo somatic hypermutation and affinity maturation. Interestingly, TCR diversity is greatly reduced in patients with some infections or myelomas [179]. In the future, highly multiplex immunoproteomic approaches may enable the identification of the dominant epitope-specific MHCs, assembly of the synthetic peptide-MHC complexes, and their use as baits to enrich specific TCRs for their de novo sequencing. TCR sequencing may revolutionize the development of efficient immunotherapies including CAR-T and autologous cellular immunotherapies [180].
Conclusions
Serological assays were traditionally implemented as indirect immunoassays and their design has not changed for decades. The straightforward setup, speed of manufacturing, and affordability of indirect immunoassays were leveraged by their qualitative or semi-quantitative measurements, non-specific binding, cross-reactivity, lack of reference standards, and challenges with multiplexed measurements of isotypes and subclasses. Immunoaffinity proteomics provides an innovative platform for serological diagnostics to complement conventional indirect immunoassays and resolve their limitations. Clinical needs discussed in this review represent the areas for potential immediate improvement of serological testing. Even small improvements in diagnostic sensitivity and specificity of these assays may provide significant diagnostic benefits due to the large number of serological tests performed in clinical labs. The proposed immunoaffinity proteomic approaches may evolve into a routine serological testing platform and revolutionize serological diagnostics to the same extent as MALDI-TOF revolutionized clinical microbiology testing. Finally, novel approaches for fast and accurate de novo sequencing of human antibodies and TCRs isolated directly from patient samples may facilitate rapid and cost-effective development of high-affinity therapeutic antibodies and cellular immunotherapies, saving millions of lives worldwide.
Data Availability
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ELISA:
-
Enzyme-linked immunosorbent assay
- IA-MS:
-
Immunoaffinity-mass spectrometry
- MHC:
-
Major Histocompatibility Complex
- SRM:
-
Selected reaction monitoring
References
Wassermann, Av. Bruck CGv. Experimentelle Studien über die Wirkung von Tuberkelbacillen-Präparaten auf den tuberculös erkrankten Organismus. Dtsch Med Wochenschr. 1906;32:449–54.
Allard-Chamard H, Boire G. Serologic diagnosis of rheumatoid arthritis. Clin Lab Med. 2019;39(4):525–37.
Lindfors K, Ciacci C, Kurppa K, Lundin KEA, Makharia GK, Mearin ML, et al. Coeliac disease. Nat Reviews Disease Primers. 2019;5(1):3.
Yang W-L, Lu Z, Guo J, Fellman BM, Ning J, Lu KH, et al. Human epididymis protein 4 antigen-autoantibody complexes complement cancer antigen 125 for detecting early-stage ovarian cancer. Cancer. 2020;126(4):725–36.
Brownstein NC, Chen YA. Predictive values, uncertainty, and interpretation of serology tests for the novel coronavirus. Sci Rep. 2021;11(1):5491.
Stages in ELISA. In: The ELISA Guidebook. edn. Edited by Crowther JR. Totowa, NJ: Humana Press. ; 2009: 43–78.
Deane KD. Preclinical rheumatoid arthritis (autoantibodies): an updated review. Curr Rheumatol Rep. 2014;16(5):419.
Zaenker P, Gray ES, Ziman MR. Autoantibody Production in Cancer—The Humoral Immune Response toward Autologous Antigens in Cancer Patients. Autoimmun rev. 2016;15(5):477–83.
Finn OJ. Immune Response as a Biomarker for Cancer Detection and a Lot more. N Engl J Med. 2005;353(12):1288–90.
Fu Z, Rais Y, Dara D, Jackson D, Drabovich AP. Rational design and development of SARS-CoV-2 Serological Diagnostics by immunoprecipitation-targeted proteomics. Anal Chem. 2022;94(38):12990–9.
Snow TM, Coble M. Maternal prenatal screening and serologies. Adv Neonatal Care. 2018;18(6):431–7.
Trépo C, Chan HLY, Lok A. Hepatitis B virus infection. The Lancet. 2014;384(9959):2053–63.
Busch MP, Bloch EM, Kleinman S. Prevention of transfusion-transmitted infections. Blood. 2019;133(17):1854–64.
Prasidthrathsint K, Stapleton JT. Laboratory diagnosis and monitoring of viral Hepatitis. Gastroenterol Clin N Am. 2019;48(2):259–79.
Manns MP, Buti M, Gane E, Pawlotsky J-M, Razavi H, Terrault N, et al. Hepatitis C virus infection. Nat Reviews Disease Primers. 2017;3(1):17006.
Kamar N, Izopet J, Pavio N, Aggarwal R, Labrique A, Wedemeyer H, et al. Hepatitis E virus infection. Nat Reviews Disease Primers. 2017;3(1):17086.
Hwang JP, Feld JJ, Hammond SP, Wang SH, Alston-Johnson DE, Cryer DR, et al. Hepatitis B Virus Screening and Management for patients with Cancer Prior to Therapy: ASCO Provisional Clinical Opinion Update. J Clin Oncol. 2020;38(31):3698–715.
Chen LH, Wilson ME. Yellow fever control: current epidemiology and vaccination strategies. Trop Dis Travel Med Vaccines. 2020;6(1):1.
Waggoner JJ, Rojas A, Pinsky BA. Yellow fever virus: Diagnostics for a persistent arboviral threat. J Clin Microbiol. 2018;56(10):e00827–00818.
Masuet-Aumatell C, Atouguia J. Typhoid fever infection – antibiotic resistance and vaccination strategies: a narrative review. Travel Med Infect Dis. 2021;40:101946.
Crump John A, Mintz Eric D. Global Trends in Typhoid and Paratyphoid Fever. Clin Infect Dis. 2010;50(2):241–6.
Jegaskanda S, Reading PC, Kent SJ. Influenza-specific antibody-dependent Cellular cytotoxicity: toward a Universal Influenza Vaccine. J Immunol. 2014;193(2):469–75.
