
Abstract
Rheumatoid arthritis (RA) is an autoimmune disease involving T and B lymphocytes. Autoantibodies contribute to joint deterioration and worsening symptoms. Adenosine deaminase (ADA), an enzyme in purine metabolism, influences adenosine levels and joint inflammation. Inhibiting ADA could impact RA progression. Intracellular ATP breakdown generates adenosine, which increases in hypoxic and inflammatory conditions. Lymphocytes with ADA play a role in RA. Inhibiting lymphocytic ADA activity has an immune-regulatory effect. Synovial fluid levels of ADA are closely associated with the disease’s systemic activity, making it a useful parameter for evaluating joint inflammation. Flavonoids, such as quercetin (QUE), are natural substances that can inhibit ADA activity. QUE demonstrates immune-regulatory effects and restores T-cell homeostasis, making it a promising candidate for RA therapy. In this review, we will explore the impact of QUE in suppressing ADA and reducing produced the inflammation in RA, including preclinical investigations and clinical trials.
Graphical Abstract

Similar content being viewed by others


Rheumatoid arthritis (RA)
Rheumatoid arthritis (RA) is a chronic inflammation of the joints. The activation of synovial tissue in the joint capsule, cartilage and bone invasion, as well as increasing joint dysfunction are the key hall marks of RA [1]. The epigenetic and environmental variables have a role in the genesis and progression of RA. Furthermore, non-genetic variables such as sex hormones, smoking, periodontal infection, and microbiota, as well as autoantibodies, cytokines, chemokines, and proteases, are engaged in the inflammatory processes that assault the cartilage and bone, resulting in joint dysfunction [2] (Fig. 1). The over-activation of T and B lymphocytes, synovial-like fibroblasts, and macrophages, as well as the significant production of proinflammatory cytokines like tumor necrosis factor alpha (TNF-α) and interleukin 6 (IL-6), are the main inflammatory processes that result in ongoing inflammation and joint degeneration, as mentioned by Huang et al. [1].

Factors that contribute to the progression of RA
Epidemiology
RA is present worldwide, its prevalence varies among countries, regions, and ethnic groups [3]. The prevalence of RA is higher in Africa and the Middle East, usually ranging from 0.25% to 3.4% in most countries, according to Finckh et al. [4]. Also, Abdel Fattah et al. [5] estimated that the RA disease prevalence in Egypt is about 5%. These estimates are based on older populations, self-reported patients, and clinic or hospital-based studies [6], and urban populations tend to be higher than those based on data from the Global Burden of Disease (GBD) study, as mentioned by Riedmann et al. [7] (Fig. 2).

The epidemiology of rheumatoid arthritis
Causes of RA
While the cause of RA is unknown, there is evidence that both hereditary and environmental factors have a role in the disease’s progression [4] (Fig. 3). The patient’s genetic predisposition, which results in the production of auto reactive T and B cells, and a triggering event, such as a viral or bacterial infection or tissue injury, are the two independent factors that lead to the initial cause of RA. Furthermore, RA is most likely caused by a stochastic event caused by a combination of genetic variation, epigenetic changes, and environmental variables in people who are genetically vulnerable to the disorder [8]. Moreover, the cause of RA has been linked to lung microbiota, periodontal disease (periodontitis), and infections [9]. Identical twins had higher concordance risk rates than unrelated control groups and non-identical twins, suggesting that genetic factors have a role in the development of RA. A family history of RA raises the likelihood of developing the disease by three to five times [10].

Causes of rheumatoid arthritis
Another cause of RA is the discovery of over 100 loci related with disease progression in genome-wide association studies using single nucleotide polymorphisms (SNPs) [11]. Many of these loci are seen in other chronic inflammatory diseases and are implicated in the regulation, activation, and maintenance of immune responses [12]. Human leukocyte antigen (HLA) alleles, which are linked to an increased risk of developing RA, are among these sites [13]. Furthermore, HLA variants have been associated to more severe bone deterioration and higher death rates [14].
Path mechanism of RA
Synovitis, an inflammation of the joint capsule that affects the accompanying bones, the synovial membrane, and the synovial fluid, is an indicator of autoimmune tissue destruction in RA [15]. A variety of dendritic cell subtypes, T cells, macrophages, B cells, neutrophils, fibroblasts, and osteoclasts collaborate to initiate and sustain joint inflammation [16]. Due to the frequency of RA-specific autoantigens and the difficulty to totally remove them, continuing immune cell activation leads to a self-perpetuating inflammatory state in the joint, causing pain and joint swelling in afflicted individuals [17] (Fig. 4). Pannus, which is a swelling of the synovial membrane that invades the periarticular bone at the cartilage–bone interface, is caused by the arthritic joint’s continuing inflammatory milieu and results in bone loss and cartilage deterioration [18].

Path mechanism of rheumatoid arthritis
Dendritic cells (DC) and RA
DCs are crucial for RA inflammation maintenance and promotion [19]. DCs, or antigen-presenting cells, play an important role in the initiation of immune responses by capturing and presenting antigens to T cells [20]. DCs of the myeloid (mDCs) and plasmacytoid (pDCs) subtypes have been found in the synovial tissue of patients with RA [21]. The high density of DCs in the synovium of patients with RA synovium shows that DCs play an important role in the etiology of RA [22]. There is evidence that T-cell responses in the synovium are improved by DCs, which may contribute to the pathophysiology of RA [23]. DCs are present in both inflammatory and homeostatic tissue. DCs are drawn from the blood into the synovium in RA [24]. The proinflammatory cytokines TNF-α, interleukin (IL)-1, and IL-6 are generated by both inflamed synovial lining cells and invading immune cells and can further encourage DCs to cause inflammation [16].
The generation of autoantibodies is aided by DCs’ involvement in B-cell activation. DCs also present antigen, release cytokines, and increase co-stimulatory molecules, which all serve to excite B cells. Autoantibodies are produced because of the increased B cell activity, which helps to cause RA [25]. Through interactions with adhesion molecules produced on endothelial cells, DCs can aid in the attraction of other immune cells, such as T cells. In RA synovial tissue, DCs have also been demonstrated to be resistant to apoptosis [17]. This ability to resist apoptosis enables DCs to stay in the synovium, helping to maintain synovitis and encouraging the release of proinflammatory cytokines. In addition, it has been demonstrated that DCs express more toll-like receptors (TLRs) in RA synovial tissue than in normal control tissue [26].
DCs generate cytokines and chemokines and exhibit surface chemicals that regulate the immune system’s induction, activation, and maintenance of tolerance [27]. However, due to the changes in DC activity and distribution, RA and other autoimmune diseases can also result in autoimmune inflammation [28]. Changes in DCs are thought to be the primary cause of RA, increasing DC migration to the inflamed joint [29]. The upregulation of CCR6, a chemokine CCL20 receptor on DCs, is assumed to be the source of DC attraction to synovial tissue [30]. Once they have grown in the joint, DCs produce cytokines including IL-12 and IL-23 (Fig. 5), encourage antigen-specific Th17 responses and result in an imbalance of Th1, Th2, and Th17 responses [31].

Dendritic cells the main cause for maintenance of inflammation in rheumatoid arthritis
Cytokines and inflammation
In the course of developing inflammation in RA, cytokines are significant molecules [32]. They act as a link between skin cells, immune cells, and tissue cells. Joint inflammation depends on key effector cytokines generated by T cells, including TNF-α, IL-17A, interferons (IFN), and receptor activator of nuclear factor kappa-β ligand (RANK-L) [33]. Between skin cells, immune cells, and tissue cells, they serve as a bridge. TNF-α, IL-17A, interferons, and RANK-L are important effector cytokines produced by T cells that are necessary for joint inflammation [34]. Uncontrolled inflammation and bone and cartilage degeneration are brought on by TNF-α upregulation [35]. Additionally, TNF-α induces osteocytes to produce RANK-L, which encourages osteoclastogenesis and causes cells from the monocyte/macrophage lineage to differentiate into osteoclasts [36]. TNF-α may draw leukocytes to the synovium and induce inflammation by inducing the release of inflammatory cytokines like IL-1 and IL-6 [37].
