
Abstract
Ni nanoparticle catalysts embedded in ZrO2 porous spheres and ZrO2 porous composite spheres, SiO2-ZrO2, MgO-ZrO2, and Y2O3-ZrO2, with 83−115 nm diameter and 167–269 m2/g specific surface area were prepared by a one-pot and one-step solvothermal reaction from precursor solutions consisting of Ni(NO3)2‧6H2O, Zr(OnBu)4, and acetylacetone in moist ethanol combined with either Si(OEt)4, magnesium acetylacetate, or Y(OiPr)3. The obtained Ni catalysts have high specific surface areas of 130–196 m2/g, even after high-temperature reduction by H2 at 450 °C for 2 h. They were utilized as catalysts for low-temperature dry reforming of methane (DRM) at 550 °C to suppress carbon deposition on Ni nanoparticles. The Ni catalysts embedded in SiO2-ZrO2 and Y2O3-ZrO2 demonstrated high catalytic activity and long stability in the reaction. Moreover, carbon deposition on Ni nanoparticles in the DRM reaction was effectively suppressed in when using the SiO2-ZrO2 and Y2O3-ZrO2 composites.
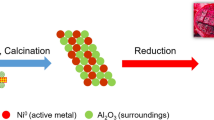
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Metal nanoparticles (NPs) have excellent catalytic activity in heterogeneous catalysis [1]. Numerous methodologies have been developed to enhance their stabilities and achieve long lifetimes for catalytic active sites by optimizing the combination of metal NP size and catalyst support [2]. However, the metal NPs on the surface of the catalyst supports still tend to aggregate and sinter at high temperatures and after prolonged practical use. Much effort has been devoted to preventing the aggregation and sintering of metal NPs on the support surface [3]. Embedding or encapsulation of NPs into porous metal oxides is an effective approach for improving catalyst stability by physical separation of the metal NPs [4]. Several approaches for embedding or encapsulating noble metal NPs in metal oxide supports have been reported. For example, Liu et al. reported a Pt NP catalyst embedded in wide-mouthed compartments tailored on a SiO2 support by a reduction method as a sintering resistance NP system [5]. Liu et al. prepared an encapsulation strategy for Au NPs in a permeable TiO2 thin layer by the deposition–precipitation method, resulting in excellent activity and stability for catalytic CO oxidation [6]. Xiao et al. prepared an embedded NP alloy from intermetallic PtFe alloys supported on carbon [7].
In this context, our group has developed a simple synthetic method for submicron-sized monodisperse porous metal oxides such as SiO2, TiO2, ZrO2, and CeO2 by a one-pot and single-step solvothermal approach. These materials are called micro-/meso-porously architected roundly integrated metal oxides (MARIMOs) [8]. For example, the TiO2 MARIMO consists of ca. 5 nm primary particles with large specific surface areas exceeding 300 m2/g, concomitant with an almost perfect spherical morphology [9]. The MARIMOs were used as a support for the Au NP catalysts. The Au NP catalyst prepared by the deposition–precipitation method (Au/TiO2) demonstrated excellent heat tolerance and durability for highly exothermic CO oxidation, while the nano-concave-convex surface of TiO2 MARIMO prevented well-dispersed Au NPs from migrating and sintering. As a result, the Au/TiO2 MARIMO catalyst exhibited low-temperature activity and long-term stability compared to that of Au NP catalysts supported on commercially available TiO2. We have also succeeded in synthesizing perfectly monodisperse ZrO2 porous spheres with large specific surface areas (244 m2/g) using a similar one-pot and single-step solvothermal approach [10]. The ZrO2 porous spheres were converted to a Ni/ZrO2 catalyst that demonstrated superior catalytic activity and heat tolerance for eminently exothermic CO2 methanation by the impregnation method. Similarly, composite metal oxide porous spheres were easily prepared by slightly modified one-pot and single-step solvothermal reactions [11,12,13]. When a Ru catalyst was applied on a SiO2-CeO2 support composite (Ru/SiO2-CeO2) prepared by impregnation, it exhibited low-temperature activity as well as long-term stability for CO2 methanation [14].
Meanwhile, the dry reforming of methane (DRM, Eq. 1) is a promising reaction for recycling CO2 [15]. Ni catalysts supported on (metal) oxides are commonly used in the DRM, although carbon deposition on Ni NPs is a serious disadvantage. To avoid the formation of carbon species, the reaction is usually performed at high temperatures, such as 700 °C. However, the sintering and aggregation of the Ni NPs as well as the (metal) oxide supports occurred concurrently, which was a serious drawback. Furthermore, according to the mechanism of carbon deposition in the DRM, it is possible for the exposed Ni NPs on the flat surface of the support to react with the excess methane molecules during the reaction, resulting in the accumulation of carbon atoms as nickel carbide, causing carbon deposition.