Khetarpal N, Khanna I, Dengue Fever. Causes, Complications, and Vaccine Strategies. Journal of Immunology Research. 2016; 2016:1–14.
St John AL, Rathore APS. Adaptive immune responses to primary and secondary dengue virus infections. Nat Rev Immunol. 2019;19(4):218–30.
Rossi SL, Ross TM, Evans JD. West Nile Virus. Clin Lab Med. 2010;30(1):47–65.
Sood N, Simon P, Ebner P, Eichner D, Reynolds J, Bendavid E, et al. Seroprevalence of SARS-CoV-2-Specific antibodies among adults in Los Angeles County, California, on April 10–11, 2020. JAMA. 2020;323(23):2425–7.
Rosenberg ES, Tesoriero JM, Rosenthal EM, Chung R, Barranco MA, Styer LM, et al. Cumulative incidence and diagnosis of SARS-CoV-2 infection in New York. Ann Epidemiol. 2020;48:23–9. e24.
Qi H, Liu B, Wang X, Zhang L. The humoral response and antibodies against SARS-CoV-2 infection. Nat Immunol. 2022;23(7):1008–20.
Isho B, Abe KT, Zuo M, Jamal AJ, Rathod B, Wang JH et al. Persistence of serum and saliva antibody responses to SARS-CoV-2 spike antigens in COVID-19 patients. Sci Immunol. 2020; 5(52).
Silman AJ, Pearson JE. Epidemiology and genetics of rheumatoid arthritis. Arthritis Res Therapy. 2002;4(3):265.
Gajendran M, Loganathan P, Catinella AP, Hashash JG. A comprehensive review and update on Crohn’s disease. Dis Mon. 2018;64(2):20–57.
Marakhouski Y, Staliarova T, Gorgun J, Beliauskaya S, Stahievich V, Ruksha K et al. Helicobacter pylori (Hp) infection and upper gastrointestinal mucosal changes in Crohn’s disease patients from the population with high prevalence of Hp. Japan J Res. 2020; 2(1).
Reese GE, Constantinides VA, Simillis C, Darzi AW, Orchard TR, Fazio VW, et al. Diagnostic precision of anti-Saccharomyces cerevisiae antibodies and perinuclear antineutrophil cytoplasmic antibodies in inflammatory bowel disease. Am J Gastroenterol. 2006;101(10):2410–22.
Horn MP, Peter AM, Righini Grunder F, Leichtle AB, Spalinger J, Schibli S, et al. PR3-ANCA and panel diagnostics in pediatric inflammatory bowel disease to distinguish ulcerative colitis from Crohn’s disease. PLoS ONE. 2018;13(12):e0208974.
Papp M, Lakatos P. Serological studies in inflammatory bowel disease: how important are they? Curr Opin Gastroenterol. 2014;30(4):359–64.
Toh B-H, Chan J, Kyaw T, Alderuccio F. Cutting Edge issues in Autoimmune Gastritis. Clin Rev Allergy Immunol. 2012;42(3):269–78.
Toh BH, van Driel IR, Gleeson PA. Pernicious anemia. N Engl J Med. 1997;337(20):1441–8.
Green PH, Jabri B. Coeliac disease. The Lancet. 2003;362(9381):383–91.
Lewis NR, Scott BB. Meta-analysis: deamidated gliadin peptide antibody and tissue transglutaminase antibody compared as screening tests for coeliac disease. Aliment Pharmacol Ther. 2010;31(1):73–81.
Browne P, Chandraratna D, Angood C, Tremlett H, Baker C, Taylor BV, et al. Atlas of multiple sclerosis 2013: a growing global problem with widespread inequity. Neurology. 2014;83(11):1022–4.
Matute-Blanch C, Montalban X, Comabella M. Chap. 5 - Multiple sclerosis, and other demyelinating and autoimmune inflammatory diseases of the central nervous system. In: Handbook of Clinical Neurology. Volume 146, edn. Edited by Deisenhammer F, Teunissen CE, Tumani H: Elsevier; 2018: 67–84.
Thompson AJ, Baranzini SE, Geurts J, Hemmer B, Ciccarelli O. Multiple sclerosis. The Lancet. 2018;391(10130):1622–36.
Cervera R, Khamashta MA, Hughes GRV. The Euro-lupus project: epidemiology of systemic lupus erythematosus in Europe. Lupus. 2009;18(10):869–74.
Petri M, Orbai A-M, Alarcón GS, Gordon C, Merrill JT, Fortin PR, et al. Derivation and validation of the systemic Lupus International collaborating clinics classification criteria for systemic lupus erythematosus. Arthr Rhuem. 2012;64(8):2677–86.
Arbuckle MR, McClain MT, Rubertone MV, Scofield RH, Dennis GJ, James JA, et al. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med. 2003;349(16):1526–33.
Mascarenhas MN, Flaxman SR, Boerma T, Vanderpoel S, Stevens GA. National, Regional, and global Trends in Infertility Prevalence since 1990: a systematic analysis of 277 health surveys. PLoS Med. 2012;9(12):e1001356.
Bieniek JM, Drabovich AP, Lo KC. Seminal biomarkers for the evaluation of male infertility. Asian J Androl. 2016;18(3):426–33.
Schiza CG, Jarv K, Diamandis EP, Drabovich AP. An emerging role of TEX101 protein as a male infertility biomarker. Electron J Int Federation Clin Chem Lab Med. 2014;25(1):9–26.
Jarvi K, Schlegel P, Schiza C, Drabovich A, Lau S, Soosaipillai A, et al. Semen biomarker TEX101 predicts sperm retrieval success for men with testicular failure. F1000Research. 2021;10:569.
Drabovich AP, Saraon P, Jarvi K, Diamandis EP. Seminal plasma as a diagnostic fluid for male reproductive system disorders. Nat Reviews Urol. 2014;11(5):278–88.
Leathersich S, Hart RJ. Immune infertility in men. Fertil Steril. 2022;117(6):1121–31.