Th17 cells produce IL-17A, which causes localized inflammation and hastens the development of RA illness by accelerating the loss of cartilage, bone resorption, and angiogenesis (Fig. 6) [38]. IL-17A plays a significant role in the development of RA by stimulating the synthesis of RANK-L through osteoblasts and synoviocytes leading to decreased bone development and increased bone degradation [39]. Also, facilitating the formation of matrix metalloproteinase (MMP-1) by synoviocytes [40] and promoting both endothelial cell migration and angiogenesis [41]. Inflammation and joint degeneration brought on by activated neutrophils are exacerbated by proinflammatory cytokines produced by synovial activated macrophages that attract and excite other innate immune cells [42].

Cytokines and inflammation in rheumatoid arthritis disease
Activated fibroblasts also contribute to local joint injury by developing RANK-L and MMPs and migrating between joints [1]. Overall, the RA joint inflammation is a unique tissue reaction that combines local fibroblasts with active proinflammatory phenotypes, matrix modulation, osteoclast formation, and invasive properties [43].
Neovascularization and RA
The RA prevascular stage is brief before progressing to a major vascular stage with an obvious rise in vessel formation [44]. An increase in macrophages and fibroblast synoviocytes is an indication of the prevascular stage in the lining layer [45]. The destructive and invasive front known as the synovial pannus develops when the cluster of differentiation 4 (CD4+) T cells, B cells, and macrophages enter the sublining layer (Fig. 7). This pannus acts like a regional cancer, invading and damaging bone and cartilage [46].

Mechanical pathway for the role of synovial neovascularization in rheumatoid arthritis
In both inflamed and non-inflamed joints, there is an increase in the synovial lining layer’s thickness and mononuclear cell infiltration [47]. The lining layer of RA synovium is chronically hypoxic despite increased vessel density associated to active endothelial growth and EC survival [48] (Fig. 7). Direct oxygen tension measurements are a spike in synovial hypoxic metabolites that supported the occurrence of reduced oxygen levels in the RA synovium [15]. The RA synovium’s vasculature is further put in danger by the mobility and accumulation of synovial fluid, which exacerbates hypoxia in a preischemic environment. The concurrent spike in metabolic demand and hypoxia serves as a strong signal for the emergence of new vascular tissue [49].
Angiogenic
The proinflammatory and hypoxic microenvironment leads to the production of a wide array of growth factors, cytokines, and chemokines in the RA synovium. These components cause endothelial cells (ECs) to arise from preexisting arteries, to proliferate, and to move into inflamed regions, which starts the RA vascular stage [50] (Fig. 8).

Mode of action of ADA in rheumatoid arthritis
Increased angiogenesis is also linked to morphological defects in newly formed capillaries in rheumatoid arthritis. Mural cells that are positive for smooth muscle actin (α-SMA) are absent from this subgroup of immature, dilated, and leaky neoangiogenic arteries. It is believed that chronic vascular endothelial growth factor (VEGF) overexpression is due to the imbalance between EC proliferation and the absence of concomitant pericyte production. Most often found in the sublining layer, these small capillaries are surrounded by inflammatory infiltrates (Fig. 7). Unexpectedly, RA activity and progression are connected to the density of immature vasculature, which is the only vascular component to regress in response to anti-TNF therapy [51].
Vasculogenesis
Is the process through which blood vessels grow organically. This process begins in the mammalian embryonic yolk sac and continues later during the embryo’s development [52]. In the synovial membrane area, these cells were previously shown to be present in cell clusters next to CD133+ cells. Alpha-chemokine receptor specific for stromal-derived factor 1 (CXCR4) was expressed in large amounts by CD34+ progenitor cells, whereas VEGF receptor 2 (VEGFR-2) was expressed by CD34+ and CD133+ cells. Moreover [53], CD34+ cells were cultured in the presence of granulocyte-macrophage colony-stimulating factor (GM–CSF) and stem cell factor after being extracted from the bone marrow of 13 patients with active RA and 9 controls. Von Willebrand factor-positive cells (vWF+) and CD31+/vWF+ cells were produced by RA bone marrow-derived CD34+ cells in much higher amounts compared to control samples. Consequently, bone marrow CD34+ cells may aid in the neovascularization of the synovium and may be responsible for the etiology of RA by supplying endothelial precursor cells [54].
Clinical biomarkers and diagnosis of RA
Clinically
There are several typical RA symptoms, including stiff and tender joints, morning joint pain, widespread nausea, and inconsistent lab results [55]. Early identification is essential in the treatment of RA since it may often stop the disease’s course in patients, preventing damage to the joints, irreversible disease progression, and early injury. RA biomarkers can be assessed at the molecular, biochemical, or cellular level and is a quantifiable sign of a particular biochemical, physiological, or morphological state. The past 5 years have seen the identification of novel biomarkers, particularly genomics, which are the fields that follow from the study of proteins (proteomics) and metabolites (metabolomics). When treating patients with RA, these indicators help the doctor choose the best course of action [56].
Typically
To diagnose RA, a variety of methods are utilized, including the assessment of risk factors, family history, joint ultrasound sonography, assessment of laboratory indicators including high C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR) in blood, discovery of RA-specific autoantibodies, and others [57]. CRP and ESR are frequently used as clinical indicators to assess the overall inflammatory status of patients with RA [58]. The acute phase reactant, often known as CRP, is composed of five 23 kDa pentraxin protein subunits. If tissue injury, inflammation, or infection are present, the serum concentration will rise by three or more log steps [59]. Increased neutrophil influx and phagocytosis are caused by CRP, which also activates the classical complement system and serves as an immune effector [60]. The release of monocyte chemoattractant protein 1 (MCP-1) and macrophage colony-stimulating factor (M–CSF), as well as the development of proinflammatory cytokines and the subsequent amplification of inflammation, have all been shown to be additional ways that CRP promotes macrophage survival and proliferation [61].
Clinical indications such morning stiffness, pain, nausea, grip strength, articular index, and impairment were all shown to positively correlate with CRP levels, as were the prevalence of the disorder, synovial histology improvements, and radiographic advancement [62]. To diagnose RA, monitor the course of the condition, and forecast the prognosis of joint injuries, CRP has been found to be a helpful marker [63]. ESR is a common laboratory test that gauges how quickly the patient’s blood erythrocytes are settling inside a test tube. Red blood cell coagulation results from elevated amounts of fibrinogen in the blood during inflammatory responses, tumors, and autoimmune diseases [64].
To identify the diagnosis and monitor the development of the condition, patients with RA have also been prescribed ultrasound and magnetic resonance imaging (MRI) [65]. The power of grayscale with ultrasound examination of inflammatory joints, allows Doppler imaging of synovial proliferation, active inflammation, and neo-angiogenesis [66]. Furthermore, ultrasound can detect bone erosions [67], as well as subclinical synovitis, which can lead to radiographic disease worsening even when the patient appears to be in clinical remission [68].
The advantages of ultrasonography include its availability, affordability, lack of side effects, and non-invasive real-time imaging capabilities [69]. Also, MRI techniques, on the other hand, are a highly sensitive diagnostic tool for detecting synovial hypertrophy and pannus formation prior to the onset of bone erosion [70].
Medication of RA
Traditional synthetic disease-modifying antirheumatic drugs (DMARDs) include TNF antagonists, anti-B cell, anti-T cell, and anti-IL6 antibodies, as well as methotrexate (MTX), sulfasalazine, leflunomide, and hydroxychloroquine [71]. MTX is the most often given drug for RA despite having an immunosuppressive effect and antiinflammatory qualities, and is reasonably priced. However, MTX has several restrictions because of toxicity. MTX has been related to immunological toxicity, cardiovascular toxicity, gastrointestinal toxicity, developmental toxicity, urinary toxicity, integumentary toxicity, and neurotoxicity [72]. Thus, the search for better and less expensive RA therapeutic agents is needed, so we will focus on this point in our review.