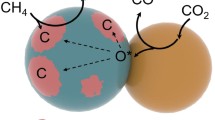
To avoid the carbon deposition, several approaches have been reported: Liu et al. prepared a core-shell catalyst with Ni-ZrO2@microporous SiO2 structure [16], Peng et al. fabricated a unique catalyst with Ni NPs confined on a dendric mesoporous SiO2 [17], Lin et al. also developed a core-shell catalyst with Ni-CeO2@microporous SiO2 structure [18], and Liu et al. reported an intermetallic alloy nano catalyst (InxNi@SiO2) [19]. In this context, we hypothesized that metal NPs embedded into support (metal) oxides would physically suppress the formation of carbon species (Fig. 1). To demonstrate our hypothesis, we designed a new synthetic approach to embed Ni NPs into (composite) metal oxide supports by means of a new one-pot and single-step solvothermal technique.

Schematic of embedded nanometal in gaps of primary (metal) oxide support nanoparticles
In this study, ZrO2 porous spheres were selected as catalyst supports because of their high heat tolerance. A SiO2-ZrO2 composite was selected as a catalyst support to extend the surface area, the MgO-ZrO2 composite was chosen as a catalyst support in anticipation of positive acid–base interactions between CO2 and MgO [20, 21], and Y2O5-ZrO2 functioned as a long-lifetime catalyst support by suppressing the sintering of the catalyst support.
Methods
Reagents
85% zirconium butoxide 1-butanol solution, nickel (II) nitrate hexahydrate, and tetraethoxysilane were purchased from Fujifilm Wako Chemical Corporation. Yttrium isopropoxides, acetylacetone, and magnesium acetylacetonate were purchased from Tokyo Chemical Industry Corporation. Ethanol was purchased from Kishida Chemical Corporation. UEP-100 ZrO2 NPs were obtained from Daiichi Kigenso Kagaku Kogyo Co., Ltd. All reagents were used without further purification.
Catalyst Preparation
A precursor solution including 85 wt% zirconium butoxide in 1-butanol (3.20 g, 7.09 mmol), acetylacetone (50 mL, 0.48 mmol), and Ni(NO3)2·6H2O (500 mg, 1.68 mmol) in ethanol (35 mL) was heated in an SUS-316 stainless steel autoclave (MMS-5000, OM LAB-TECH Co., Ltd.) at 250 °C for 60 min, and the quantity of Ni salt was adjusted to Ni/support metal oxide(s) = 10/90 (wt/wt). The reactor was then cooled to room temperature. The obtained solid was centrifuged at 10,000 rpm for 15 min at 25 °C and washed three times with methanol. The solid was dried under vacuum for 20 h at room temperature to yield a powdery product. The product, denoted L-Ni@ZrO2, was calcined at 300 °C for 2 h in air. “L” and “@” represent “large” and “obtained by one-pot solvothermal method,” respectively. Ni catalysts supported on SiO2-ZrO2, MgO-ZrO2, and Y2O3-ZrO2 composite porous spheres were prepared as follows: tetraethoxysilane (156 μL, 0.703 mmol), magnesium acetylacetonate (155 μL, 0.699 mmol), or yttrium isopropoxide (18.6 mg, 0.700 mmol) was dissolved in ethanol (85 mL). 85 wt% zirconium butoxide solution in 1-butanol (2.84 g, 6.29 mmol), acetylacetone (50 mL, 0.48 mmol), and an ethanol (35 mL) solution of Ni(NO3)2·6H2O (445 mg, 1.53 mmol) were added to the solution successively, to produce a precursor solution, in which the Si, Mg, or Y content was adjusted to (Si, Mg, or Y)/Zr = 1/9 (mol/mol). Similar solvothermal treatment of the precursor solutions afforded Ni catalysts supported on the porous composite spheres, SiO2-ZrO2, MgO-ZrO2, and Y2O3-ZrO2, which are denoted L-Ni@SiO2-ZrO2, L-Ni@MgO-ZrO2, and L-Ni@Y2O3-ZrO2, respectively.