Zhang J, Kanoatov M, Jarvi K, Gauthier-Fisher A, Moskovtsev SI, Librach C, et al. Germ cell-specific proteins AKAP4 and ASPX facilitate identification of rare spermatozoa in non-obstructive azoospermia. Mol Cell Proteom. 2023;22(6):100556.
Barbonetti A, Castellini C, D’Andrea S, Cordeschi G, Santucci R, Francavilla S, et al. Prevalence of anti-sperm antibodies and relationship of degree of sperm auto-immunization to semen parameters and post-coital test outcome: a retrospective analysis of over 10 000 men. Hum Reprod. 2019;34(5):834–41.
Saraon P, Drabovich AP, Jarvi KA, Diamandis EP. Mechanisms of androgen-independent prostate Cancer. Electron J Int Federation Clin Chem Lab Med. 2014;25(1):42–54.
Monroy-Iglesias MJ, Crescioli S, Beckmann K, Le N, Karagiannis SN, Van Hemelrijck M et al. Antibodies as biomarkers for cancer risk: a systematic review. Clin Exp Immunol. 2022:uxac030.
Henderson MC, Silver M, Tran Q, Letsios EE, Mulpuri R, Reese DE, et al. A noninvasive blood-based Combinatorial Proteomic Biomarker Assay to detect breast Cancer in women over age 50 with BI-RADS 3, 4, or 5 Assessment. Clin Cancer Res. 2019;25(1):142–9.
Sullivan FM, Mair FS, Anderson W, Armory P, Briggs A, Chew C et al. Earlier diagnosis of lung cancer in a randomised trial of an autoantibody blood test followed by imaging. Eur Respir J. 2021; 57(1).
Choi MY, Clarke AE, Urowitz M, Hanly J, St-Pierre Y, Gordon C, et al. Longitudinal analysis of ANA in the systemic Lupus International collaborating clinics (SLICC) inception cohort. Annals of Rheumatic Diseases. 2022;81(8):1143–50.
Dalmau J, Rosenfeld MR. Paraneoplastic syndromes of the CNS. Lancet Neurol. 2008;7(4):327–40.
Titulaer MJ, Soffietti R, Dalmau J, Gilhus NE, Giometto B, Graus F, et al. Screening for tumours in paraneoplastic syndromes: report of an EFNS task force. Eur J Neurol. 2011;18(1):19–e13.
Akahoshi M, Arinobu Y, Kashiwado Y, Omoto A, Ayano M, Mitoma H, et al. IgG4-related disease presenting as a paraneoplastic syndrome: report of two cases and literature review. Mod Rheumatol Case Rep. 2021;5(2):371–6.
Yamamoto M, Takahashi H, Tabeya T, Suzuki C, Naishiro Y, Ishigami K, et al. Risk of malignancies in IgG4-related disease. Mod Rheumatol. 2012;22(3):414–8.
Sharonov GV, Serebrovskaya EO, Yuzhakova DV, Britanova OV, Chudakov DM. B cells, plasma cells and antibody repertoires in the tumour microenvironment. Nat Rev Immunol. 2020;20(5):294–307.
Volkov M, Coppola M, Huizinga R, Eftimov F, Huizinga TWJ, van der Kooi AJ, et al. Comprehensive overview of autoantibody isotype and subclass distribution. J Allergy Clin Immunol. 2022;150(5):999–1010.
Bernal F, Shams’ili S, Rojas I, Sanchez-Valle R, Saiz A, Dalmau J, et al. Anti-tr antibodies as markers of paraneoplastic cerebellar degeneration and Hodgkin’s disease. Neurology. 2003;60(2):230–4.
Aalberse RC, Stapel SO, Schuurman J, Rispens T. Immunoglobulin G4: an odd antibody. Clin Experimental Allergy. 2009;39(4):469–77.
Karagiannis P, Gilbert AE, Josephs DH, Ali N, Dodev T, Saul L, et al. IgG4 subclass antibodies impair antitumor immunity in melanoma. J Clin Invest. 2013;123(4):1457–74.
Wang H, Xu Q, Zhao C, Zhu Z, Zhu X, Zhou J et al. An immune evasion mechanism with IgG4 playing an essential role in cancer and implication for immunotherapy. J Immunother Cancer. 2020; 8(2).
Crescioli S, Correa I, Karagiannis P, Davies AM, Sutton BJ, Nestle FO, et al. IgG4 characteristics and functions in Cancer Immunity. Curr Allergy Asthma Rep. 2016;16(1):7.
Go RS, Rajkumar SV. How I manage monoclonal gammopathy of undetermined significance. Blood. 2018;131(2):163–73.
Kyle RA, Rajkumar SV. Management of monoclonal gammopathy of undetermined significance (MGUS) and smoldering multiple myeloma (SMM). Oncol (Williston Park). 2011;25(7):578–86.
Zajec M, Langerhorst P, VanDuijn MM, Gloerich J, Russcher H, van Gool AJ, et al. Mass Spectrometry for Identification, Monitoring, and minimal residual disease detection of M-Proteins. Clin Chem. 2020;66(3):421–33.
Engvall E. The ELISA, enzyme-linked immunosorbent assay. Clin Chem. 2010;56(2):319–20.
Hornbeck P. Enzyme-Linked Immunosorbent Assays. Current Protocols in Immunology. 1992; 1(1):2.1.1–2.1.22.
Perkmann T, Mucher P, Osze D, Muller A, Perkmann-Nagele N, Koller T, et al. Comparison of five Anti-SARS-CoV-2 antibody assays across three doses of BNT162b2 reveals insufficient standardization of SARS-CoV-2 serology. J Clin Virol. 2023;158:105345.
Tighe PJ, Ryder RR, Todd I, Fairclough LC. ELISA in the multiplex era: potentials and pitfalls. Proteom Clin Appl. 2015;9(3–4):406–22.