Adenosine deaminase (ADA) a key inflammatory enzyme
The primary enzyme responsible for the RA pathway’s inflammation, ADA (EC 3.5.4.4), is a small, monomeric 40 kDa enzyme with 363 amino acid residues [73]. Three different isoforms of ADA exist in humans: ADA-1, ADA-2, and ADA-3. ADA-1 also exists in a low molecular weight form in addition to a complex with the ADA-binding protein CD26. The enzymatic mechanism of ADA-2 and ADA-1 are the same. It has a complex multi-domain design and is a homodimer of 114 kDa [74]. ADA-2 has only been found in eukaryotic and multicellular organisms, and it has been dubbed an ADA growth factor (ADGF). ADA-2 is mostly localized in extracellular space. The protein ADA adopts a triose phosphate isomerase (TIM) barrel structure, folding up and including eight periphery helices, according to X-ray crystallographic research. Five additional helices cause the barrel’s regularity to be decreased.
The enzyme’s active site is located and firmly embedded on the C-terminal side of the barrel. In the deepest portion of what seems to be a funnel-shaped pocket, a catalytic zinc ion is tightly bound. The zinc ion is needed by the enzyme’s mechanism, which permits the addition elimination reaction of ADA catalyzes. The intermediate tetrahedral transition state is produced when the water molecule transfers to its hydroxyl group stereo specifically in the C6 position of adenosine. The inosine product is produced following the intermediate’s subsequent ammonia has been lost. Additionally, the zinc ion has a significant structural role, since its absence causes structural changes that spread throughout the ADA and significantly reduce its stability (Fig. 8).
The regulating role of ADA in immune system activity was of interest as evidence that the major cause of reduced T- and B-cell function is a congenital deficit of this enzyme. It affects 20–30% of people with severe combined immunodeficiency disease (SCID). These results highlighted the critical role of ADA in the development and function of the immune system, as seen in Fig. 7, along with significant research activities targeted at clarifying the role of purine metabolism in immune cell activity. As a result, it has been discovered that ADA regulates a variety of immune cell types, including neutrophils, macrophages, lymphocytes, and dendritic cells [75].
Role of ADA in inflammatory disorders
The function of ADA in the pathogenesis of RA illness has gained attention because of its unique immunological characteristics. The circulating mononuclear cells from patients with RA had far lower levels of ADA than cells from healthy individuals. On the other hand, the synovial effusions of those with RA contained high quantities of this enzyme activity [56].
As well as this, synovial fluid from those with reactive arthritis, juvenile chronic arthritis, chronic seronegative polyarthritis, and seropositive RA showed signs of ADA activation. The presence of ADA in synovial fluid exhibited a strong correlation with the disease’s systemic activity, as measured by hemoglobin concentration and erythrocyte sedimentation rate. This finding suggests that ADA activity should be evaluated as an additional criterion for judging the severity of joint inflammation. The enzyme activity was highest in the lymphocytes and monocytes of individuals with RA, and ADA-2 was the isoform that was only expressed in monocytes. Additionally, any possible connections between enzymatic activity in synovial fluid and the amount of matrix metalloproteinase-9 (MMP-9) present were elucidated, as well as the consequences of patients with RA having elevated ADA isozyme activity [76].
These results were confirmed by the fact that patients with RA had increased ADA activity in their synovial fluid and by the fact that their data showed strong positive correlations between MMP-9 and ADA isoforms [77]. The effects of MTX on a variety of enzymes, including ADA, hypoxanthine–guanine phosphoribosyltransferase, purine nucleoside phosphorylase, and 5′-nucleotidase. After taking the drugs, they saw a considerable reduction in all purine’s enzymatic activity. The appraisal of these measures as useful biochemical markers in patients with RA is further supported by prior research, which demonstrated a robust and proportional association between total blood ADA and ADA-2 activity and the degree of inflammation [78].
Inhibitors of ADA activity
ADA inhibitors come in four main varieties: transition state, ground state, non-nucleoside, and plant extracts. The tetrahedral intermediate produced by the ADA-catalyzed deamination process shares structural similarities with transition-state inhibitors [79]. The third class of derivatives, known as non-nucleoside inhibitors, was specifically composed of a set of imidazole-4-carboxamides produced and synthesized by Terasaka and colleagues at Fujisawa Pharmaceutical Company, which are equivalent to ground-state compounds [80]. Additional chemicals that successfully inhibit ADA activity include a wide range of medicines and phenolic compounds found in plants, such as flavonoids. The kinds of ADA inhibitors and the compounds that prevent ADA from interacting with cell surface proteins will be discussed in the review’s subsequent sections, as seen in Fig. 9.

The main classes of adenosine deaminase inhibitors
Transition-state inhibitors
Coformycin and deoxycoformycin analogs
In the sections that follow, we will go through the various ADA inhibitor classes and compounds that prevent ADA from interacting with cell surface proteins (Fig. 9) [81]. The two substances that effectively limit ADA activity the most frequently are the transition-state inhibitors. Their extraordinarily lengthy, practically irreversible, and tightly binding interactions with the enzyme are thought to be the reason for their efficiency [82]. The tetrahedral carbon (C8) in both variants has a hydroxyl group attached to it. The stereochemistry here has a big impact on potency; the 8R-diastereomer is almost 107 times stronger than the 8S equivalent [83].
Ground-state compounds
Deaza- and dideazaadenosine derivatives
Compared with 7-deaza (tubercidin) and 1,7-dideazaadenosine, which are absolutely inert, 3-deaza and 1,3-dideazaadenosine are only weak inhibitors [84]. Although 1-deazaadenosine retains all molecular recognition properties when used as a substrate for ADA, it is not deaminated because it lacks the catalytically required N1-protonation [81]. A chlorine atom at position 2 decreased the inhibitory action of ADA. The compounds became more ADA resistant when a chlorine atom was added to the substrates in this location [85]. The 20-deoxyribose derivatives produced good inhibitory effects when hydroxyl, methyl, and cyclopropyl groups were substituted at the N6 position [86].
Erythro-9-(2-hydroxy-3-nonyl) adenine compounds
When adenine is connected at the N9 position to a chiral hydroxy nonyl chain, erythro-9-(2-hydroxy-3-nonyl) adenine (EHNA) is produced. The ADA inhibitor is a semi-tight one that first exhibits conventional competitive inhibition before successively rearranging the enzyme and tightly form ADA-inhibitor complex [81].
Non-nucleoside inhibitors
Despite the above-mentioned drugs’ success in preventing ADA activity, their poor pharmacokinetics continue to prevent their broad use. However, fresh non-nucleoside ADA inhibitors have recently been created, such as a family of imidazole-4-carboxamides [81]. The molecular interactions between 1-deazaadenosine and murine ADA were the focus of these newly discovered compounds [87]. Additionally, a 1-(1-hydroxy-4-phenylbutan-2-yl)-1H-imidazole-4-carboxamide with good pharmacokinetics (oral bioavailability) was created [84].
Plant extracts
In the typical person’s diet, flavonoids and phenolic compounds are found in plants, fruits, and vegetables. Numerous pharmacological effects of these plant phenolic and flavonoid compounds to modestly decrease of ADA activity had been studied [88]. The increase in endogenous adenosine that emerges from these drugs’ ability to inhibit competitive ADA may have some positive effects. Studies on the structure–activity correlation of these medicines and ADA indicate that the inhibitory effect requires the presence of the hydroxyl group at the three positions of the chromone molecule. There is a suggestion that the hydroxyl groups on the side of the phenyl ring are also significant [80].
Natural product
Natural remedies have traditionally been used to treat infectious diseases and are now accepted cures for a wide range of illnesses [89]. Over the past 10 years, natural treatment has become widely accepted and the public has become more interested in it. As a result, herbal medications are now sold not only in drugstores but also in supermarkets and grocery shops. In Africa and other underdeveloped nations, almost 80% of people still use traditional herbal treatments to cure illnesses because they are more readily available and less expensive than manufactured drugs [90]. They also have antiinflammatory, spasmolytic, antioxidants sedative, antimicrobial, disinfectants, anti-diabetic, and immunostimulant properties against a variety of health problems [91].
