
Abstract
Objective
Lymph node metastasis (LNM) and lymphatic vasculature space infiltration (LVSI) in cervical cancer patients indicate a poor prognosis, but satisfactory methods for diagnosing these phenotypes are lacking. This study aimed to find new effective plasma biomarkers of LNM and LVSI as well as possible mechanisms underlying LNM and LVSI through data-independent acquisition (DIA) proteome sequencing.
Methods
A total of 20 cervical cancer plasma samples, including 7 LNM-/LVSI-(NC), 4 LNM-/LVSI + (LVSI) and 9 LNM + /LVSI + (LNM) samples from a cohort, were subjected to DIA to identify differentially expressed proteins (DEPs) for LVSI and LNM. Subsequently, Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses were performed for DEP functional annotation. Protein–protein interaction (PPI) and weighted gene coexpression network analysis (WGCNA) were used to detect new effective plasma biomarkers and possible mechanisms.
Results
A total of 79 DEPs were identified in the cohort. GO and KEGG analyses showed that DEPs were mainly enriched in the complement and coagulation pathway, lipid and atherosclerosis pathway, HIF-1 signal transduction pathway and phagosome and autophagy. WGCNA showed that the enrichment of the green module differed greatly between groups. Six interesting core DEPs (SPARC, HPX, VCAM1, TFRC, ERN1 and APMAP) were confirmed to be potential plasma diagnostic markers for LVSI and LNM in cervical cancer patients.
Conclusion
Proteomic signatures developed in this study reflected the potential plasma diagnostic markers and new possible pathogenesis mechanisms in the LVSI and LNM of cervical cancer.
Similar content being viewed by others


Introduction
Cervical cancer is one of the most common malignant gynaecological tumours. It is also one of the leading causes of morbidity and mortality among females worldwide [1]. The prognosis of cervical cancer patients is highly related to postoperative pathological factors. Lymph node metastasis (LNM) is a high-risk factor, and lymphatic vasculature space infiltration (LVSI) is a moderate-risk factor. Given this, the International Federation of Gynaecology & Obstetrics (FIGO) staging system underwent revision in 2018 [2]. The new staging system expanded the stage III classifications to include stage IIIC: All cases with lymph node involvement were classified as stage IIIC; those with pelvic or para-aortic lymph node involvement were classified as stage IIIC1 or stage IIIC2. The classification was also based on whether diagnosis was made radiographic assessment only (r) or pathological confirmation (p). The stage was classified as IIIC1r if imaging showed pelvic lymph node metastasis. If confirmed by pathological results, the stage was IIIC1p. However, the latest meta-analysis showed that imaging examinations are barely satisfactory. The false-negative rate of PET-CT and MRI or CT imaging for the detection of para-aortic lymph node metastasis reaches 11–21%. The sensitivity of transvaginal ultrasonography (TVUS) in diagnosing LNM in cervical cancer is only 52% [3, 4]. Hence, there is a need to identify other modalities with better sensitivity and specificity to act as effective screening and diagnostic tools for the diagnosis of LNM in cervical cancer.
Serological detection of squamous cell carcinoma antigen (SCC-Ag) is currently widely used for advanced cervical cancer. Some studies have shown that SCC-Ag is related to recurrence and survival but cannot be used as an indicator of lymph node metastases [5,6,7]. At present, data-independent acquisition (DIA)-based quantitative proteomics technology can be used for protein identification and quantification. Compared with traditional data-dependent acquisition (DDA) mass spectrometry technology, DIA performed the full scanning range of the mass spectrum, and all ions are detected and crushed. Thus, the information of all ions in the sample can be obtained without omission or difference. It has been applied in biomarker screening, fundamental research, and other fields [8,9,10]. It has many advantages, such as high accuracy, deep proteome coverage and high quantitative reproducibility. In this study, we performed DIA to provide full-scale protein profiles from plasma to further reveal the pathogenesis process of LVSI and LNM in cervical cancer and to reveal candidate biomarkers.