Vignali DA. Multiplexed particle-based flow cytometric assays. J Immunol Methods. 2000;243(1–2):243–55.
Koczula Katarzyna M, Gallotta A. Lateral flow assays. Essays Biochem. 2016;60(1):111–20.
Peto T, Team UC-LFO. COVID-19: Rapid antigen detection for SARS-CoV-2 by lateral flow assay: a national systematic evaluation of sensitivity and specificity for mass-testing. EClinicalMedicine. 2021;36:100924.
Lynch HE, Sanchez AM, D’Souza MP, Rountree W, Denny TN, Kalos M, et al. Development and implementation of a proficiency testing program for Luminex bead-based cytokine assays. J Immunol Methods. 2014;409:62–71.
Falkowski P, Lukaszewski Z, Gorodkiewicz E. Potential of surface plasmon resonance biosensors in cancer detection. J Pharm Biomed Anal. 2021;194:113802.
Lazcka O, Campo FJD, Muñoz FX. Pathogen detection: a perspective of traditional methods and biosensors. Biosens Bioelectron. 2007;22(7):1205–17.
Calvo-Lozano O, Sierra M, Soler M, Estevez MC, Chiscano-Camon L, Ruiz-Sanmartin A, et al. Label-free Plasmonic Biosensor for Rapid, quantitative, and highly sensitive COVID-19 serology: implementation and clinical validation. Anal Chem. 2022;94(2):975–84.
Jung S-H, Jung J-W, Suh I-B, Yuk JS, Kim W-J, Choi EY, et al. Analysis of C-Reactive protein on Amide-Linked N -Hydroxysuccinimide – dextran arrays with a spectral surface Plasmon Resonance Biosensor for Serodiagnosis. Anal Chem. 2007;79(15):5703–10.
Thoren KL, Pasi B, Delgado JC, Wu AHB, Lynch KL. Quantitation of Infliximab and Detection of Antidrug antibodies in serum by Use of Surface Plasmon Resonance. J Appl Lab Med. 2018;2(5):725–36.
Rhea K. Determining the binding kinetics of peptide macrocycles using Bio-Layer Interferometry (BLI). Methods Mol Biol. 2022;2371:355–72.
Guo Z, Wilson JR, York IA, Stevens J. Biosensor-based epitope mapping of antibodies targeting the hemagglutinin and neuraminidase of influenza a virus. J Immunol Methods. 2018;461:23–9.
Brady T, Zhang T, Tuffy KM, Haskins N, Du Q, Lin J, et al. Qualification of a Biolayer Interferometry Assay to support AZD7442 resistance monitoring. Microbiol Spectr. 2022;10(5):e0103422.
Dzimianski JV, Lorig-Roach N, O’Rourke SM, Alexander DL, Kimmey JM, DuBois RM. Rapid and sensitive detection of SARS-CoV-2 antibodies by biolayer interferometry. Sci Rep. 2020;10(1):21738.
Luo YR, Chakraborty I, Lazar-Molnar E, Wu AHB, Lynch KL. Development of label-free Immunoassays as Novel Solutions for the measurement of monoclonal antibody drugs and Antidrug antibodies. Clin Chem. 2020;66(10):1319–28.
Luo YR, Chakraborty I, Zuk RF, Lynch KL, Wu AHB. A thin-film interferometry-based label-free immunoassay for the detection of daratumumab interference in serum protein electrophoresis. Clin Chim Acta. 2020;502:128–32.
Luo YR, Yun C, Chakraborty I, Wu AHB, Lynch KL. A SARS-CoV-2 label-free surrogate virus neutralization test and a longitudinal study of antibody characteristics in COVID-19 patients. J Clin Microbiol. 2021;59(7):e0019321.
Liu H, Yang A, Song J, Wang N, Lam P, Li Y, et al. Ultrafast, sensitive, and portable detection of COVID-19 IgG using flexible organic electrochemical transistors. Sci Adv. 2021;7(38):eabg8387.
Zhang Z, Li Q, Du X, Liu M. Application of electrochemical biosensors in tumor cell detection. Thorac Cancer. 2020;11(4):840–50.
Esteves-Villanueva JO, Trzeciakiewicz H, Martic S. A protein-based electrochemical biosensor for detection of tau protein, a neurodegenerative disease biomarker. Analyst. 2014;139(11):2823–31.
Mahshid SS, Flynn SE, Mahshid S. The potential application of electrochemical biosensors in the COVID-19 pandemic: a perspective on the rapid diagnostics of SARS-CoV-2. Biosens Bioelectron. 2021;176:112905.
Haab BB, Dunham MJ, Brown PO. Protein microarrays for highly parallel detection and quantitation of specific proteins and antibodies in complex solutions. Genome Biol. 2001;2(2):research00040001.
Robinson WH, DiGennaro C, Hueber W, Haab BB, Kamachi M, Dean EJ, et al. Autoantigen microarrays for multiplex characterization of autoantibody responses. Nat Med. 2002;8(3):295–301.
Wang H, Wu X, Zhang X, Hou X, Liang T, Wang D, et al. SARS-CoV-2 Proteome microarray for Mapping COVID-19 antibody interactions at amino acid resolution. ACS Cent Sci. 2020;6(12):2238–49.
Venkataraman A, Yang K, Irizarry J, Mackiewicz M, Mita P, Kuang Z, et al. A toolbox of immunoprecipitation-grade monoclonal antibodies to human transcription factors. Nat Methods. 2018;15(5):330–8.
Pan J, Yu L, Wu Q, Lin X, Liu S, Hu S, et al. Integration of IgA and IgG autoantigens improves performance of Biomarker Panels for early diagnosis of Lung Cancer. Mol Cell Proteomics. 2020;19(3):490–500.
Syu G-D, Dunn J, Zhu H. Developments and applications of functional protein microarrays. Mol Cell Proteom. 2020;19(6):916–27.