Ni catalysts supported on small porous spheres were obtained according to a procedure similar to that mentioned above, except for the addition of water (3 mL) to the precursor solutions. The prepared small catalysts are referred to as S-Ni@ZrO2, S-Ni@SiO2 − ZrO2, S-Ni@MgO − ZrO2, and S-Ni@Y2O3 − ZrO, in which “S” represents “small.” Reference Ni catalysts supported on UEP-100 ZrO2 and ZrO2 porous spheres were prepared by the impregnation method. A mixture of 131 mg (0.451 mmol) Ni(NO3)2·6H2O and 500 mg UEP-100 ZrO2 or ZrO2 porous spheres in 12.5 mL of water was mixed well using a planetary centrifugal mixer (THINKY AR-100) for 2 h. The obtained powder was dried in air at 80 °C for 2 h and then calcined in air at 300 °C for 2 h to produce a Ni catalyst, U-Ni/ZrO2 or L-Ni/ZrO2, where "U" and “/” represent "UEP-100" and “obtained by impregnation method,” respectively.
Catalyst Characterization
Transmission electron microscopy (TEM) images were obtained using a JEOL JEM-2100F microscope. Energy-dispersive X-ray (EDX) spectroscopy was performed using an Oxford INCA X-Max 80 EDX spectrometer with the TEM instrument. Scanning electron microscopy (SEM) images were obtained using a Hitachi SU8020 FE-SEM. Inductively coupled plasma optical emission spectroscopy (ICP-OES) was performed using a Hitachi High-Tech Science PS3520UV-DD spectrometer. Elemental analysis was carried out on a Malvern Panalytical Epsilon 1 X-ray fluorescence (XRF) spectrometer. Nitrogen adsorption/desorption experiments were conducted using a BEL BELSORP mini II instrument. The specific surface area was calculated using the Brunauer–Emmett–Teller method using the obtained nitrogen adsorption–desorption isotherms. The crystalline phases of the catalysts were identified by X-ray diffraction (XRD) on a Rigaku SmartLab diffractometer using Cu-Kα radiation. The data were recorded over a 2θ range of 10 to 90°.
Catalytic Reaction
Catalytic activity tests for the DRM were performed using a flow-type reactor, MicrotracBEL BELCAT II. Prior to reaction, the catalyst (100 mg) was loaded in a tubular reactor (inner diameter = 7.5 mm) and fixed with quartz wool on both sides. Then, the catalyst was reduced by H2 at 30 mL/min flow at 450 °C for 2 h. The DRM was conducted at 550 °C under a mixed gas stream of CO2/CH4/N2 = 10/10/5 (v/v/v) with a total gas flow rate of 25 mL/min. The temperature of the reactor was maintained at 550 °C for 15 h. The gaseous products were analyzed using a gas chromatograph (GL Sciences GC3200) with an active carbon column equipped with a TCD.
The CH4 and CO2 turnover frequencies (TOFs) were calculated by the moles of CH4 or CO2 converted per second per the moles of catalysts with the following equation:
where N (gas), N (catalyst), and conversion represent gas flow rate (mol/s), amount of catalyst (mol), and conversion of reactant gas, respectively.
Results and Discussion
Catalyst Preparation
The impregnation method is a simple and representative preparation technique that affords supported nanometal catalysts. In addition, it is a versatile technique, applicable to almost all types of supports. We applied this technique to obtain a Ni catalyst supported on commercially available UEP-100 ZrO2 NPs, yielding U-Ni/ZrO2. A ZrO2 porous sphere supported Ni catalyst (L-Ni/ZrO2) was also prepared by the impregnation method [22]. The estimated secondary particle sizes, specific surface areas, pore volumes, and elemental content of the obtained materials are summarized in Table 1. The Ni contents of the catalysts (U-Ni/ZrO2: 8.2 wt%) and (L-Ni/ZrO2: 8.4 wt%) were close to the Ni contents in the 10.0 wt% precursor solutions. Judging from the SEM and TEM images (Fig. 2e), U-Ni/ZrO2 was a simple aggregate of primary NPs with a specific surface area of 76 m2/g after calcination in air, while L-Ni/ZrO2 exhibited a spherical morphology (Fig. 2f) with a specific surface area of 20 m2/g. The XRD patterns of U-Ni/ZrO2 (Fig. 4i) and L-Ni/ZrO2 (Fig. 4j) correspond to monoclinic and cubic ZrO2, respectively, and no peaks corresponding to cubic Ni were observed. The broad peak width of the ZrO2 signals of L-Ni/ZrO2 clearly indicate that the ZrO2 crystallite sizes in L-Ni/ZrO2 are very small (< 5 nm, according to the Scherrer equation). In the case of L-Ni/ZrO2, high-temperature H2 reduction at 450 °C for 2 h caused narrowing of the ZrO2 peaks, and a new peak corresponding to the cubic Ni phase appeared (Fig. 4Bj). These results clearly indicate that sintering of the ZrO2 primary particles and Ni particles occurred. Meanwhile, the coprecipitation technique is an alternative way to prepare noble metal catalysts supported on base metal oxides [23]. Typically, a mixed solution of a noble metal salt and a base metal salt is neutralized by an alkaline solution to yield a coprecipitate containing mixed hydroxides of both metals. The coprecipitate is calcined to produce a noble metal catalyst supported on a base metal oxide. We applied the fundamentals of the coprecipitation technique to the solvothermal reaction. A precursor solution containing Ni(NO3)2 and Zr(OnBu)4 in ethanol was treated solvothermally at 250 °C to yield L-Ni@ZrO2, in which a Ni catalyst was supported on ZrO2 porous spheres. The product was calcined at 300 °C for 2 h in air to remove organic residues. Our proposed mechanism of the solvothermal reaction is as follows: Ni(NO3)2 and Zr(OnBu)4 can be hydrolyzed by H2O, which is generated through the condensation of ethanol at high temperature, to yield hydroxides of both metals. At elevated temperatures, they condense with each other, yielding a mixture of corresponding metal oxides at the nanoscale. Then, nickel hydroxide and/or nickel oxide in the mixed oxides can be reduced by high-temperature ethanol under solvothermal conditions, yielding embedded Ni metal NPs in ZrO2 porous sphere supports.