Quercetin
Quercetin is the name of the major polyphenolic flavonoid that may be found in berries, lovage, capers, cilantro, dill, apples, and onions. It belongs to one of the six subclasses of flavonoids [92]. It is completely soluble in lipids and alcohol and is colored yellow. It is, however, hardly soluble in hot water and insoluble in cold water. The word “quercetin” is derived from the Latin word “quercetum,” which means “oak forest.” Additionally, it belongs to the flavanol class, which the human body does not make [93]. The designations C15H10O7 and 2,3,5,7-trihydroxy-2,(3,4-dihydroxyphenyl)chromen-4-one, respectively, were assigned to the quercetin by the International Union of Pure and Applied Chemistry (IUPAC). One of the most important plant chemicals, quercetin is used medicinally to treat a variety of conditions, including arthritis, rheumatoid arthritis, allergic arthritis, metabolic illnesses, and inflammatory diseases [94].
Antiinflammatory effects of quercetin
Recently, quercetin proved its antiinflammatory activity through direct inhibition to ADA in RA rat model [95]. Moreover, several in vitro studies elucidated that quercetin could inhibit the generation of TNF-α, which is mediated by lipopolysaccharide (LPS) in macrophages and IL-8-induced LPS in lung A549 cells [96]. The capacity of quercetin to inhibit TNF-α and IL-1 levels of LPS-generated mRNA results in a reduced degree of apoptotic neuronal cell death produced by microglial activation. Quercetin prevents the synthesis of inflammatory enzymes such as cyclooxygenase (COX) and lipoxygenase (LOX) [97].
Additionally, it may limit the generation of tryptase, histamine, and proinflammatory cytokines by mast cells created from human umbilical cord blood; this protection is most likely brought about by the reduction of calcium influx and the suppression of phosphoprotein kinase C (PKC) [98] (Fig. 10).

Antiinflammatory effects of quercetin
Quercetin has demonstrated potent antiinflammatory activity with higher absorption through the skin’s surface in rats [99]. According to numerous studies, quercetin blocks the expression of vascular cell adhesion molecules (VCAM-1), intracellular cell adhesion molecules (ICAM-1), and E-selectin in human umbilical vein endothelial cells, as well as the secretion of iNOS, IL-1, and TNF induced by bacterial lipopolysaccharide (LPS) in macrophages, and RAW2647 cells. In non-alcoholic steatohepatitis (NASH) mice, quercetin and its glycoside rutin were shown to reduce TNF-α and IL-6 inflammatory markers [100] (Fig. 10).
Olabiyi et al., revealed that, with an IC50 value of about 0.00400005 mg/ml, quercetin had the strongest effect in inhibiting ADA [101]. The histological study supports all quercetin dosages’ efficacy in lowering edema development and the inflammatory response. This is in line with other studies that discovered quercetin reduced inflammatory effects on neutrophil activation and synovial cell activity [102].
Quercetin exerts antiinflammatory properties by regulating inflammatory cytokine production mediated by macrophages and T lymphocytes, as demonstrated by the finding that doses of quercetin (20 M and 40 M) could lower IFN levels in supernatants from activated Th cells cultured with either rutin or quercetin [103].
Inhibitory mechanism of adenosine deaminase (ADA) by quercetin (QUE) for RA treatment
RA is an autoimmune disease. It is well known that T and B lymphocytes are essential to the etiology and progression of RA [104]. Furthermore, it has been demonstrated that joint deterioration and an aggravation of clinical symptoms are associated with autoantibodies in patients with RA [105, 106].
Adenosine deaminase (ADA) is an essential enzyme in purine metabolism, it converts adenosine to inosine to control intra- and extracellular adenosine concentrations [107, 108]. Adenosine is a significant purine that interacts with receptors and controls a wide range of physiological processes [109, 110]. Adenosine and subsequently adenosine deaminase could have either pro- or antiinflammatory effects on joints tissues [111]. Extracellular adenosine concentrations are typically less than 1 μM (30–200 nM) under normal settings, but they can rise to 100 μM in hypoxic and inflammatory situations [112]. Under low energy charge conditions, intracellular ATP breakdown is the primary source of extracellular adenosine [113, 114], which is subsequently stored and exported out of cells via equilibrate nucleoside transporters instead of being deaminated to inosine right away [115]. Moreover, adenosine nucleotides released into extracellular space can hydrolyze to form additional cellular adenosine under cellular stress conditions as shown in Fig. 11 [116]. Furthermore, it has been documented that adenosine deaminase activity increases in several disorders inside the body [117]. Therefore, the inhibition of this inflammatory key enzyme can significantly affect the clinical progression treatment of numerous diseases especially RA [118, 119].

Mechanistic role of quercetin as inhibitor for adenosine deaminase enzyme for the treatment of rheumatoid arthritis
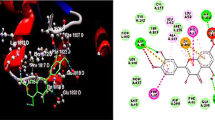
The pathophysiology of RA involves lymphocytes that contain ADA with abnormal activity [120, 121]. According to the previous literature point of view, QUE has an inhibitory effect on the activity of lymphocytic ADA activity [122,123,124] as shown in Fig. 11.
This inhibitory effect is dieted by reduced the adenosine elevated levels through restoration of T-cell homeostasis [125,126,127,128], regulation of Th17 cell differentiation [129], regulation of Th17/Treg-related cytokine levels, reduction of autoantibody production, and regulation of nucleoside triphosphate diphosphohydrolase (NTPDase) activities [128, 130]. Collectively, QUE established the immune-regulatory effect and is considered one of the most important natural candidates that can used for RA therapy [131, 132] as mentioned in Fig. 11.
Conclusion
Rheumatoid arthritis (RA) is an autoimmune disease that involves the immune system, particularly T and B lymphocytes, in its etiology and progression. Joint deterioration and worsening clinical symptoms in patients with RA are associated with autoantibodies. Adenosine deaminase (ADA), an essential enzyme in purine metabolism, plays a role in controlling adenosine concentrations and can have pro- antiinflammatory effects on joint tissues. Inhibition of ADA can potentially impact the clinical progression and treatment of RA. Intracellular ATP breakdown is primary source of extracellular adenosine, which increases in hypoxic and inflammatory condition. The pathophysiology of RA involves lymphocytes that contain ADA. Therefore, inhibition of lymphocytic ADA activity has been shown to have an immune-regulatory effect on such diseases. Quercetin (QUE) is considered an important natural candidate for RA therapy due to its immune-regulatory effect, as it inhibits lymphocyte ADA activity and reduce elevated adenosine. Also, QUE has the potential effect in restoring T cell homeostasis, regulating Th17 cell differentiation, and reducing autoantibody production. Ultimately QUE can be used as a potent candidate in the treatment of RA disease.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ADA:
-
Adenosine deaminase
- CD4+ :
-
Cluster of differentiation 4
- COX:
-
Cyclooxygenase
- CRP:
-
C-reactive protein
- CXCR4:
-
Alpha-chemokine receptor specific for stromal-derived factor 1
- DC:
-
Dendritic cell
- DMARDs:
-
Synthetic disease-modifying antirheumatic drugs
- ESR:
-
Erythrocyte sedimentation rate
- GBD:
-
Global Burden of Disease
- GM-CSF:
-
Granulocyte-macrophage colony-stimulating factor
- ICAM-1:
-
Intracellular cell adhesion molecules
- IFN:
-
Interferons
- IL-1:
-
Interleukin 1
- IL-6:
-
Interleukin 6
- LOX:
-
Lipoxygenase
- LPS:
-
Lipopolysaccharide
- MCP-1:
-
Monocyte chemoattractant protein 1
- M-CSF:
-
Macrophage colony-stimulating factor
- MMP-1:
-
Matrix metalloproteinase 1
- MMP-9:
-
Matrix metalloproteinase 9
- MRI:
-
Magnetic resonance imaging
- MTX:
-
Methotrexate
- PKC:
-
Phosphoprotein kinase C
- RA:
-
Rheumatoid arthritis
- RANK-L:
-
Receptor activator of nuclear factor kappa-β ligand
- SCID:
-
Severe combined immunodeficiency disease
- SNPs:
-
Single nucleotide polymorphisms
- TIM:
-
Triose phosphate isomerase
- TLRs:
-
Toll-like receptors
- TNF-α:
-
Tumor necrosis factor alpha
- VCAM-1:
-
Vascular cell adhesion molecules
- VEGF:
-
Vascular endothelial growth factor
References
Huang J-B, Chen Z-R, Yang S-L, Hong F-F. Nitric oxide synthases in rheumatoid arthritis. Molecules. 2023;28:4414.