Saraon P, Musrap N, Cretu D, Karagiannis GS, Batruch I, Smith C, et al. Proteomic profiling of androgen-independent prostate cancer cell lines reveals a role for protein S during the development of high grade and castration-resistant prostate cancer. J Biol Chem. 2012;287(41):34019–31.
Konvalinka A, Zhou J, Dimitromanolakis A, Drabovich AP, Fang F, Gurley S, et al. Determination of an angiotensin II-regulated proteome in primary human kidney cells by stable isotope labeling of amino acids in cell culture (SILAC). J Biol Chem. 2013;288(34):24834–47.
Giansanti P, Tsiatsiani L, Low TY, Heck AJ. Six alternative proteases for mass spectrometry-based proteomics beyond trypsin. Nat Protoc. 2016;11(5):993–1006.
Han X, Aslanian A, Yates JR. 3rd. Mass spectrometry for proteomics. Curr Opin Chem Biol. 2008;12(5):483–90.
Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422(6928):198–207.
Zubarev RA, Makarov A. Orbitrap mass spectrometry. Anal Chem. 2013;85(11):5288–96.
Liebler DC, Zimmerman LJ. Targeted quantitation of proteins by mass spectrometry. Biochemistry. 2013;52(22):3797–806.
Zhurov KO, Fornelli L, Wodrich MD, Laskay UA, Tsybin YO. Principles of electron capture and transfer dissociation mass spectrometry applied to peptide and protein structure analysis. Chem Soc Rev. 2013;42(12):5014–30.
Drabovich AP, Jarvi K, Diamandis EP. Verification of male infertility biomarkers in seminal plasma by multiplex selected reaction monitoring assay. Mol Cell Proteom. 2011;10(12):M110004127.
Drabovich AP, Pavlou MP, Schiza C, Diamandis EP. Dynamics of protein expression reveals primary targets and secondary messengers of estrogen receptor alpha signaling in MCF-7 breast cancer cells. Mol Cell Proteom. 2016;15(6):2093–107.
Schiza C, Korbakis D, Panteleli E, Jarvi K, Drabovich AP, Diamandis EP. Discovery of a human testis-specific protein complex TEX101-DPEP3 and selection of its disrupting antibodies. Mol Cell Proteom. 2018;17(12):2480–95.
Drabovich AP, Diamandis EP. Combinatorial peptide libraries facilitate development of multiple reaction monitoring assays for low-abundance proteins. J Proteome Res. 2010;9(3):1236–45.
Prakash A, Rezai T, Krastins B, Sarracino D, Athanas M, Russo P, et al. Platform for establishing interlaboratory reproducibility of selected reaction monitoring-based mass spectrometry peptide assays. J Proteome Res. 2010;9(12):6678–88.
Cho CK, Drabovich AP, Batruch I, Diamandis EP. Verification of a biomarker discovery approach for detection of Down syndrome in amniotic fluid via multiplex selected reaction monitoring (SRM) assay. J Proteom. 2011;74(10):2052–9.
Begcevic I, Brinc D, Drabovich AP, Batruch I, Diamandis EP. Identification of brain-enriched proteins in the cerebrospinal fluid proteome by LC-MS/MS profiling and mining of the human protein atlas. Clin Proteomics. 2016;13:11.
Cho CK, Drabovich AP, Karagiannis GS, Martinez-Morillo E, Dason S, Dimitromanolakis A, et al. Quantitative proteomic analysis of amniocytes reveals potentially dysregulated molecular networks in Down syndrome. Clin Proteomics. 2013;10(1):2.
Konvalinka A, Batruch I, Tokar T, Dimitromanolakis A, Reid S, Song X, et al. Quantification of angiotensin II-regulated proteins in urine of patients with polycystic and other chronic kidney diseases by selected reaction monitoring. Clin Proteomics. 2016;13:16.
Dimitrakopoulos L, Prassas I, Diamandis EP, Nesvizhskii A, Kislinger T, Jaffe J, et al. Proteogenomics: Opportunities and Caveats. Clin Chem. 2016;62(4):551–7.
Drabovich AP, Martínez-Morillo E, Diamandis EP. Chap. 3 - Protein Biomarker Discovery: An Integrated Concept. In: Proteomics for Biological Discovery 2nd Edition edn. Edited by Veenstra TD, Yates III JR. Hoboken, NJ2019: Wiley-Blackwell; 2019: 63–88.
Saraon P, Cretu D, Musrap N, Karagiannis GS, Batruch I, Drabovich AP, et al. Quantitative proteomics reveals that enzymes of the ketogenic pathway are associated with prostate cancer progression. Mol Cell Proteom. 2013;12(6):1589–601.
Cox J, Mann M, Quantitative. High-resolution proteomics for Data-Driven Systems Biology. Annu Rev Biochem. 2011;80(1):273–99.
Martinez-Morillo E, Cho CK, Drabovich AP, Shaw JL, Soosaipillai A, Diamandis EP. Development of a multiplex selected reaction monitoring assay for quantification of biochemical markers of down syndrome in amniotic fluid samples. J Proteome Res. 2012;11(7):3880–7.
Martinez-Morillo E, Nielsen HM, Batruch I, Drabovich AP, Begcevic I, Lopez MF, et al. Assessment of peptide chemical modifications on the development of an accurate and precise multiplex selected reaction monitoring assay for apolipoprotein e isoforms. J Proteome Res. 2014;13(2):1077–87.
Begcevic I, Brinc D, Dukic L, Simundic AM, Zavoreo I, Basic Kes V, et al. Targeted mass spectrometry-based assays for relative quantification of 30 brain-related proteins and their clinical applications. J Proteome Res. 2018;17(7):2282–92.
Drabovich AP, Pavlou MP, Dimitromanolakis A, Diamandis EP. Quantitative analysis of energy metabolic pathways in MCF-7 breast cancer cells by selected reaction monitoring assay. Mol Cell Proteom. 2012;11(8):422–34.