Materials and methods
Study design and collection of clinical samples
The study design is shown in Fig. 1. In this study, the main inclusion criteria were as follows: (1) Diagnosis of cervical cancer by two independent pathologists; (2) No surgery or neoadjuvant chemotherapy was performed prior to plasma collection. Exclusion criteria: (1) The patients had other primary tumours; (2) The postoperative pathology showed no tumour cells. Finally, a total of 25 cervical cancer plasma samples, including LNM−/LVSI− (named NC), LNM−/LVSI + (named LVSI) and LNM + /LVSI + (named LNM) samples, were collected from the Department of Obstetrics and Gynaecology, Qilu Hospital of Shandong University from January 2019 to June 2020. 5 cervical cancer patients were excluded. 1 patient was excluded for she had both cervical and lung cancer, 4 patients were excluded due to the neoadjuvant chemotherapy before we collected the plasma samples. Among the other 20 specimens, 7 NC, 4 LVSI and 9 LNM plasma samples were analysed by DIA to investigate the protein changes in these three states.

Schematic workflow of the study
For plasma collection, blood specimens were immediately handled according to the protocol suggested by the HUPO Plasma Proteome Project [8], and informed consent was obtained from all patients. The study was approved by the ethics committee of the Qilu Hospital of Shandong University (Ethics number: KYLL-2017–560).
Sample preparation and fractionation for DDA library generation
The plasma pools were separated from the most abundant proteins using a Human 14 Multiple Affinity Removal System Column following the manufacturer’s protocol (Agilent Technologies). The remaining steps were the same as in a previous report [9].
Data-dependent acquisition (DDA) mass spectrometry assay
All fractions for DDA library generation were analysed by a Thermo Scientific QExactive HF X mass spectrometer connected to an Easy nLC 1200 chromatography system (Thermo Scientific). The peptide (1.5 μg) was first loaded onto an EASY-SprayTMC18 Trapcolumn (Thermo Scientific, P/N 164946, 3 µm, 75 um*2 cm), then separated on an EASY-SprayTM C18 LC Analytical Column (Thermo Scientific, ES802, 2 µm, 75 um*25 cm) with a linear gradient of buffer B (84% acetonitrile and 0.1% Formic acid) at a flow rate of 250 nl/min over 120 min. MS detection method was positive ion, the scan range was 300–1800 m/z, the resolution for the MS1 scan was 60,000 at 200 m/z, the target automatic gain control (AGC) was 3e6, the maximum IT was 25 ms, and the dynamic exclusion parameter was 30.0 s. Each full MS–SIM scan followed 20 ddMS2 scans. The resolution for the MS2 scan was 15,000, the AGC target was 5e4, the maximum IT was 25 ms, and the normalized collision energy was 30 eV.
Mass spectrometry assay for DIA
The peptides from each sample were analysed by LC‒MS/MS operating in the DIA mode by Shanghai Applied Protein Technology Co., Ltd. Each DIA cycle contained one full MS–SIM scan, and 30 DIA scans covered a mass range of 350–1800 m/z with the following settings: SIM full scan resolution, 120,000 at 200 m/z; AGC, 3e6; maximum IT, 50 ms; profile mode, DIA scan parameters were set as follows: resolution, 15,000; AGC target, 3e6; max IT, auto; and normalized collision energy, 30 eV. The run time was 120 min with a linear gradient of buffer B (84% acetonitrile and 0.1% formic acid) at a flow rate of 250 nl/min. QC samples (pooled sample from an equal aliquot of each sample in the experiment) were injected in DIA mode at the beginning of the MS study and after every 6 injections throughout the experiment, which was used to monitor the MS performance.
Mass spectrometry data analysis
For DDA library data, the FASTA sequence database was searched with Spectronaut TM14.4.200727.47784 software. The database was the UniProt human database, and the iRT peptide sequence was added (Biognosys|iRTKit|). DIA data were analysed with Spectronaut TM14.4.200727.47784 by searching the above constructed spectral library. The main software parameters were set as follows: retention time prediction type was dynamic iRT, interference on MS2 level correction was enabled, and cross-run normalization was enabled. All results were filtered based on a Q value cut-off of 0.01 (equivalent to FDR < 1%).
Bioinformatic and statistical analysis
Basic bioinformatics analysis
The subcellular localization, domain annotation, GO annotation, KEGG annotation, enrichment analysis, and protein‒protein interaction (PPI) analysis steps were the same as those in a previous report [10].