Mishra R, Mohanty S, Mahapatra S, Prasad P. Overview of microbial therapeutics in immunological disorders. In: Microbiome Therapeutics, Elsevier, 2023, 289–353.
Gheita TA, Raafat HA, El-Bakry SA, Elsaman A, El-Saadany HM, Hammam N, El-Gazzar II, Samy N, Elsaid NY, Al-Adle SS. Rheumatoid arthritis study of the Egyptian College of Rheumatology (ECR): nationwide presentation and worldwide stance. Rheumatol Int. 2023;43:667.
Finckh A, Gilbert B, Hodkinson B, Bae S-C, Thomas R, Deane KD, Alpizar-Rodriguez D, Lauper K. Global epidemiology of rheumatoid arthritis. Nat Rev Rheumatol. 2022;18:591–602.
Abdel Fattah MA, Barghouth MH, Wassel MO, Deraz OH, Khalil AE, Sarsik HM, Mohsen AMA, Qenawy AS, Abou El Fadl RK. Epidemiology of dental caries in permanent dentition: evidence from a population-based survey in Egypt. BMC Public Health. 2022;22:1–11.
Tsao CW, Aday AW, Almarzooq ZI, Anderson CA, Arora P, Avery CL, Baker-Smith CM, Beaton AZ, Boehme AK, Buxton AE. Heart disease and stroke statistics—2023 update: a report from the American Heart Association. Circulation. 2023;147:e93–621.
Riedmann J, Solonavalona AF, Rakotozafy AR, Ralamboson S, Endres M, Siegerink B, Siebert E, Knauss S, Emmrich JV. Proportion of stroke types in Madagascar: a tertiary-level hospital-based case series. PLoS ONE. 2022;17:e0276199.
Laborde CM, Larzabal L, González-Cantero Á, Castro-Santos P, Díaz-Peña R. Advances of genomic medicine in psoriatic arthritis. J Personalized Med. 2022;12:35.
Kozak M, Pawlik A. The role of the oral microbiome in the development of diseases. Int J Mol Sci. 2023;24:5231.
Cho MH, Hobbs BD, Silverman EK. Genetics of chronic obstructive pulmonary disease: understanding the pathobiology and heterogeneity of a complex disorder. Lancet Respir Med. 2022;10:485.
Hettiarachchi G, Komar AA. GWAS to Identify SNPs Associated with Common Diseases and Individual Risk: Genome Wide Association Studies (GWAS) to Identify SNPs Associated with Common Diseases and Individual Risk. in: Single Nucleotide Polymorphisms: Human Variation and a Coming Revolution in Biology and Medicine, Springer, 2022, 51–76.
Zhang HG, McDermott G, Seyok T, Huang S, Dahal K, L’Yi S, Lea-Bonzel C, Stratton J, Weisenfeld D, Monach P. Identifying shared genetic architecture between rheumatoid arthritis and other conditions: a phenome-wide association study with genetic risk scores. EBioMedicine. 2023;92:104581.
Schor D, Porto LC, Roma EH, Castro-Alves J, Villela AP, Araújo AQ, Glória Bonecini-Almeida M. Putative role of HLA polymorphism among a Brazilian HTLV-1-associated myelopathy/tropical spastic paraparesis (HAM/TSP) population. Sci Reports. 2023;13:7659.
Andreev D, Kachler K, Schett G, Bozec A. Rheumatoid arthritis and osteoimmunology: the adverse impact of a deregulated immune system on bone metabolism. Bone. 2022;162:116468.
Wang X, Fan D, Cao X, Ye Q, Wang Q, Zhang M, Xiao C. The role of reactive oxygen species in the rheumatoid arthritis-associated synovial microenvironment. Antioxidants. 2022;11:1153.
Ding Q, Hu W, Wang R, Yang Q, Zhu M, Li M, Cai J, Rose P, Mao J, Zhu YZ. Signaling pathways in rheumatoid arthritis: implications for targeted therapy. Signal Transduct Target Ther. 2023;8:68.
Fearon U, Hanlon MM, Floudas A, Veale DJ. Cellular metabolic adaptations in rheumatoid arthritis and their therapeutic implications. Nat Rev Rheumatol. 2022;18:398–414.
Peng X, Wang Q, Li W, Ge G, Peng J, Xu Y, Yang H, Bai J, Geng D. Comprehensive overview of microRNA function in rheumatoid arthritis. Bone Res. 2023;11:8.
Mrid RB, Bouchmaa N, Ainani H, El Fatimy R, Malka G, Mazini L. Anti-rheumatoid drugs advancements: new insights into the molecular treatment of rheumatoid arthritis. Biomed Pharmacother. 2022;151:113126.
Del Prete A, Salvi V, Soriani A, Laffranchi M, Sozio F, Bosisio D, Sozzani S. Dendritic cell subsets in cancer immunity and tumor antigen sensing. Cell Mol Immunol. 2023;20:432–47.
Suszczyk D, Skiba W, Pawłowska A, Polak G, Tarkowski R, Wertel I. Expression of Gal-9 on dendritic cells and soluble forms of TIM-3/Gal-9 in patients suffering from endometriosis. Int J Mol Sci. 2023;24:5948.
Mantel I, Fein MR, Donlin LT. Emerging synovial cell states in rheumatoid arthritis as potential therapeutic targets. Curr Opinion Rheumatol. 2023;35:249–54.
Petrelli F, Mariani FM, Alunno A, Puxeddu I. Pathogenesis of rheumatoid arthritis: one year in review 2022. Clin Exp Rheumatol. 2022;40:475–82.
Keller CW, Adamopoulos IE, Lünemann JD. Autophagy pathways in autoimmune diseases. J Autoimmunity. 2023;136:103030.
Rastogi I, Jeon D, Moseman JE, Muralidhar A, Potluri HK, McNeel DG. Role of B cells as antigen presenting cells. Front Immunol. 2022;13:954936.
Li Y, Shao Y, He Y, Li Q, Duan L. Potential role of interleukin-33 in systemic lupus erythematosus by regulating toll like receptor 4. Eur J Inflamm. 2022;20:1721727X221094455.
Xu Y-D, Cheng M, Shang P-P, Yang Y-Q. Role of IL-6 in dendritic cell functions. J Leukocyte Biol. 2022;111:695–709.
Zeng L, Yu G, Yang K, Xiang W, Li J, Chen H. Efficacy and safety of mesenchymal stem cell transplantation in the treatment of autoimmune diseases (rheumatoid arthritis, systemic lupus erythematosus, inflammatory bowel disease, multiple sclerosis, and ankylosing spondylitis): a systematic review and meta-analysis of randomized controlled trial. Stem Cells Int. 2022;2022:20.
Dagar S, Singh J, Saini A, Kumar Y, Chhabra S, Minz RW, Rani L. Gut bacteriome, mycobiome and virome alterations in rheumatoid arthritis. Front Endocrinol. 2023;13:1044673.
Gómez-Melero S, Caballero-Villarraso J. CCR6 as a potential target for therapeutic antibodies for the treatment of inflammatory diseases. Antibodies. 2023;12:30.
Deng J, Lu C, Zhao Q, Chen K, Ma S, Li Z. The Th17/Treg cell balance: crosstalk among the immune system, bone and microbes in periodontitis. J Periodontal Res. 2022;57:246–55.
Gargano G, Oliva F, Oliviero A, Maffulli N. Small interfering RNAs in the management of human rheumatoid arthritis. Br Med Bull. 2022;142:34–43.
Bala N, McGurk AI, Zilch T, Rup AN, Carter EM, Leddon SA, Fowell DJ. T cell activation niches—optimizing T cell effector function in inflamed and infected tissues. Immunol Rev. 2022;306:164–80.
Weivoda MM, Bradley EW. Macrophages and bone remodeling. J Bone Mineral Res. 2023;38:359–69.