Korbakis D, Brinc D, Schiza C, Soosaipillai A, Jarvi K, Drabovich AP, et al. Immunocapture-selected reaction monitoring screening facilitates the development of ELISA for the measurement of native TEX101 in Biological Fluids. Mol Cell Proteom. 2015;14(6):1517–26.
Korbakis D, Schiza C, Brinc D, Soosaipillai A, Karakosta TD, Legare C, et al. Preclinical evaluation of a TEX101 protein ELISA test for the differential diagnosis of male infertility. BMC Med. 2017;15(1):60.
Prakash A, Rezai T, Krastins B, Sarracino D, Athanas M, Russo P, et al. Interlaboratory reproducibility of selective reaction monitoring assays using multiple upfront analyte enrichment strategies. J Proteome Res. 2012;11(8):3986–95.
Drabovich AP, Dimitromanolakis A, Saraon P, Soosaipillai A, Batruch I, Mullen B, et al. Differential diagnosis of azoospermia with proteomic biomarkers ECM1 and TEX101 quantified in seminal plasma. Sci Transl Med. 2013;5(212):212ra160.
Rais Y, Fu Z, Drabovich AP. Mass spectrometry-based proteomics in basic and translational research of SARS-CoV-2 coronavirus and its emerging mutants. Clin Proteom. 2021;18(1):19.
Thompson NJ, Rosati S, Heck AJR. Performing native mass spectrometry analysis on therapeutic antibodies. Methods. 2014;65(1):11–7.
Ladwig PM, Barnidge DR, Snyder MR, Katzmann JA, Murray DL. Quantification of serum IgG subclasses by Use of Subclass-Specific tryptic peptides and Liquid Chromatography–Tandem Mass Spectrometry. Clin Chem. 2014;60(8):1080–8.
Drabovich AP, Martinez-Morillo E, Diamandis EP. Toward an integrated pipeline for protein biomarker development. Biochim Biophys Acta. 2015;1854(6):677–86.
Drabovich AP, Saraon P, Drabovich M, Karakosta TD, Dimitromanolakis A, Hyndman ME, et al. Multi-omics biomarker pipeline reveals elevated levels of protein-glutamine Gamma-glutamyltransferase 4 in seminal plasma of prostate cancer patients. Mol Cell Proteom. 2019;18(9):1807–23.
Drabovich AP, Pavlou MP, Batruch I, Diamandis EP. Chap. 2 - Proteomic and mass spectrometry technologies for biomarker discovery. In: Proteomic and Metabolomic Approaches to Biomarker Discovery edn. Edited by Issaq HJ, Veenstra TD. Waltham, MA: Academic Press (Elsevier); 2013: 17–37.
Nelson RW, Krone JR, Bieber AL, Williams P. Mass spectrometric immunoassay. Anal Chem. 1995;67(7):1153–8.
Anderson NL, Jackson A, Smith D, Hardie D, Borchers C, Pearson TW. SISCAPA peptide enrichment on magnetic beads using an in-line bead trap device. Mol Cell Proteomics. 2009;8(5):995–1005.
Fu Z, Rais Y, Bismar TA, Hyndman ME, Le XC, Drabovich AP. Mapping isoform abundance and interactome of the endogenous TMPRSS2-ERG Fusion protein by Orthogonal Immunoprecipitation-Mass spectrometry assays. Mol Cell Proteom. 2021;20:100075.
Moore JL, Patterson NH, Norris JL, Caprioli RM. Prospective on imaging Mass Spectrometry in Clinical Diagnostics. Mol Cell Proteomics. 2023:100576.
Schiza C, Korbakis D, Jarvi K, Diamandis EP, Drabovich AP. Identification of TEX101-associated proteins through Proteomic Measurement of Human Spermatozoa homozygous for the missense variant rs35033974. Mol Cell Proteom. 2019;18(2):338–51.
Karakosta TD, Soosaipillai A, Diamandis EP, Batruch I, Drabovich AP. Quantification of human kallikrein-related peptidases in Biological Fluids by Multiplatform targeted Mass Spectrometry assays. Mol Cell Proteom. 2016;15(9):2863–76.
Liang X, Sun R, Wang J, Zhou K, Li J, Chen S, et al. Proteomics Investigation of Diverse serological patterns in COVID-19. Mol Cell Proteom. 2023;22(2):100493.
Doykov I, Baldwin T, Spiewak J, Gilmour KC, Gibbons JM, Pade C, et al. Quantitative, multiplexed, targeted proteomics for ascertaining variant specific SARS-CoV-2 antibody response. Cell Rep Methods. 2022;2(9):100279.
Carr SA, Abbatiello SE, Ackermann BL, Borchers C, Domon B, Deutsch EW, et al. Targeted peptide measurements in biology and medicine: best practices for mass spectrometry-based assay development using a fit-for-purpose approach. Mol Cell Proteomics. 2014;13(3):907–17.
CLSI. C64 - Quantitative measurement of proteins and peptides by mass spectrometry.https://clsi.org/standards/products/clinical-chemistry-and-toxicology/documents/c64.
Nikolaev EN, Indeykina MI, Brzhozovskiy AG, Bugrova AE, Kononikhin AS, Starodubtseva NL, et al. Mass-Spectrometric detection of SARS-CoV-2 Virus in Scrapings of the epithelium of the nasopharynx of infected patients via Nucleocapsid N protein. J Proteome Res. 2020;19(11):4393–7.
Sarkar D, Sinclair E, Lim SH, Walton-Doyle C, Jafri K, Milne J, et al. Paper Spray Ionization Ion mobility Mass Spectrometry of Sebum classifies biomarker classes for the diagnosis of Parkinson’s Disease. JACS Au. 2022;2(9):2013–22.
Oviano M, Bou G. Matrix-assisted laser desorption ionization-time of Flight Mass Spectrometry for the Rapid detection of Antimicrobial Resistance Mechanisms and Beyond. Clin Microbiol Rev. 2019; 32(1).
Carter PJ, Lazar GA. Next generation antibody drugs: pursuit of the ‘high-hanging fruit’. Nat Rev Drug Discovery. 2018;17(3):197–223.