Weighted gene coexpression network analysis (WGCNA)
The WGCNA package in R (Version 1.69) was used to identify distinct protein modules among all identified proteins. A weighted protein coexpression network was generated using the log2 protein abundance sample matrix.
Statistical analysis
GraphPad Prism version 5.01 (GraphPad Software Inc., San Diego, CA, USA) was used for statistical analysis. In the present study, data are expressed as the means with standard deviations (SDs), and statistical comparisons were performed using Student's t test or ANOVA. P < 0.05 was considered to indicate a statistically significant result.
Results
Quality evaluation and identification of differentially expressed proteins (DEPs)
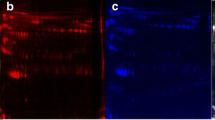
The clinical characteristics of the 20 cervical cancer patients are summarized in Table 1. Quality control (QC) analysis was performed to evaluate the DIA proteomic information. The results suggested that there was enough protein, and the QC samples appeared to have a strong correlation (Fig. 2A, B). The principal component analysis (PCA) of the proteins showed no significant difference among these three groups (Fig. 2C). In total, 12,915 peptides associated with 1357 proteins were identified in our study (Fig. 2D). A total of 1066, 1030, and 1054 proteins were identified in NC, LVSI, and LNM, respectively. A total of 972 proteins were shared by all three groups (Fig. 2E). The DIA protein heatmap is also shown in Fig. 2F. For the three-group comparison by one-way ANOVA, proteins with a fold change (FC) ≥ 1.5 or ≤ 0.67 and P value < 0.05 were defined as DEPs. In total, 79 DEPs of the three groups were identified in this research (Fig. 3A). The LNM group had a total of 24 DEPs, including 14 upregulated proteins and 10 downregulated proteins, compared with the NC group (Fig. 3A, B; Additional file 1: Table S1). There were 23 DEPs (7 upregulated proteins and 16 downregulated proteins) in the LNM group compared with the LVSI group (Fig. 3A, C; Additional file 1: Table S1). There were 32 DEPs (25 upregulated proteins and 7 downregulated proteins) in the LVSI group compared with the NC group (Fig. 3A, D; Additional file 1: Table S1).

Data quality control and general overview of protein identification. A Protein abundance. B Intensity correlation of QC samples. C PCA score plot. Samples from the NC (n = 7), LVSI (n = 4) and LNM (n = 9) groups are plotted along the three principal components. D The identified peptides and proteins of the three groups. E Venn diagram showing the number of proteins common and unique to NC, LVSI and LNM cervical cancer patients. F DIA quantitative heatmap of total protein

Expression of differentially expressed proteins (DEPs). A Bar chart of protein fold change values. B Volcano plot of DEPs between the LNM and NC groups. C Volcano plot of DEPs between the LNM and LVSI groups. D Volcano plot of DEPs between the LVSI and NC groups
Functional annotation analysis
To better understand the function of these DEPs, subcellular localization, domain analysis, gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were conducted. Extracellular proteins were most abundant in the subcellular localization analysis (Fig. 4A). Domain enrichment analysis showed that haemopexin and immunoglobulin had the largest changes in both protein numbers and P values (Fig. 4B). GO analysis (Fig. 5A) showed that the DEPs between the three groups were mainly enriched in the metabolic process in the biological process (BP) category, binding in the molecular function (MF) category, and cell part on the cellular component (CC) category. Moreover, KEGG analysis revealed that the DEPs were mainly enriched in the lipid and atherosclerosis pathways, which are related to cardiovascular disease; other pathways, such as the HIF-1 signal transduction pathway, phagosome and autophagy and ferroptosis pathway, were also of interest (Fig. 5B). Notably, the results above are from ANOVA of the three groups. The results for pairwise comparisons between each pair of groups are provided in the supplementary materials (Additional file 1: Figure S1–7).

Subcellular localization and domain enrichment analysis of DEPs. A Pie chart of subcellular localization of DEPs. B Domain enrichment analysis of DEPs

GO and KEGG pathway analysis of DEPs between the NC, LVSI and LNM groups. A GO analysis. The results for the biological process (BP), molecular function (MF) and cellular component (CC) categories are presented. B KEGG pathway analysis
Protein‒protein interaction (PPI) network
The 79 DEPs were submitted to the STRING 11.0 database via Cytoscape 3.8.0 software to obtain the PPI network diagram. In the PPT network, the nodes represent the difference in protein expression, and the lines represent the connection degree. A high fold change and a high connectivity degree are two characteristics of hub proteins. We identified six hub proteins, which are summarized in Table 2 and Fig. 6.