Ahmad SF, Nadeem A, Ansari MA, Bakheet SA, Alomar HA, Al-Mazroua HA, Ibrahim KE, Alshamrani AA, Al-Hamamah MA, Alfardan AS. CXCR3 antagonist NBI-74330 mitigates joint inflammation in collagen-induced arthritis model in DBA/1J mice. Int Immunopharmacol. 2023;118:110099.
Sadek KM, El Moshy S, Radwan IA, Rady D, Abbass MM, El-Rashidy AA, Dörfer CE, Fawzy El-Sayed KM. Molecular basis beyond interrelated bone resorption/regeneration in periodontal diseases: a concise review. Int J Mol Sci. 2023;24:4599.
Jakovljevic A, Nikolic N, Holtzman LP, Tournier P, Gaudin A, Cordaro L, Milinkovic I. Involvement of the Notch signaling system in alveolar bone resorption. Jpn Dental Sci Rev. 2023;59:38–47.
Juma SN, Liao J, Huang Y, Vlashi R, Wang Q, Wu B, Wang D, Wu M, Chen G. Osteoarthritis versus psoriasis arthritis: physiopathology, cellular signaling, and therapeutic strategies. Genes Dis. 2023.
da Fonseca LF, Lana JF, Visoni SBC, Lana AVS, Irlandini E, Azzini GOM. “Preparing the Soil”: optimizing metabolic management in regenerative medicine procedures joint function preservation: a focus on the osteochondral unit. 2022, 63–74.
Nowaczyk A, Szwedowski D, Dallo I, Nowaczyk J. Overview of first-line and second-line pharmacotherapies for osteoarthritis with special focus on intra-articular treatment. Int J Mol Sci. 2022;23:1566.
Connors JP, Stelzer JW, Garvin PM, Wellington IJ, Solovyova O. The role of the innate immune system in wear debris-induced inflammatory peri-implant osteolysis in total joint arthroplasty. Bioengineering. 2022;9:764.
Gao D, Gao X, Yang F, Wang Q. Neuroimmune crosstalk in rheumatoid arthritis. Int J Mol Sci. 2022;23:8158.
Kurowska-Stolarska M, Alivernini S. Synovial tissue macrophages in joint homeostasis, rheumatoid arthritis and disease remission. Nat Rev Rheumatol. 2022;18:384–97.
Kumar A, Sood A, Singhmar R, Mishra YK, Thakur VK. Manufacturing of functional hydrogels for inducing angiogenic-osteogenic coupled progressions in hard tissue repair: prospects and challenges. Biomaterials Sci. 2022;10:5472.
Neumayer G, Torkelson J, Li S, McCarthy K, Zhen H, Vangipuram M, Jackow J, Rami A, Hansen C, Guo Z. A scalable, GMP-compatible, autologous organotypic cell therapy for Dystrophic Epidermolysis Bullosa bioRxiv. 2023; 2023.2002. 2028.529447.
Behl T, Chadha S, Sehgal A, Singh S, Sharma N, Kaur R, Bhatia S, Al-Harrasi A, Chigurupati S, Alhowail A. Exploring the role of cathepsin in rheumatoid arthritis. Saudi J Biol Sci. 2022;29:402–10.
Sanchez-Lopez E, Coras R, Torres A, Lane NE, Guma M. Synovial inflammation in osteoarthritis progression. Nat Rev Rheumatol. 2022;18:258–75.
Olivotto E, Belluzzi E, Pozzuoli A, Cigolotti A, Scioni M, Goldring SR, Goldring MB, Ruggieri P, Ramonda R, Grigolo B. Do synovial inflammation and meniscal degeneration impact clinical outcomes of patients undergoing arthroscopic partial meniscectomy? A histological study. Int J Mol Sci. 2022;23:3903.
Kondo N, Kanai T, Okada M. Rheumatoid arthritis and reactive oxygen species: a review. Curr Issues Mol Biol. 2023;45:3000–15.
Vasdev N, Pawar B, Gupta T, Mhatre M, Tekade RK. A bird’s eye view of various cell-based biomimetic nanomedicines for the treatment of arthritis. Pharmaceutics. 2023;15:1150.
Luzo ÂCM, Leme KC, Fávaro WJ, Durán N, Bíscaro GG, de Oliveira ALR, Boumediene K, Hammad M, Baugé C. Platelet-rich plasma, their growth factors, cytokines and clinical use. in: Nanotechnology and Regenerative Medicine, Elsevier, 2023, 265–314.
Zarychta E, Ruszkowska-Ciastek B. Cooperation between angiogenesis, vasculogenesis, chemotaxis, and coagulation in breast cancer metastases development: pathophysiological point of view. Biomedicines. 2022;10:300.
Farahmand Y, Tehrany PM, Nazari A, Nava ZH, Alsaffar MF, Yazdani O, Adili A, Esbati R, Ghafouri K. A comprehensive survey into the role of exosomes in pancreatic cancer; from the origin of cancer to the progress and possibility of diagnosis and treatment. Pathol Res Pract. 2023;245:154465.
Pan Y, Li Y, Dong W, Jiang B, Yu Y, Chen Y. Role of nano-hydrogels coated exosomes in bone tissue repair. Front Bioeng Biotechnol. 2023;11:1167012.
d Sousa BFT. Anti-inflammatory diet and rheumatoid arthritis: overview on the current evidence. [sn], 2022.
Sharma A, Goel A. Pathogenesis of rheumatoid arthritis and its treatment with anti-inflammatory natural products. Mol Biol Rep. 2023;50:4687–706.
Ahmed R, Soliman N. Disease Activity Score (DAS) correlation to serum prolidase as a collagen turnover marker in comparison to some pro-inflammatory markers in rheumatoid arthritis patients: clinical and sonographic study. J Adv Med Med Res. 2023;35:14–24.
Elhelaly M, Nassar DK, Nassar MK, Abdelalim KT, Tharwat S. Evaluation of CXCL2 and autophagy genes expression in rheumatoid arthritis patients and its relation to cardiovascular diseases. Mansoura Med J. 2023;52:1–25.
Meurman J, Celec P, Gutierrez AM, Sjöwall C. Pronounced diurnal pattern of salivary c-reactive protein (crp) with modest associations diagnostic and therapeutic applications of pentraxin and pentraxin-associated proteins. 2022.
Behrens LM, van Egmond M, van den Berg TK. Neutrophils as immune effector cells in antibody therapy in cancer. Immunol Rev. 2023;314:280–301.
Tahmasebi S, Alimohammadi M, Khorasani S, Rezaei N. Pro-tumorigenic and anti-tumorigenic roles of pro-inflammatory cytokines in cancer. in: Handbook of Cancer and Immunology, Springer, 2022, 1–25.
Koh JH, Yoon SJ, Kim M, Cho S, Lim J, Park Y, Kim H-S, Kwon SW, Kim W-U. Lipidome profile predictive of disease evolution and activity in rheumatoid arthritis. Exp Mol Med. 2022;54:143–55.
Wu D, Luo Y, Li T, Zhao X, Lv T, Fang G, Ou P, Li H, Luo X, Huang A. Systemic complications of rheumatoid arthritis: focus on pathogenesis and treatment. Front Immunol. 2022;13:1051082.
Kahar MA. Erythrocyte sedimentation rate (with its inherent limitations) remains a useful investigation in contemporary clinical practice. Ann Pathol Lab Med. 2022;9:R9.
Bird A, Oakden-Rayner L, McMaster C, Smith LA, Zeng M, Wechalekar MD, Ray S, Proudman S, Palmer LJ. Artificial intelligence and the future of radiographic scoring in rheumatoid arthritis: a viewpoint. Arthritis Res Ther. 2022;24:1–10.
Carotti M, Filippucci E, Salaffi F, Martino F. Therapy efficacy evaluation in synovitis. In: Musculoskeletal ultrasound in orthopedic and rheumatic disease in adults, Springer, 2022, 233–248.
Draghi F, Ferrozzi G, Ballerini D, Bortolotto C. Psoriatic arthritis: Ultrasound peculiarities with particular emphasis on enthesitis. J Clin Ultrasound. 2022;50:556–60.