SEM, TEM, and STEM/EDX images of as-prepared Ni catalysts embedded in large ZrO2-based supports and TEM images of them after hydrogen reduction: a L-Ni@ZrO2, b L-Ni@SiO2-ZrO2, c L-Ni@MgO-ZrO2, and d L-Ni@Y2O3-ZrO2. SEM and TEM images of Ni catalysts prepared by impregnation method: e U-Ni/ZrO2 and f L-Ni/ZrO2
SEM and TEM images of the products revealed that L-Ni@ZrO2 had an almost perfect spherical morphology with a diameter of 602 ± 63 nm (Fig. 2a). STEM/EDX (Additional file 1: Fig. S1) and XRF analyses indicate perfect dispersion of Ni atoms throughout the spheres with 9.5 wt% content. The XRD spectrum of L-Ni@ZrO2 indicates that ZrO2 has a cubic crystal phase with a very small crystallite size (< 5 nm, according to the Scherrer equation), while no peaks related to Ni species were observed, indicating the small particle size of Ni (Fig. 4Aa). H2 reduction of L-Ni@ZrO2 at 450 °C for 2 h resulted in narrower peaks for ZrO2, and a peak corresponding to the cubic Ni phase (Fig. 4Ba). The specific surface area changed from 71 to 4 m2/g, which, unfortunately, indicates that H2 reduction at high temperature caused considerable sintering of the ZrO2 NPs and Ni NPs.
According to similar solvothermal reactions, Ni catalysts embedded in ZrO2 composites, L-Ni@SiO2-ZrO2, L-Ni@MgO-ZrO2, and L-Ni@Y2O3-ZrO2, were easily obtained from the precursor solutions including additional tetraethoxy silane, magnesium acetylacetonate, and yttrium isopropoxide, respectively. SEM and TEM images show that the obtained secondary particles have spherical morphology with 600–700 nm diameters (Fig. 2b–d). The STEM/EDX results indicate that Ni, Si, Mg, and Y atoms were dispersed homogeneously throughout the network of porous spheres (Additional file 1: Fig. S1). XRF or ICP-OES analysis revealed that the embedded Ni contents of the catalysts obtained by the solvothermal method were more than 8 wt% in the cases of L-Ni@SiO2-ZrO2 and L-Ni@MgO-ZrO2, while L-Ni@Y2O3-ZrO2 was slightly smaller, at 6.5 wt%. The Si content was ca. 6 wt%, and the Mg and Y content was 9–12 wt%, which is similar to the corresponding content (10 wt%) in the precursor solutions. Moreover, L-Ni@SiO2-ZrO2, L-Ni@MgO-ZrO2, and L-Ni@Y2O3-ZrO2 exhibited higher specific surface areas (110–240 m2/g) than that of the prototype L-Ni@ZrO2 (71 m2/g). However, the high specific surface areas were drastically reduced to 2–6 m2/g, when the catalysts were reduced by H2 at 450 °C for 2 h. Unlike the results for L-Ni@MgO-ZrO2, in the L-Ni@SiO2-ZrO2 and L-Ni@Y2O3-ZrO2 results exhibited almost no peaks related to Ni species after high-temperature reduction, indicating that the ZrO2 and Ni particles remained small even after H2 reduction at 450 °C.