The DIA expression pattern of six hub proteins
WGCNA
WGCNA is a method for analysing molecular expression patterns of multiple samples. It can cluster molecules with similar expression patterns and analyse the association between modules and specific traits or phenotypes. Therefore, WGCNA is widely used in the study of diseases and other traits. We performed WGCNA on these three groups of samples. Based on the optimal soft-power threshold β (Fig. 7A), the protein expression correlation coefficients were calculated. Then, protein hierarchical clustering trees and network heatmap plots were constructed to describe the different coexpression modules (Fig. 7B, C). A total of nine coexpression modules were constructed (Fig. 7D). Based on the criteria (Pearson r ≥ 0.4 or r ≤ − 0.4, P value ≤ 0.05), the green module of 84 proteins appeared statistical difference. The coexpressed protein network of the green module was visualized (Fig. 7E, F).

WGCNA of DEPs. A Scale-free fit metrics of the network topology obtained using soft threshold analysis. B Hierarchical clustering tree diagram. C Heatmap of the visualized gene network. The heatmap depicts the TOM between all DEPs in the analysis. Light colours indicate low overlap, and dark colours indicate high overlap. D A total of nine coexpression modules were constructed, and each colour represents one module of the protein coexpression network constructed by WGCNA. The top numbers in each cell represent Pearson r, and the bottom numbers represent P values. E Trend of green module protein expression. F The scatter distribution of gene significance and module membership
Functional analysis of the green module from WGCNA
To study the green module’s hub proteins and molecular mechanisms from a systematic perspective, we constructed a PPI network and performed GO and KEGG analysis. We identified hub proteins such as adipocyte plasma membrane-associated protein (APMAP, Q9HDC9), insulin-like growth factor-binding protein (IGF-BP2, P18065) and α-1-acid-glycoprotein 1 (ORM1, P02763). They are all summarized in Fig. 8. In addition, the GO enrichment analysis showed that vesicle-mediated transport and the innate immune response were important in the BP category, signalling receptor binding and protein binding were important in the MF category, and extracellular region was important in the CC category. KEGG enrichment analysis suggested that the green module DEPs were mainly enriched in complement and coagulation cascades, while other KEGG pathways, such as platelet activation, the PI3K-Akt signalling pathway and the HIF-1 signalling pathway, were also enriched (Fig. 9).

DEP coexpression network of the green module. The higher the number of connecting lines is, the larger the size of the node and the higher the importance in the protein‒protein interaction (PPI) network

GO and KEGG pathway analysis of the proteins in the green module. A GO analysis. The BP, MF and CC categories are presented. B KEGG pathway analysis
Discussion
Disease stage and lymph node involvement are two of the most important prognostic factors in cervical cancer patients. According to the latest meta-analysis, the LNM incidence rate in the pelvic region ranged from 2% (stage IA2) to 14–36% (IB), 38–51% (IIA) and 47% (IIB), and in the para-aortic region, the LNM rate ranged from 2 to 5% (stage IB), 10–20% (IIA), 9% (IIB), 13–30% (III) and 50% (IV) [11]. Notably, these data were based on the 2009 FIGO staging system. Despite all the progress of imaging technologies such as TVUS/CT/MRI/PET, as well as the widely used SCC biomarker in squamous cell cervical cancer, the prediction rates are not robust enough to make a safe decision on whether to perform surgery in the pelvic or para-aortic region. Additionally, the mechanisms of LNM in cervical cancer are still not clear, and some studies have focused on changes in signalling pathways related to EMT, STAT3/p-STAT3, and immune escape [12,13,14]. Therefore, this study was designed to identify new candidate biomarkers and possible pathogenic mechanisms for cervical cancer. LVSI is a prerequisite and risk factor for LNM, and previous reports suggested that tumours with LVSI had a 9.3–12.0% LNM rate [15]. Therefore, LVSI was included as a separate study group in our study.