Wang Y-W, Chen J-F, Ko C-H, Cheng T-T, Chiu W-C, Yu S-F, Hsu C-Y, Chen Y-C. Factors associated with subclinical inflammation of wrist joints in rheumatoid arthritis patients with low or no disease activity-a RA ultrasound registry study. BMC Musculoskelet Disord. 2023;24:438.
Aouad K, Gossec L. Defining and managing flares in axial spondyloarthritis. Curr Opinion Rheumatol. 2022;34:195–202.
Dnofrio B, van der Helm-van Mil A, Huizinga TWJ, van Mulligen E. Inducibility or predestination? Queries and concepts around drug-free remission in rheumatoid arthritis. Expert Rev Clin Immunol. 2023;19:217.
Han R, Ren HC, Zhou S, Gu S, Gu Y-Y, Sze DM-Y, Chen M-H. Conventional disease-modifying anti-rheumatic drugs combined with Chinese Herbal Medicines for rheumatoid arthritis: a systematic review and meta-analysis. J Tradit Complement Med. 2022;12:437–46.
Yazbeck V, Alesi E, Myers J, Hackney MH, Cuttino L, Gewirtz DA. An overview of chemotoxicity and radiation toxicity in cancer therapy. Adv Cancer Res. 2022;155:1–27.
Lackie J. A dictionary of biomedicine, Oxford Quick Reference, 2010.
Antonioli L, Colucci R, La Motta C, Tuccori M, Awwad O, Da Settimo F, Blandizzi C, Fornai M. Adenosine deaminase in the modulation of immune system and its potential as a novel target for treatment of inflammatory disorders. Curr Drug Targets. 2012;13:842–62.
Herman-de-Sousa C, Costa MA, Silva RP, Ferreirinha F, Ribeiro S, Correia-de-Sá P. A2A receptor-induced overexpression of pannexin-1 channels indirectly mediates adenosine fibrogenic actions by favouring ATP release from human subcutaneous fibroblasts. Life Sci. 2022;310:121080.
M. Abedsaeidi, F. Hojjati, A. Tavassoli and A. Sahebkar Biology of Tenascin C and Its Role in Physiology and Pathology Current Medicinal Chemistry (2023).
Sanges S, Guerrier T, Duhamel A, Guilbert L, Hauspie C, Largy A, Balden M, Podevin C, Lefèvre G, Jendoubi M. Soluble markers of B cell activation suggest a role of B cells in the pathogenesis of systemic sclerosis-associated pulmonary arterial hypertension. Front Immunol. 2022;13:4196.
Saffari F, Jafarzadeh A. Development of anti-rituximab antibodies in rituximab-treated patients: related parameters & consequences. Indian J Med Res. 2022;155:335–46.
Venugopal PP, Chakraborty D. Molecular mechanism of inhibition of COVID-19 main protease by β-adrenoceptor agonists and adenosine deaminase inhibitors using in silico methods. J Biomol Struct Dyn. 2022;40:5112–27.
Monroy-Mora A, de Lourdes Mora-García M, Mora KAM, Hernández-Montes J, García-Rocha R, Don-López CA, Weiss-Steider B, Montesinos-Montesinos JJ, Monroy-García A. Inhibition of adenosine deaminase activity reverses resistance to the cytotoxic effect of high adenosine levels in cervical cancer cells. Cytokine. 2022;158:155977.
Kutryb-Zajac B, Mierzejewska P, Slominska EM, Smolenski RT. Therapeutic perspectives of adenosine deaminase inhibition in cardiovascular diseases. Molecules. 2020;25:4652.
Minnow YV. Purine metabolism in plasmodium falciparum as a drug target for malaria. (Ed.^, Eds.), Albert Einstein College of Medicine, 2022.
Malki Y, Martinez J, Masurier N. 1, 3-Diazepine: a privileged scaffold in medicinal chemistry. Med Res Rev. 2021;41:2247–315.
Matyugina ES, Kochetkov SN, Khandazhinskaya AL. Synthesis and biological activity of aza and deaza analogues of purine nucleosides. Russian Chem Rev. 2021;90:1454.
Zhang M, Dai X, Xiang Y, Xie L, Sun M, Shi J. Advances in CD73 inhibitors for immunotherapy: antibodies, synthetic small molecule compounds, and natural compounds. Eur J Med Chem. 2023;258:115546.
Plé C, Tam H-K, Vieira Da Cruz A, Compagne N, Jiménez-Castellanos J-C, Müller RT, Pradel E, Foong WE, Malloci G, Ballée A. Pyridylpiperazine-based allosteric inhibitors of RND-type multidrug efflux pumps. Nat Commun. 2022;13:115.
Man S, Lu Y, Yin L, Cheng X, Ma L. Potential and promising anticancer drugs from adenosine and its analogs. Drug Discovery Today. 2021;26:1490–500.
Anantachoke N, Duangrat R, Sutthiphatkul T, Ochaikul D, Mangmool S. Kombucha beverages produced from fruits, vegetables, and plants: a review on their pharmacological activities and health benefits. Foods. 2023;12:1818.
Salatino A. Perspectives for uses of propolis in therapy against infectious diseases. Molecules. 2022;27:4594.
Aware CB, Patil DN, Suryawanshi SS, Mali PR, Rane MR, Gurav RG, Jadhav JP. Natural bioactive products as promising therapeutics: a review of natural product-based drug development. South Afr J Bot. 2022;151:512.
Mishra B. Assessment of plant diversity: a medicinal, conservational and environmental study. Asian J Biol. 2023;18:39–57.
Alzate-Yepes T, Pérez-Palacio L, Martínez E, Osorio M. Mechanisms of action of fruit and vegetable phytochemicals in colorectal cancer prevention. Molecules. 2023;28:4322.
Bisht A, Sharma P, Agarwal G. An insight into physiochemical property, bioavailability and pharmacology of Quercetin: a bioflavonoid. Group. 2023;8:9.
Yi Y-S. Regulatory roles of flavonoids in caspase-11 non-canonical inflammasome-mediated inflammatory responses and diseases. Int J Mol Sci. 2023;24:10402.
Di Cristo F, Valentino A, De Luca I, Peluso G, Bonadies I, Di Salle A, Calarco A. Polylactic acid/poly (vinylpyrrolidone) co-electrospun fibrous membrane as a tunable quercetin delivery platform for diabetic wounds. Pharmaceutics. 2023;15:805.
Kianmehr M, Behdadfard M, Hedayati-Moghadam M, Khazdair MR. Effects of herbs and derived natural products on lipopolysaccharide-induced toxicity: a literature review. Oxidative Med Cell Longevity. 2023;2023:1.
Guang Q, Zhang L, Tang X, Li J, Cao C, Chen H, Qiu L. Quercetin alleviates inflammation induced by porcine reproductive and respiratory syndrome virus in MARC-145 cells through the regulation of arachidonic acid and glutamine metabolism. 2023.
Koraganji DV, Mounika A, Sushanth P, Kandra P. Effect of plant-derived immunomodulators on the immune system. In: Nutraceuticals and functional foods in immunomodulators, Springer, 2023, 109–120.
Shrivastava AK, Sahu PK, Cecchi T, Shrestha L, Shah SK, Gupta A, Palikhey A, Joshi B, Gupta PP, Upadhyaya J. An emerging natural antioxidant therapy for COVID-19 infection patients: current and future directions. Food Front. 2023;4:1179.
Nweze CC, Tseaa W, Ekpe IP. Anti-inflammatory properties of quercetin: a review. J Drug Delivery Ther. 2022;12:205–10.
Olabiyi AA, AlliSmith YR, Ukwenya VO. Quercetin enhances sexual behavior and improves ectonucleotidases activity in the hypothalamus of rats treated with cyclosporine. J Food Biochem. 2021;45:e13864.
Zarenezhad E, Abdulabbas HT, Kareem AS, Kouhpayeh SA, Barbaresi S, Najafipour S, Mazarzaei A, Sotoudeh M, Ghasemian A. Protective role of flavonoids quercetin and silymarin in the viral-associated inflammatory bowel disease: an updated review. Arch Microbiol. 2023;205:252.