Thus, we thought that the porous spheres consisting of slightly larger ZrO2 primary particles would tolerate sintering even at high temperatures, and smaller (spherical) secondary particles would be effective for gas diffusion into and out of porous sphere catalysts. Therefore, the reaction conditions, such as solvent, reaction temperature, and reaction time, were optimized to produce small (spherical) secondary particles with slightly larger primary particle. Finally, we found that the addition of a small volume of water to the precursor solutions dramatically reduced the secondary particle sizes from to 600–700 nm to 60–120 nm (Figs. 2 and 3). The Ni catalysts embedded in the small porous supports are denoted S-Ni@ZrO2, S-Ni@SiO2-ZrO2, S- Ni@MgO-ZrO2, and S-Ni@Y2O3-ZrO2. Judging from the SEM and TEM images, all the catalysts exhibited small secondary particle sizes of 63–120 nm (Fig. 3). The Ni atom is dispersed homogeneously throughout the (composite) supports in all cases (Additional file 1: Fig. S2) at 6.5–9.0 wt%.As shown in Fig. 4Aa–h, the XRD peaks for ZrO2 in the small porous spheres, S-Ni@ZrO2, S-Ni@SiO2-ZrO2, S-Ni@MgO-ZrO2, and S-Ni@Y2O3-ZrO2, are somewhat sharper than those of the large porous spheres, L-Ni@ZrO2, L-Ni@SiO2-ZrO2, L-Ni@MgO-ZrO2, and L-Ni@Y2O3-ZrO2, indicating that the crystallite size of the ZrO2 primary particles in the small porous spheres slightly increased as expected. Clear Ni peaks were also observed for S-Ni@ZrO2 and S-Ni@MgO-ZrO2 (Fig. 4Ae,g), while almost no Ni peaks were observed in the cases of S-Ni@SiO2-ZrO2 and S-Ni@Y2O3-ZrO2 (Fig. 4Af,h). Notably, the XRD profiles of the four samples were very broad (Fig. 4Ae–h), and a cubic Ni phase was observed in the cases of S-Ni@ZrO2 and S-Ni@MgO-ZrO2. Almost no Ni peaks were observed in the cases of S-Ni@SiO2-ZrO2 (Fig. 4Bf) and S-Ni@Y2O3-ZrO2 (Fig. 4Bh), even after H2 reduction at 450 °C for 2 h, suggesting that the small catalysts are high-temperature-resistant catalysts.

SEM, TEM, and STEM/EDX images of as-prepared Ni catalysts embedded in small ZrO2-based supports and TEM images of them after hydrogen reduction: a S-Ni@ZrO2, b S-Ni@SiO2-ZrO2, c S-Ni@MgO-ZrO2, and d L-Ni@Y2O3-ZrO2

XRD patterns of Ni catalysts embedded in ZrO2-based supports: a) L-Ni@ ZrO2, b) L-Ni@SiO2-ZrO2, c) L-Ni@MgO-ZrO2, d) L-Ni@Y2O3-ZrO2, e) S-Ni@ZrO2, f) S-Ni@SiO2-ZrO2, g) S-Ni@MgO-ZrO2, h) S-Ni@Y2O3-ZrO2, i) U-Ni/ZrO2, and j) L-Ni/ZrO2. A Before H2 reduction and B after H2 reduction
DRM Reaction
To elucidate the effect of the embedded morphology of Ni NPs on ZrO2-based porous spheres, the DRM was selected as a probe reaction. Generally, the DRM is performed at temperatures over 700 °C to suppress carbon deposition on the catalyst metal NPs. However, here, we intentionally selected a lower temperature (550 °C) to investigate the suppression effects resulting from the particular morphology of our Ni NPs in their catalyst framework on carbon deposition.
Figure 5 shows the results of the DRM reaction catalyzed by U-Ni/ZrO2, L-Ni/ZrO2, L-Ni@ZrO2, and S-Ni@ZrO2. Notably, U-Ni/ZrO2 prepared by impregnation from commercially available UEP-100 ZrO2 demonstrated the highest catalytic ability in both CO2 and CH4 conversions, and the highest CO and H2 yields at the very beginning of the reaction. However, the catalytic ability was rapidly lost (within 5 h). L-Ni/ZrO2 and L-Ni@ZrO2 were prepared by impregnation from ZrO2 porous spheres and solvothermal method, respectively. Those catalysts exhibited relatively lower catalytic abilities in both CO2 and CH4 conversions, and CO and H2 yields. However, small S-Ni@ZrO2 particles exhibited high activity in CH4 and CO2 conversions as well as high CO and H2 yields compared to those of the impregnated L-Ni/ZrO2 and embedded L-Ni@ZrO2. These differences can be ascribed to the much higher specific surface area of S-Ni@ZrO2 (140 m2/g, after H2 reduction) than those of L-Ni/ZrO2 (3 m2/g, after H2 reduction) and L-Ni@ZrO2 (4 m2/g, after H2 reduction). Turn over frequencies (TOFs) of the catalysts are summarized in Table 2. The values were comparable to a representative low-temperature DRM catalyzed by Ni catalyst supported on SiO2-ZrO2 obtained by co-impregnation method [24].