Snapkov I, Chernigovskaya M, Sinitcyn P, Lê Quý K, Nyman TA, Greiff V. Progress and challenges in mass spectrometry-based analysis of antibody repertoires. Trends Biotechnol. 2022;40(4):463–81.
Hershberg U, Luning Prak ET. The analysis of clonal expansions in normal and autoimmune B cell repertoires. Philos Trans R Soc Lond B Biol Sci. 2015; 370(1676).
Lefranc MP, Giudicelli V, Duroux P, Jabado-Michaloud J, Folch G, Aouinti S, et al. IMGT(R), the international ImMunoGeneTics information system(R) 25 years on. Nucleic Acids Res. 2015;43(Database issue):D413–422.
Soto C, Bombardi RG, Branchizio A, Kose N, Matta P, Sevy AM, et al. High frequency of shared clonotypes in human B cell receptor repertoires. Nature. 2019;566(7744):398–402.
Bondt A, Hoek M, Tamara S, de Graaf B, Peng W, Schulte D, et al. Human plasma IgG1 repertoires are simple, unique, and dynamic. Cell Syst. 2021;12(12):1131–1143e1135.
van Rijswijck DMH, Bondt A, Hoek M, van der Straten K, Caniels TG, Poniman M, et al. Discriminating cross-reactivity in polyclonal IgG1 responses against SARS-CoV-2 variants of concern. Nat Commun. 2022;13(1):6103.
Wec AZ, Wrapp D, Herbert AS, Maurer DP, Haslwanter D, Sakharkar M, et al. Broad neutralization of SARS-related viruses by human monoclonal antibodies. Science. 2020;369(6504):731–6.
Boutz DR, Horton AP, Wine Y, Lavinder JJ, Georgiou G, Marcotte EM. Proteomic identification of monoclonal antibodies from serum. Anal Chem. 2014;86(10):4758–66.
Cheung WC, Beausoleil SA, Zhang X, Sato S, Schieferl SM, Wieler JS, et al. A proteomics approach for the identification and cloning of monoclonal antibodies from serum. Nat Biotechnol. 2012;30(5):447–52.
Hieu TN, Xianglilan Z, Lei X, Baozhen S, Ming L. De novo peptide sequencing by deep learning. Proceedings of the National Academy of Sciences. 2017; 114(31):8247–8252.
Tran NH, Qiao R, Xin L, Chen X, Liu C, Zhang X, et al. Deep learning enables de novo peptide sequencing from data-independent-acquisition mass spectrometry. Nat Methods. 2019;16(1):63–6.
Drabovich AP, Berezovski M, Okhonin V, Krylov SN. Selection of smart aptamers by methods of kinetic capillary electrophoresis. Anal Chem. 2006;78(9):3171–8.
Drabovich AP, Berezovski MV, Musheev MU, Krylov SN. Selection of smart small-molecule ligands: the proof of principle. Anal Chem. 2009;81(1):490–4.
Berezovski M, Drabovich A, Krylova SM, Musheev M, Okhonin V, Petrov A, et al. Nonequilibrium capillary electrophoresis of equilibrium mixtures: a universal tool for development of aptamers. J Am Chem Soc. 2005;127(9):3165–71.
Berezovski M, Musheev M, Drabovich A, Krylov SN. Non-SELEX selection of aptamers. J Am Chem Soc. 2006;128(5):1410–1.
Drabovich A, Berezovski M, Krylov SN. Selection of smart aptamers by equilibrium capillary electrophoresis of equilibrium mixtures (ECEEM). J Am Chem Soc. 2005;127(32):11224–5.
Drabovich AP, Okhonin V, Berezovski M, Krylov SN. Smart aptamers facilitate multi-probe affinity analysis of proteins with ultra-wide dynamic range of measured concentrations. J Am Chem Soc. 2007;129(23):7260–1.
Berezovski MV, Musheev MU, Drabovich AP, Jitkova JV, Krylov SN. Non-SELEX: selection of aptamers without intermediate amplification of candidate oligonucleotides. Nat Protoc. 2006;1(3):1359–69.
Melani RD, Des Soye BJ, Kafader JO, Forte E, Hollas M, Blagojevic V, et al. Next-generation serology by Mass Spectrometry: readout of the SARS-CoV-2 antibody repertoire. J Proteome Res. 2022;21(1):274–88.
Wang Y, Yuan M, Lv H, Peng J, Wilson IA, Wu NC. A large-scale systematic survey reveals recurring molecular features of public antibody responses to SARS-CoV-2. Immunity. 2022;55(6):1105–1117e1104.
Robinson WH. Sequencing the functional antibody repertoire—diagnostic and therapeutic discovery. Nat Rev Rheumatol. 2015;11(3):171–82.
Janetzki S, Price L, Schroeder H, Britten CM, Welters MJP, Hoos A. Guidelines for the automated evaluation of Elispot assays. Nat Protoc. 2015;10(7):1098–115.
Britten CM, Gouttefangeas C, Welters MJP, Pawelec G, Koch S, Ottensmeier C, et al. The CIMT-monitoring panel: a two-step approach to harmonize the enumeration of antigen-specific CD8 + T lymphocytes by structural and functional assays. Cancer Immunol Immunother. 2008;57(3):289–302.
Croft NP, Purcell AW, Tscharke DC. Quantifying epitope presentation using mass spectrometry. Mol Immunol. 2015;68(2):77–80.
Davis MM, Boniface JJ, Reich Z, Lyons D, Hampl J, Arden B, et al. Ligand recognition by αβ T cell receptors. Annu Rev Immunol. 1998;16(1):523–44.
Susac L, Vuong MT, Thomas C, von Bulow S, O’Brien-Ball C, Santos AM, et al. Structure of a fully assembled tumor-specific T cell receptor ligated by pMHC. Cell. 2022;185(17):3201–3213e3219.