Zhou H-F, Yang C, Li J-Y, He Y-Y, Huang Y, Qin R-J, Zhou Q-L, Sun F, Hu D-S, Yang J. Quercetin serves as the major component of Xiang-lian Pill to ameliorate ulcerative colitis via tipping the balance of STAT1/PPARγ and dictating the alternative activation of macrophage. J Ethnopharmacol. 2023;313:116557.
Li ZY, Cai ML, Qin Y, Chen Z. Age/autoimmunity-associated B cells in inflammatory arthritis: an emerging therapeutic target. Front Immunol. 2023;14:1103307.
Ding Q, Hu W, Wang R, Yang Q, Zhu M, Li M, et al. Signaling pathways in rheumatoid arthritis: implications for targeted therapy. Signal Transduct Target Ther. 2023;8(1):68.
Karami Fath M, Azami J, Jaafari N, Akbari Oryani M, Jafari N, Karim Poor A, et al. Exosome application in treatment and diagnosis of B-cell disorders: leukemias, multiple sclerosis, and arthritis rheumatoid. Cell Mol Biol Lett. 2022;27(1):74.
Zhulai G, Oleinik E, Shibaev M, Ignatev K. Adenosine-metabolizing enzymes, adenosine kinase and adenosine deaminase, in cancer. Biomolecules. 2022;12(3):418.
Yegutkin GG, Boison D. ATP and adenosine metabolism in cancer: exploitation for therapeutic gain. Pharmacol Rev. 2022;74(3):799–824.
Kutryb-Zajac B, Kawecka A, Caratis F, Urbanowicz K, Braczko A, Furihata T, et al. The impaired distribution of adenosine deaminase isoenzymes in multiple sclerosis plasma and cerebrospinal fluid. Front Mol Neurosci. 2022;15:998023.
Geiger JD, Nagy JI. Distribution of adenosine deaminase activity in rat brain and spinal cord. J Neurosci. 1986;6(9):2707–14.
Qian X, Jiang Y, Luo Y, Jiang Y. The anti-hyperuricemia and anti-inflammatory effects of Atractylodes macrocephala in hyperuricemia and gouty arthritis rat models. Comb Chem High Throughput Screening. 2023;26(5):950–64.
Yin X, Vesvoranan O, Andreopoulos F, Dauer EA, Gu W, Huang CYC. Analysis of extracellular ATP distribution in the intervertebral disc. Ann Biomed Eng. 2023;1–14.
Gessner P, Lum J, Frenguelli BG. The mammalian purine salvage pathway as an exploitable route for cerebral bioenergetic support after brain injury. Neuropharmacology. 2023;224:109370.
Dutta N, Deb I, Sarzynska J, Lahiri A. Inosine and its methyl derivatives: occurrence, biogenesis, and function in RNA. Prog Biophys Mol Biol. 2022;169:21–52.
Marucci G, Buccioni M, Varlaro V, Volpini R, Amenta F. The possible role of the nucleoside adenosine in countering skin aging: a review. BioFactors. 2022;48(5):1027–35.
Fernandez CA. Pharmacological strategies for mitigating anti-TNF biologic immunogenicity in rheumatoid arthritis patients. Curr Opin Pharmacol. 2023;68: 102320.
Abed AH, Altaee MF. Immunological assessment of human adenosine deaminase activity in Iraqi female with thyroid autoimmune disease. Egypt J Hosp Med. 2023;90(2):2303–7.
Avram-Shperling A, Kopel E, Twersky I, Gabay O, Ben-David A, Karako-Lampert S, et al. Identification of exceptionally potent adenosine deaminases RNA editors from high body temperature organisms. PLoS Genet. 2023;19(3):e1010661.
Castillo C, Hernandez J, Sotillo J, Muiño R, Benedito JL, Montes A, et al. Is adenosine deaminase (ADA) activity in saliva and serum a more accurate disease detection tool than traditional redox balance parameters in early-lactating dairy cows? Vet Res Commun. 2023;47:1255.
Ledderose C, Valsami EA, Junger WG. Optimized HPLC method to elucidate the complex purinergic signaling dynamics that regulate ATP, ADP, AMP, and adenosine levels in human blood. Purinergic Signalling. 2022;18(2):223–39.
Yang L, Zhou Y, Zhang L, Wang Y, Zhang Y, Xiao Z. Aryl hydrocarbon receptors improve migraine-like pain behaviors in rats through the regulation of regulatory T cell/T-helper 17 cell-related homeostasis. Headache J Head Face Pain. 2023;63(8):1045–60.
El-Said KS, Atta A, Mobasher MA, Germoush MO, Mohamed TM, Salem MM. Quercetin mitigates rheumatoid arthritis by inhibiting adenosine deaminase in rats. Mol Med. 2022;28(1):24.
Tang M, Zeng Y, Peng W, Xie X, Yang Y, Ji B, Li F. Pharmacological aspects of natural quercetin in rheumatoid arthritis. Drug Design Dev Ther. 2022;16:2043–53.
Thammavongsa V, Schneewind O, Missiakas DM. Enzymatic properties of Staphylococcus aureus adenosine synthase (AdsA). BMC Biochem. 2011;12:1–11.
Stellrecht CM, Vangapandu HV, Le XF, Mao W, Shentu S. ATP directed agent, 8-chloro-adenosine, induces AMP activated protein kinase activity, leading to autophagic cell death in breast cancer cells. J Hematol Oncol. 2014;7(1):1–13.
Khan MT, Khan MIUR, Ahmad E, Yousaf MR, Oneeb M. Synergistic effect of extracellular adenosine triphosphate and quercetin on post-thaw quality and fertilization potential of Lohi ram sperm. Cryobiology. 2023;113: 104593.
Akinyemi AJ, Thome GR, Morsch VM, Stefanello N, da Costa P, Cardoso A, et al. Effect of dietary supplementation of ginger and turmeric rhizomes on ectonucleotidases, adenosine deaminase and acetylcholinesterase activities in synaptosomes from the cerebral cortex of hypertensive rats. J Appl Biomed. 2016;14(1):59–70.
Yuan K, Zhu Q, Lu Q, Jiang H, Zhu M, Li X, et al. Quercetin alleviates rheumatoid arthritis by inhibiting neutrophil inflammatory activities. J Nutr Biochem. 2020;84:108454.
Faria A, Pestana D, Teixeira D, Azevedo J, Freitas V, Mateus N, Calhau C. Flavonoid transport across RBE4 cells: a blood-brain barrier model. Cell Mol Biol Lett. 2010;15(2):234–41.
Saccol RDSP, da Silveira KL, Adefegha SA, Manzoni AG, da Silveira LL, Coelho APV, Leal DBR. Effect of quercetin on E-NTPDase/E-ADA activities and cytokine secretion of complete Freund adjuvant–induced arthritic rats. Cell Biochem Funct. 2019;37(7):474–85.
Kour G, Choudhary R, Anjum S, Bhagat A, Bajaj BK, Ahmed Z. Phytochemicals targeting JAK/STAT pathway in the treatment of rheumatoid arthritis: is there a future? Biochem Pharmacol. 2022;197: 114929.
Joshua PE, Yahaya J, Ekpo DE, Ogidigo JO, Odiba AS, Asomadu RO, Oka SA, Adeniyi OS. Modulation of immunological responses by aqueous extract of Datura stramonium L. seeds on cyclophosphamide-induced immunosuppression in Wistar rats. BMC Immunol. 2022;23:50.
Acknowledgements
The authors gratefully acknowledge Biochemistry Division, Chemistry Department, Faculty of Science, Tanta University.
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB). The authors declare that no funds, grants, or other support were received during the preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection, analysis, and first draft of the review manuscript were performed by A.A., M.M.S., K.S.E.S., and T.M.M. All authors commented on the previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors have no relevant financial or non-financial interests to disclose.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Atta, A., Salem, M.M., El-Said, K.S. et al. Mechanistic role of quercetin as inhibitor for adenosine deaminase enzyme in rheumatoid arthritis: systematic review. Cell Mol Biol Lett 29, 14 (2024). https://doi.org/10.1186/s11658-024-00531-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s11658-024-00531-7