CH4 and CO2 conversions; CO and H2 yields of DRM reactions catalyzed by U-Ni/ZrO2, L-Ni/ZrO2, L-Ni@ZrO2, and S-Ni@Y2O3-ZrO2
Carbon Suppression Behavior in DRM
We abandoned the large catalysts and selected the small catalysts for the DRM. As shown in Fig. 6, the Ni catalysts embedded in the ZrO2 composite (S-Ni@SiO2-ZrO2, S-Ni@MgO-ZrO2, and S-Ni@Y2O3-ZrO2) exhibited better performance than the prototype S-Ni@ZrO2. The results can be classified into two groups: One group includes S-Ni@ZrO2 and S-Ni/MgO-ZrO2, which exhibit lower performance, and the other includes S-Ni@SiO2-ZrO2 and S-Ni@Y2O3-ZrO2, which exhibit higher performance. The conversion of CH4 and CO2 and the CO and H2 yields in the reactions catalyzed by S-Ni@SiO2-ZrO2 and S-Ni@Y2O3-ZrO2 were stable for 15 h, while those catalyzed by S-Ni@ZrO2 and S-Ni/MgO-ZrO2 slowly decreased over time.

CH4 and CO2 conversions; CO and H2 yields of DRM reactions catalyzed by S-Ni@ZrO2, S-Ni@SiO2-ZrO2, S-Ni@MgO- ZrO2, and S-Ni@Y2O3-ZrO2
The high catalytic activity and long-term stability of S-Ni@SiO2-ZrO2 and S-Ni@Y2O3-ZrO2 can be ascribed to the sintering resistance of the catalysts. To directly clarify the carbon deposition on the catalysts, SEM and TEM images of the spent catalysts were obtained (Fig. 7). As expected, the formation of a large number of carbon nanotubes (CNTs) was observed in the case of prototype S-Ni@ZrO2 (Fig. 7a) and S-Ni@MgO-ZrO2 (Fig. 7c). In contrast, only a small number of CNT were observed in the cases of S-Ni@SiO2-ZrO2 (Fig. 7b) and S-Ni@Y2O3-ZrO2 (Fig. 7d).

SEM and TEM images of spent Ni catalysts embedded in small ZrO2-based supports, a S-Ni@ZrO2, b S-Ni@SiO2-ZrO2, c S-Ni@MgO-ZrO2, d S-Ni@Y2O3-ZrO2, and e U-Ni/ZrO2
The exact quantity of deposited CNTs was estimated by TG analysis of the spent catalysts (Fig. 8). In the cases of S-Ni@ZrO2 and S-Ni@MgO-ZrO2, weight loss started at approximately 400 °C and was almost complete at 600 °C, with weight losses corresponding to 30 and 19%, respectively. However, in the cases of S-Ni@SiO2-ZrO2 and S-Ni@Y2O3-ZrO2, weight losses started at a lower temperature, approximately 350 °C, and had almost completed by 550 or 600 °C, with weight losses corresponding to 15 and 5%, respectively.

TGA profiles of spent catalysts for S-Ni@ZrO2, S-Ni@SiO2-ZrO2, S-Ni@MgO-ZrO2, and S-Ni@Y2O3-ZrO2
The different decomposition initiation temperatures suggest that the former and latter groups resulted in the formation of different carbon species. The lower decomposition temperature suggests the formation of amorphous carbon, and the higher decomposition temperature indicates the formation of CNT [25]. Thus, Ni catalysts embedded among small SiO2-ZrO2 and Y2O3-ZrO2 porous sphere supports effectively suppressed the formation of carbon species in the DRM reaction.
XRD spectra of the spent catalysts are shown in Fig. 9. Almost no changes were observed in the cases of S-Ni@SiO2-ZrO2 and S-Ni@Y2O3-ZrO2, where the profiles left broad even after DRM. However, sharper peaks corresponding to cubic ZrO2 and cubic Ni were recognized in the cases of S-Ni@ZrO2 and S-Ni@MgO-ZrO2. These results clearly indicate that sintering of the Ni particles and ZrO2 primary particles were suppressed effectively in the cases of S-Ni@SiO2-ZrO2 and S-Ni@Y2O3-ZrO2. Confinement effect of Ni NPs and ZrO2 primary particles by embedded SiO2 and Y2O3 can be an additional reason for the high catalytic performance of them [24].