Our results show that the occurrence of LNM and LVSI is a process involving multiple systems and multiple signalling pathways. In addition to the complement and coagulation cascades we mentioned above, we found that the lipid and atherosclerosis pathways were significantly differentially enriched in the LNM vs. NC group as well as in the green module KEGG analysis. Considering these results and the results of the GO analysis, we propose that lipid metabolism may play an important role in lymph node metastasis. Some new published studies have focused on this topic and explored the regulatory mechanisms in cervical cancer [16, 17]. The HIF-1 signal transduction pathway changed greatly in the LVSI vs. NC group as well as in the green module, which was also proven by cytological and animal model experiments in some studies [18, 19]. In addition to this commonly studied pathway, we found some pathways worthy of more attention, including the phagosome and autophagy pathway; the modulation of this pathway in the context of hepatocellular carcinoma cell migration through EMT has only been reported in one study [20]. The ferroptosis pathway could promote the metastasis of cancer cells in melanoma, oesophageal squamous cell carcinoma, colon adenocarcinoma and prostate cancer [21,22,23,24]; however, its has not been well studies in the context of cervical cancer metastasis, so this will be the focus of our next study.
In total, six candidate biomarkers were screened out: SPARC, HPX, VCAM1, TFRC, ERN1 and APMAP. Among them, four proteins (SPARC, HPX, ERN, APMAP) were significantly upregulated in the LNM/NC group, three proteins (VCAM1, TFRC, APMAP) were upregulated in the LVSI/NC group, and one protein (APMAP) was upregulated in the LNM/LVSI group.
SPARC is a calcium-binding matricellular glycoprotein that is able to interact with various extracellular matrix macromolecules and regulate cell adhesion, proliferation and migration [25]. It has been widely reported to be associated with EMT in cervical cancer [26,27,28], and its methylation frequency increases with the severity of the underlying cervical lesion [29]. The serum level of SPARC was higher in cervical cancer patients than in healthy controls and CIN patients [28], and high expression of SPARC was correlated with the LNM of cervical carcinomas, which is consistent with the results of our DIA proteome sequencing study. However, we have yet to see its widespread application in clinical work.
VCAM-1 appears to be functionally important in the immune response and vascularization in high-grade cervical intraepithelial lesions and cervical cancer patients in several reports [30,31,32], and the study of its role as a molecular marker in the diagnosis of cervical diseases has been limited to histological and cytological specimens [31, 33, 34]. The detection of serum or plasma in patients with cervical disease has not been reported. TFRC was reported to be related to the prognosis of cervical cancer patients by participating in the JAK-STAT pathway and HIF-1 signalling pathway in some bioinformatics analysis articles [35, 36]. APMAP promotes the EMT and metastasis of cervical cancer cells by activating the Wnt/β-catenin pathway, and ERN1 is related to the apoptosis of cervical cancer cells [37, 38]. Unlike the molecules mentioned above, according to the results of our literature search, HPX has rarely been reported in the context of cervical disease. However, its diagnostic value has been proven in gallbladder carcinoma, hepatocellular carcinoma, breast cancer, prostate cancer [39,40,41], and so on. Therefore, we believe that these five protein molecules (including SPARC) have the potential to be plasma biomarkers in cervical cancer patients.
Conclusions
In this study, we assessed the whole-protein profiles to identify factors related to LVSI and LNM in cervical cancer through plasma DIA-based quantitative proteomics. Through the analysis of 79 DEPs identified by bioinformatics methods, we found six new candidate biomarkers and a series of pathways (such as the phagosome and autophagy pathway) that are involved in the whole pathogenic process, and further functional studies and validation studies would be worthwhile.
Availability of data and materials
The data used and/or analysis during the current study are available from the corresponding author on reasonable request.
References
World Cancer Report, 2020 (https://publications.iarc.fr/Non-Series-Publications/World-Cancer-Reports/World-Cancer-Report-Cancer-Research-For-Cancer-Prevention-2020)
Bhatla N, Berek JS, Cuello Fredes M, Denny LA, Grenman S, Karunaratne K, Kehoe ST, Konishi I, Olawaiye AB, Prat J, et al. Revised FIGO staging for carcinoma of the cervix uteri. Int J Gynaecol Obstetrics. 2019;145(1):129–35.