Nikolich-Žugich J, Slifka MK, Messaoudi I. The many important facets of T-cell repertoire diversity. Nat Rev Immunol. 2004;4(2):123–32.
Matos TR, de Rie MA, Teunissen MBM. Research Techniques made simple: high-throughput sequencing of the T-Cell receptor. J Invest Dermatology. 2017;137(6):e131–8.
Maciocia PM, Wawrzyniecka PA, Philip B, Ricciardelli I, Akarca AU, Onuoha SC, et al. Targeting the T cell receptor β-chain constant region for immunotherapy of T cell malignancies. Nat Med. 2017;23(12):1416–23.
Hübschen JM, Bork SM, Brown KE, Mankertz A, Santibanez S, Ben Mamou M, et al. Challenges of measles and rubella laboratory diagnostic in the era of elimination. Clin Microbiol Infect. 2017;23(8):511–5.
Sundqvist VA, Linde A, Wahren B. Virus-specific immunoglobulin G subclasses in herpes simplex and varicella-zoster virus infections. J Clin Microbiol. 1984;20(1):94–8.
Yates NL, Liao HX, Fong Y, deCamp A, Vandergrift NA, Williams WT, et al. Vaccine-induced Env V1-V2 IgG3 correlates with lower HIV-1 infection risk and declines soon after vaccination. Sci Transl Med. 2014;6(228):228ra239.
Satyaputra F, Hendry S, Braddick M, Sivabalan P, Norton R. The Laboratory diagnosis of Syphilis. J Clin Microbiol. 2021;59(10):e00100–00121.
Geisler WM, Morrison SG, Doemland ML, Iqbal SM, Su J, Mancevski A, et al. Immunoglobulin-specific responses to Chlamydia elementary bodies in individuals with and at risk for genital chlamydial infection. J Infect Dis. 2012;206(12):1836–43.
Sanchez E, Vannier E, Wormser GP, Hu LT. Diagnosis, treatment, and Prevention of Lyme Disease, Human Granulocytic Anaplasmosis, and babesiosis: a review. JAMA. 2016;315(16):1767.
Mangano VD, Perandin F, Tiberti N, Guerriero M, Migliaccio F, Prato M, et al. Risk of transfusion-transmitted malaria: evaluation of commercial ELISA kits for the detection of anti-plasmodium antibodies in candidate blood donors. Malar J. 2019;18(1):17.
Jones S, Finnegan K, Wee JH, D’Argoeuves P, Roalfe L, Byrne S et al. Assignment of serotype-specific IgG1, IgG2, and IgA Weight-Based antibody units to the human pneumococcal standard reference serum, 007sp. mSphere. 2019; 4(3).
Chapuy-Regaud S, Nogueira L, Clavel C, Sebbag M, Vincent C, Serre G. IgG subclass distribution of the rheumatoid arthritis-specific autoantibodies to citrullinated fibrin. Clin Exp Immunol. 2005;139(3):542–50.
Ruthlein J, Ibe M, Burghardt W, Mossner J, Auer IO, Immunoglobulin G. (IgG), IgG1, and IgG2 determinations from endoscopic biopsy specimens in control, Crohn’s disease, and ulcerative colitis subjects. Gut. 1992;33(4):507–12.
Lahner E, Norman GL, Severi C, Encabo S, Shums Z, Vannella L et al. Reassessment of intrinsic factor and parietal cell autoantibodies in Atrophic Gastritis with respect to Cobalamin Deficiency. Am J Gastroenterol. 2009; 104(8).
Tabata T, Kamisawa T, Takuma K, Anjiki H, Egawa N, Kurata M, et al. Serum IgG4 concentrations and IgG4-related sclerosing disease. Clin Chim Acta. 2009;408(1):25–8.
Palladini G, Russo P, Bosoni T, Verga L, Sarais G, Lavatelli F, et al. Identification of Amyloidogenic Light Chains requires the combination of serum-free light chain assay with immunofixation of serum and urine. Clin Chem. 2009;55(3):499–504.
Chen Y-T, Scanlan MJ, Sahin U, Türeci Ö, Gure AO, Tsang S et al. A testicular antigen aberrantly expressed in human cancers detected by autologous antibody screening. Proceedings of the National Academy of Sciences. 1997; 94(5):1914–1918.
Lubin R, Schlichtholz B, Teillaud JL, Garay E, Bussel A, Wild CP. p53 antibodies in patients with various types of cancer: assay, identification, and characterization. Clin Cancer Res. 1995;1(12):1463–9.
Soussi T. p53 antibodies in the Sera of patients with various types of Cancer: a Review1. Cancer Res. 2000;60(7):1777–88.
Weber D, Ibn-Salem J, Sorn P, Suchan M, Holtstrater C, Lahrmann U, et al. Accurate detection of tumor-specific gene fusions reveals strongly immunogenic personal neo-antigens. Nat Biotechnol. 2022;40(8):1276–84.
Yang W, Lee KW, Srivastava RM, Kuo F, Krishna C, Chowell D, et al. Immunogenic neoantigens derived from gene fusions stimulate T cell responses. Nat Med. 2019;25(5):767–75.
Akdis M, Akdis CA. Mechanisms of allergen-specific immunotherapy: multiple suppressor factors at work in immune tolerance to allergens. J Allergy Clin Immunol. 2014;133(3):621–31.
Acknowledgements
Figures were created with BioRender.com.
Funding
This work was supported by the Canadian Institutes of Health Research COVID-19 Immunity Task Force grant (VR2-173207) and the Alberta Innovates Health Programs grant to A.P.D.
Author information
Authors and Affiliations
Contributions
J.W., Z.E. and A.P. D. wrote the main manuscript text and prepared figures and tables. All authors reviewed and revised the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Walter, J., Eludin, Z. & Drabovich, A.P. Redefining serological diagnostics with immunoaffinity proteomics. Clin Proteom 20, 42 (2023). https://doi.org/10.1186/s12014-023-09431-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12014-023-09431-y