XRD patterns of spent catalysts for a S-Ni@ZrO2, b S-Ni@SiO2-ZrO2, c S-Ni@MgO-ZrO2, and d S-Ni@Y2O3-ZrO2
Conclusion
We successfully prepared embedded Ni NP catalysts in ZrO2 porous spheres and SiO2-ZrO2, MgO-ZrO2, and Y2O3-ZrO2 composite porous spheres by simple one-pot and single-step solvothermal reactions. The porous sphere sizes were reduced from to 600–700 nm to 63–120 nm by adding a small volume of water to the precursor solutions. The low-temperature DRM was selected as a probe reaction to verify the catalyst activity and carbon-deposition-suppression ability on the Ni NPs. The embedded Ni catalysts exhibited better catalytic activity and longer-lasting stability than the Ni-impregnated commercially available ZrO2 NP catalyst and Ni-impregnated ZrO2 porous spherical catalyst. In addition, carbon deposition on Ni NPs was suppressed by the small ZrO2 composites embedded with Ni NPs. In particular, the Ni catalyst embedded in small SiO2-ZrO2 porous spheres with a high specific surface area demonstrated good activity and long stability.
Availability of data and materials
The data used and analyzed during the current study are available from the corresponding authors upon reasonable request.
Abbreviations
- DRM:
-
Dry reforming of methane
- NP:
-
Nanoparticle
- MARIMO:
-
Micro-/meso-porously architected roundly integrated metal oxides
- TEM:
-
Transmission electron microscopy
- EDX:
-
Energy-dispersive X-ray
- SEM:
-
Scanning electron microscopy
- ICP-OES:
-
Inductively coupled plasma optical emission spectroscopy
- XRF:
-
X-ray fluorescence
- XRD:
-
X-ray diffraction
- TCD:
-
Thermal conductivity detector
- CNT:
-
Carbon nanotube
References
Xu D, Lv H, Liu B (2018) Encapsulation of metal nanoparticle catalysts within mesoporous zeolites and their enhanced catalytic performances: a review. Front Chem 6:550–564
Van Deelen TW, Mejia CH, De Jong KP (2019) Control of metal-support interactions in heterogeneous catalysts to enhance activity and selectivity. Nat Catal 2:955–970
De Rogatis L, Cargnello M, Gombac V, Lorenzut B, Montini T, Fornasiero P (2010) Embedded phases: a way to active and stable catalysts. Chemsuschem 3:24–42
Vedrine JC (2017) Heterogeneous catalysis on metal oxides. Catalysts 7:341–366
Liu J, Ji Q, Imai T, Ariga K, Abe H (2017) Sintering-resistant nanoparticles in wide-mouthed compartments for sustained catalytic performance. Sci Rep 7:41773
Liu S, Xu W, Niu Y, Zhang B, Zheng L, Liu W, Li L, Wang J (2019) Ultrastable Au nanoparticles on titania through an encapsulation strategy under oxidative atmosphere. Nat Commun 10:5790–5799
Zou X, Chen S, Wang Q, Gao X, Li J, Li L, Wei Z (2019) Leaching and sintering-resistant hollow or structurally ordered intermetallic PtFe alloy catalyst for oxygen reduction reaction. Nanoscale 11:20115–20122
Wang P, Kobiro K (2014) Synthetic versatility of nanoparticles: a new, rapid, one-pot, single-step synthetic approach to spherical mesoporous (metal) oxide nanoparticles using supercritical alcohols. Pure Appl Chem 86:785–800
Duriyasat F, Irizawa A, Hayashi K, Ohtani M, Kobiro K (2018) Sintering-resistant metal catalyst supported on concave-convex surface of TiO2 nanoparticle assemblies. ChemCatChem 10:3392–3396
Kan K, Yamamoto E, Ohtani M, Kobiro K (2020) Solvothermal synthesis of monodisperse porous zirconia spheres with large surface area. Eur J Inorg Chem 47:4431–4435
Pradeep Ellawala KC, Habu T, Tooriyama H, Ohtani M, Kobiro K (2015) Ultra-simple synthetic approach to the fabrication of CeO2–ZrO2 mixed nanoparticles into homogeneous, domain, and core–shell structures in mesoporous spherical morphologies using supercritical alcohols. J Supercrit Fluids 97:217–223
Ohtani M, Muraoka T, Okimoto Y, Kobiro K (2017) Rapid one-pot solvothermal batch synthesis of porous nanocrystal assemblies composed of multiple transition-metal elements. Inorg Chem 56:11546–11551