Thelissen AAB, Jürgenliemk-Schulz IM, van der Leij F, Peters M, Gerestein CG, Zweemer RP, van Rossum PSN. Upstaging by para-aortic lymph node dissection in patients with locally advanced cervical cancer: a systematic review and meta-analysis. Gynecol Oncol. 2022;164(3):667–74.
Tian Y, Luo H. Diagnostic accuracy of transvaginal ultrasound examination for local staging of cervical cancer: a systematic review and meta-analysis. Med Ultrason. 2022;24(3):348–55.
Charakorn C, Thadanipon K, Chaijindaratana S, Rattanasiri S, Numthavaj P, Thakkinstian A. The association between serum squamous cell carcinoma antigen and recurrence and survival of patients with cervical squamous cell carcinoma: a systematic review and meta-analysis. Gynecol Oncol. 2018;150(1):190–200.
Harima Y, Ariga T. Clinical value of serum biomarkers, squamous cell carcinoma antigen and apolipoprotein C-II in follow-up of patients with locally advanced cervical squamous cell carcinoma treated with radiation: a multicenter prospective cohort study. PLoS ONE. 2021;16(11): e0259235.
Gaarenstroom KN, Kenter GG, Bonfrer JM, Korse CM, Van de Vijver MJ, Fleuren GJ, Trimbos JB. Can initial serum cyfra 21–1, SCC antigen, and TPA levels in squamous cell cervical cancer predict lymph node metastases or prognosis? Gynecol Oncol. 2000;77(1):164–70.
Rai AJ, Gelfand CA, Haywood BC, Warunek DJ, Yi J, Schuchard MD, Mehigh RJ, Cockrill SL, Scott GB, Tammen H, et al. HUPO Plasma Proteome Project specimen collection and handling: towards the standardization of parameters for plasma proteome samples. Proteomics. 2005;5(13):3262–77.
Nie L, Xin S, Zheng J, Luo Y, Zou Y, Liu X, Chen H, Lei X, Zeng X, Lai H. DIA-based proteomics analysis of serum-derived exosomal proteins as potential candidate biomarkers for intrahepatic cholestasis in pregnancy. Archives of Gynecol Obstet. 2022;308:79–89.
Cao XL, Song JY, Sun ZG. Quantitative label-free proteomic analysis of human follicle fluid to identify novel candidate protein biomarker for endometriosis-associated infertility. J Proteomics. 2022;266: 104680.
Olthof EP, van der Aa MA, Adam JA, Stalpers LJA, Wenzel HHB, van der Velden J, Mom CH. The role of lymph nodes in cervical cancer: incidence and identification of lymph node metastases-a literature review. Int J Clin Oncol. 2021;26(9):1600–10.
Liao Y, Huang J, Liu P, Zhang C, Liu J, Xia M, Shang C, Ooi S, Chen Y, Qin S, et al. Downregulation of LNMAS orchestrates partial EMT and immune escape from macrophage phagocytosis to promote lymph node metastasis of cervical cancer. Oncogene. 2022;41(13):1931–43.
Shi S, Ma HY, Zhang ZG. Clinicopathological and prognostic value of STAT3/p-STAT3 in cervical cancer: A meta and bioinformatics analysis. Pathol Res Pract. 2021;227: 153624.
Heeren AM, Koster BD, Samuels S, Ferns DM, Chondronasiou D, Kenter GG, Jordanova ES, de Gruijl TD. High and interrelated rates of PD-L1+CD14+ antigen-presenting cells and regulatory T cells mark the microenvironment of metastatic lymph nodes from patients with cervical cancer. Cancer Immunol Res. 2015;3(1):48–58.
Wenzel HHB, Van Kol KGG, Nijman HW, Lemmens V, Van der Aa MA, Ebisch RMF, Bekkers RLM. Cervical cancer with ≤5 mm depth of invasion and >7 mm horizontal spread - Is lymph node assessment only required in patients with LVSI? Gynecol Oncol. 2020;158(2):282–6.
Du Q, Liu P, Zhang C, Liu T, Wang W, Shang C, Wu J, Liao Y, Chen Y, Huang J, et al. FASN promotes lymph node metastasis in cervical cancer via cholesterol reprogramming and lymphangiogenesis. Cell Death Dis. 2022;13(5):488.
Zhang C, Liao Y, Liu P, Du Q, Liang Y, Ooi S, Qin S, He S, Yao S, Wang W. FABP5 promotes lymph node metastasis in cervical cancer by reprogramming fatty acid metabolism. Theranostics. 2020;10(15):6561–80.
Cheng Y, Chen G, Hong L, Zhou L, Hu M, Li B, Huang J, Xia L, Li C. How does hypoxia inducible factor-1α participate in enhancing the glycolysis activity in cervical cancer? Ann Diagn Pathol. 2013;17(3):305–11.
Chaudary N, Mujcic H, Wouters BG, Hill RP. Hypoxia and metastasis in an orthotopic cervix cancer xenograft model. Radiotherapy Oncol. 2013;108(3):506–10.
Su G, Feng T, Pei T, Yang F, Sun D, Yu H, Wang X, Gao W, He J, Shen Y, et al. Autophagy modulates FSS-induced epithelial-mesenchymal transition in hepatocellular carcinoma cells. Mol Carcinog. 2021;60(9):607–19.
Ubellacker JM, Tasdogan A, Ramesh V, Shen B. Lymph protects metastasizing melanoma cells from ferroptosis. Nature. 2020;585(7823):113–8.
Feng L, Zhao K, Sun L, Yin X, Zhang J, Liu C, Li B. SLC7A11 regulated by NRF2 modulates esophageal squamous cell carcinoma radiosensitivity by inhibiting ferroptosis. J Translat Med. 2021;19(1):367.
Shi C, Xie Y, Li X, Li G, Liu W, Pei W, Liu J, Yu X, Liu T. Identification of Ferroptosis-related genes signature predicting the efficiency of invasion and metastasis ability in colon adenocarcinoma. Front Cell Devel Biol. 2021;9: 815104.
Ding Y, Feng Y, Huang Z, Zhang Y, Li X, Liu R, Li H, Wang T, Ding Y, Jia Z, et al. SOX15 transcriptionally increases the function of AOC1 to modulate ferroptosis and progression in prostate cancer. Cell Death Dis. 2022;13(8):673.
Hsiao YH, Lien HC, Hwa HL, Kuo WH, Chang KJ, Hsieh FJ. SPARC (osteonectin) in breast tumors of different histologic types and its role in the outcome of invasive ductal carcinoma. Breast J. 2010;16(3):305–8.
Qu X, Gao D, Ren Q, Jiang X, Bai J, Sheng L. miR-211 inhibits proliferation, invasion and migration of cervical cancer via targeting SPARC. Oncol Lett. 2018;16(1):853–60.
Zou T, Gao Y, Qie M. MiR-29c-3p inhibits epithelial-mesenchymal transition to inhibit the proliferation, invasion and metastasis of cervical cancer cells by targeting SPARC. Annals Translat Med. 2021;9(2):125.
Shi D, Jiang K, Fu Y, Fang R, Liu XI, Chen J. Overexpression of SPARC correlates with poor prognosis in patients with cervical carcinoma and regulates cancer cell epithelial-mesenchymal transition. Oncol Lett. 2016;11(5):3251–8.
Yang N, Nijhuis ER, Volders HH, Eijsink JJ, Lendvai A, Zhang B, Hollema H, Schuuring E, Wisman GB, van der Zee AG. Gene promoter methylation patterns throughout the process of cervical carcinogenesis. Cell Oncol. 2010;32(1–2):131–43.
Coleman N, Stanley MA. Characterization and functional analysis of the expression of vascular adhesion molecules in human papillomavirus-related disease of the cervix. Cancer. 1994;74(3):884–92.
Qin R, Cao L, Ye C, Wang J. A novel prognostic prediction model based on seven immune-related RNAs for predicting overall survival of patients in early cervical squamous cell carcinoma. BMC Med Genomics. 2021;14(1):49.
Punt S, Houwing-Duistermaat JJ, Schulkens IA, Thijssen VL, Osse EM, de Kroon CD, Griffioen AW, Fleuren GJ, Gorter A, Jordanova ES. Correlations between immune response and vascularization qRT-PCR gene expression clusters in squamous cervical cancer. Mol Cancer. 2015;14:71.