Nguyen H, Habu T, Ohtani M, Kobiro K (2017) One-step direct synthesis of SiO2-TiO2 composite nanoparticle assemblies with hollow spherical morphology. Eur J Inorg Chem 24:3017–3023
Nguyen H, Kumabe Y, Ueda S, Kan K, Ohtani M, Kobiro K (2019) Highly durable Ru catalyst supported on CeO2 nanocomposites for CO2 methanation. Appl Catal A 577:33–43
Parsapur RK, Chatterjee S, Huang K (2020) The insignificant role of dry reforming of methane in CO2 emission relief. ACS Energy Lett 5:2881–2885
Liu W, Li L, Zhang X, Wang Z, Wang X, Peng H (2018) Design of Ni-ZrO2@SiO2 catalyst with ultra-high sintering and coking resistance for dry reforming of methane to prepare syngas. J CO2 Util 27:297–307
Peng H, Zhang X, Han X, You X, Lin S, Chen H, Liu W, Wang X, Zhang N, Wang Z, Wu P, Zhu H, Dai S (2019) Catalysts in coronas: a surface spatial confinement strategy for high-performance catalysts in methane dry reforming. ACS Catal 9:9072–9080
Lin S, Wang J, Mi Y, Yang S, Wang Z, Liu W, Wu D, Peng H (2021) Trifunctional strategy for the design and synthesis of a Ni-CeO2@SiO2 catalyst with remarkable low-temperature sintering and coking resistance for methane dry reforming. Chin J Catal 42:1808–1820
Liu W, Li L, Lin S, Luo Y, Bao Z, Mao Y, Li K, Wu D, Peng H (2022) Confined Ni-In intermetallic alloy nanocatalyst with excellent coking resistance for methane dry reforming. J Energy Chem 65:34–47
Aziz MAA, Jalil AA, Wongsakulphasatch S, Vo DVN (2020) Understanding the role of surface basic sites of catalysts in CO2 activation in dry reforming of methane: a short review. Catal Sci Technol 10:35–45
Gao X, Ge Z, Zhu G, Wang Z, Ashok J, Kawi S (2021) Anti-coking and anti-sintering Ni/Al2O3 catalysts in the dry reforming of methane: recent progress and prospects. Catalysts 11:1003–1021
Tanaka Y, Usui H, Domi Y, Ohtani M, Kobiro K, Sakaguchi H (2019) Mesoporous spherical aggregates consisted of Nb-doped anatase TiO2 nanoparticles for Li and Na storage materials. ACS Appl Energy Mater 2:636–643
Biswas S, Pal A, Pal T (2020) Supported metal and metal oxide particles with proximity effect for catalysis. RCS Adv 10:35449–35472
Wang Y, Yao L, Wang Y, Wang S, Zhao Q, Mao D, Hu C (2018) Low-temperature catalytic CO2 dry reforming of methane on Ni-Si/ZrO2 catalyst. ACS Catalyst 8:6495–6506
Charisiou ND, Douvartzides SL, Siakavelas GI, Tzounis L, Sebastian V, Stolojan V, Hinder SJ, Baker MA, Polychronopoulou K, Goula MA (2019) The relationship between reaction temperature and carbon deposition on nickel catalysts based on Al2O3, ZrO2 or SiO2 supports during the biogas dry reforming reaction. Catalysis 9:676
Acknowledgements
The authors thank Daiichi Kigenso Kagaku Kogyo Co., Ltd. for providing them with the UEP-100 ZrO2 nanoparticles.
Funding
This research was partially supported by the JSPS KAKENHI Grant Number 19K05143.
Author information
Authors and Affiliations
Contributions
MM, TN, EY, and KH were involved in experiments, analysis, and discussion. MM, TN, MO, and KK drafted the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
. Fig. S3 a) H2/CO mole ratio of DRM reactions catalyzed by L-Ni@ZrO2, S-Ni@ZrO2, L-Ni/ZrO2, and U-Ni/ZrO2. b) H2/CO mole ratio of DRM reactions catalyzed by S-Ni@ZrO2, S-Ni@SiO2-ZrO2, S-Ni@MgO-ZrO2, and S-Ni@Y2O3-ZrO2. Fig. S4 SEM images of Au coated as-prepared Ni catalysts of a) S-Ni@ZrO2, b) S-Ni@SiO2-ZrO2, c) S-Ni@MgO-ZrO2, and d) S-Ni@Y2O3-ZrO2.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Meiliefiana, M., Nakayashiki, T., Yamamoto, E. et al. One-Step Solvothermal Synthesis of Ni Nanoparticle Catalysts Embedded in ZrO2 Porous Spheres to Suppress Carbon Deposition in Low-Temperature Dry Reforming of Methane. Nanoscale Res Lett 17, 47 (2022). https://doi.org/10.1186/s11671-022-03683-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s11671-022-03683-7