Kayukova EV, Sholokhov LF, Ziganshin AM, Mudrov VA. Inflammatory proteins as molecular markers in the diagnosis of cervical oncopathology. Sovremennye Tekhnologii v Meditsine. 2021;13(4):64–8.
Liu M, Wei D, Nie Q, Peng L, He L, Cui Y, Ye Y. Uncovering of potential molecular markers for cervical squamous cell carcinoma (CESC) based on analysis of methylated-differentially expressed genes. Taiwan J Obstet Gynecol. 2022;61(4):663–71.
Wang Q, Vattai A, Vilsmaier T. Immunogenomic identification for predicting the prognosis of cervical cancer patients. Int J Mol Sci. 2021;22(5):2442.
Xu X, Liu T, Wu J, Wang Y, Hong Y, Zhou H. Transferrin receptor-involved HIF-1 signaling pathway in cervical cancer. Cancer Gene Ther. 2019;26(11–12):356–65.
Zhu X, Xiang Z, Zou L, Chen X, Peng X, Xu D. APMAP Promotes epithelial-mesenchymal transition and metastasis of cervical cancer cells by activating the Wnt/β-catenin pathway. J Cancer. 2021;12(20):6265–73.
Yin Q, Chen H, Ma RH, Zhang YY, Liu MM, Thakur K, Zhang JG, Wei ZJ. Ginsenoside CK induces apoptosis of human cervical cancer HeLa cells by regulating autophagy and endoplasmic reticulum stress. Food Funct. 2021;12(12):5301–16.
Chen W, Desert R, Ge X, Han H, Song Z. The Matrisome genes from hepatitis B-related hepatocellular carcinoma unveiled. Hepatol Communicat. 2021;5(9):1571–85.
Yang C, Chen J, Yu Z, Luo J, Li X, Zhou B, Jiang N. Mining of RNA methylation-related genes and elucidation of their molecular biology in gallbladder carcinoma. Front Oncol. 2021;11: 621806.
Alijaj N, Pavlovic B, Martel P, Rakauskas A. Identification of urine biomarkers to improve eligibility for prostate biopsy and detect high-grade prostate cancer. Cancers. 2022;14(5):1135.
Acknowledgements
We sincerely appreciate all participants in the study.
Funding
This work was supported by the Natural and Science Foundation Youth Project of Shandong Province (ZR2021QH044); the Jinan City “20 New Universities” independent innovation group (2021GXRC027); the National Natural Science Foundation of China (82172940); the Shandong Provincial Key R&D Program (2017CXGC1210); the second batch of science and technology planning projects of Jinan Health Commission in 2020 (2020–3-35); and the Clinical Research fund of Shandong Medical Association—Qilu Special Project (YXH2022ZX02154).
Author information
Authors and Affiliations
Contributions
Conceptualization, funding acquisition, project administration and writing original draft, SH; Data curation, XLL and JS; Formal analysis, WDM; Investigation, LG; Methodology, QWZ; Resources, JQY and YL; Software, HLL and QC; Supervision and funding acquisition, BXC; Validation, Visualization, Writing review & editing and Funding acquisition, SXW and YZZ.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Ethics Committee of the Qilu Hospital of Shandong University. All participants were recruited after providing signed informed consent.
Consent for publication
Not available.
Competing interests
There are no potential conflicts of interest to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1.
The top nine DEPs with a significant difference between every two groups. Figure S2. Subcellular localization and domain enrichment analysis of DEPs between the LNM and NC groups. Figure S3. GO and KEGG pathway analysis of DEPs between the LNM and NC groups. Figure S4. Subcellular localization and domain enrichment analysis of DEPs between the LVSI and NC groups. Figure S5. GO and KEGG pathway analysis of DEPs between the LVSI and NC groups. Figure S6. Subcellular localization and domain enrichment analysis of DEPs between the LNM and LVSI groups. Figure S7. GO and KEGG pathway analysis of DEPs between the LNM and LVSI groups.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Han, S., Liu, X., Ju, S. et al. New mechanisms and biomarkers of lymph node metastasis in cervical cancer: reflections from plasma proteomics. Clin Proteom 20, 35 (2023). https://doi.org/10.1186/s12014-023-09427-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12014-023-09427-8