
Abstract
Background
While previous genome-wide association studies (GWAS) have identified multiple risk variants for migraine, there is a lack of evidence about how these variants contribute to the development of migraine. We employed an integrative pipeline to efficiently transform genetic associations to identify causal genes for migraine.
Methods
We conducted a proteome-wide association study (PWAS) by combining data from the migraine GWAS data with proteomic data from the human brain and plasma to identify proteins that may play a role in the risk of developing migraine. We also combined data from GWAS of migraine with a novel joint-tissue imputation (JTI) prediction model of 17 migraine-related human tissues to conduct transcriptome-wide association studies (TWAS) together with the fine mapping method FOCUS to identify disease-associated genes.
Results
We identified 13 genes in the human brain and plasma proteome that modulate migraine risk by regulating protein abundance. In addition, 62 associated genes not reported in previous migraine TWAS studies were identified by our analysis of migraine using TWAS and fine mapping. Five genes including ICA1L, TREX1, STAT6, UFL1, and B3GNT8 showed significant associations with migraine at both the proteome and transcriptome, these genes are mainly expressed in ependymal cells, neurons, and glial cells, and are potential target genes for prevention of neuronal signaling and inflammatory responses in the pathogenesis of migraine.
Conclusions
Our proteomic and transcriptome findings have identified disease-associated genes that may give new insights into the pathogenesis and potential therapeutic targets for migraine.
Similar content being viewed by others


Background
Migraine is one of the most disabling diseases globally [1], characterized by recurrent, severe headaches often accompanied by a range of associated symptoms such as sensitivity to light, sound, and smell, nausea, and vomiting [2, 3]. It is a genetically complex neurological disorder, significantly influenced by genetic factors with a heritability estimated at up to 57% [4].
Several genome-wide association studies (GWAS) have been conducted to identify potential genetic risk factors for migraine. Gormley et al. applied meta-analysis to migraine GWAS to identify genomic loci [5]. Subsequent enrichment analysis revealed an association with vascular and smooth muscle tissue, supporting the vascular theory of migraine [5]. In a study involving 873,341 participants, including 102,084 cases and 771,257 controls, 123 migraine-associated loci were identified. It was found that these genes were predominantly enriched in the central nervous system and the vascular system. Transcriptome-wide association study (TWAS) is a method used to investigate the correlation between the transcriptome and each gene locus [6]. Similarly, proteome-wide association studies (PWAS) combine GWAS data with proteomic data to identify candidate genes associated with a given trait [7].
In this study, we used migraine GWAS data in conjunction with the human brain and plasma proteome for PWAS [7]. We also employed the joint-tissue imputation (JTI) prediction model across 17 tissues in migraine GWAS for TWAS [6, 8], followed by fine mapping (FOCUS) [9, 10], to identify risk genes associated with the proteome and transcriptome of migraine. Our findings provide insight into the potential biological mechanisms by which these genes contribute to the development of migraine.
Materials and methods
Migraine GWAS data
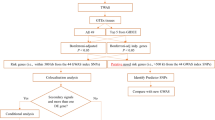
In this study, we utilized the genome-wide summary statistics from the International Headache Genetics Consortium (IHGC) to identify risk loci for migraine. The IHGC dataset consists of 48,975 cases of migraine and 540,381 controls, all of European ethnicity. This large sample size allows for robust analysis and increases the statistical power to detect significant associations. The GWAS data underwent rigorous quality control (QC) measures, including checks for genotyping errors, minor allele frequency, and Hardy–Weinberg equilibrium. The final dataset used for analysis included a specific number of single nucleotide polymorphisms (SNPs) that passed these QC measures. The exact number of SNPs used will be provided upon completion of the QC process [11]. Figure 1 summarizes the various analytical steps performed on the GWAS dataset.

The workflow of the study. PWAS, proteome-wide association study; TWAS, transcriptome-wide association study; FOCUS, Fine-mapping of causal gene sets
Proteomic data
In this study, we profiled human brain proteomes from the dorsolateral prefrontal cortex (dPFC) of post-mortem brain samples donated by participants of European descent. These samples were sourced from the Religious Orders Study and Rush Memory and Aging Project (ROS/MAP), and the Banner Sun Health Research Institute (Banner). The ROS/MAP dataset includes proteomic and genetic data from 376 subjects, with 8,356 proteins passing quality control for protein quantitative trait locus (pQTL) analysis. The Banner dataset includes data from 152 subjects, with 8,168 proteins passing quality control for pQTL analysis [12, 13]. We also utilized a plasma protein dataset consisting of 4,657 proteins from 7,213 European-Americans [14]. All of these datasets have undergone rigorous quality control to ensure accuracy and reliability of the data [15]. The ROS/MAP and Banner datasets include samples from both old and young patients, as well as controls. However, it's important to note that these datasets do not specifically indicate whether the samples were from migraine cases or what percentage of the subjects had migraine. This is a limitation of these datasets, and it could potentially impact our understanding of the relationship between genetic variants and migraine relevance at the protein and RNA level.
Proteome-wide association study
To perform the proteome-wide association study (PWAS), we adopted the functional summary-based imputation (FUSION) method to combine the genetic effect of migraine (GWAS Z-score) with protein weights. FUSION is a computational method designed to integrate functional genomic data with GWAS summary statistics, thereby enhancing the imputation of the GWAS summary statistics [16]. Initially, we employed a linkage disequilibrium (LD) reference panel downloaded from FUSION website. The purpose of this was to mitigate the influence of LD on the estimated test statistics. Following this, we estimated the SNP-based heritability for each gene, utilizing both proteomic and genetic data. We used FUSION to compute the effect of SNPs with significant heritability (p value < 0.01) on protein abundance using multiple predictive models (top1, blup, lasso, enet, and bslmm). The model that yielded the most predictive results was subsequently used for the protein weights. We used FUSION to combine the genetic effect of migraine (migraine GWAS Z-score) with the protein weights by calculating the linear sum of Z-score × weight for the independent SNPs at the locus to perform the PWAS of migraine. The results were adjusted using the Bonferroni multiple testing correction (pBonferroni < 0.05/total number of genes included in the analysis in each data). This approach allowed us to identify proteins that may be involved in the risk of developing migraine and to gain a deeper understanding of the underlying mechanisms of the disorder [7, 15].
Joint-tissue imputation (JTI) models
Joint-tissue imputation (JTI) models are pre-training models obtained on the basis of multi-tissue transcriptome data (GTEx v8), considering shared genetic effects of regulation between different tissues and unique genetic regulation in the target tissue[8]. Here, we obtained prediction models for 17 tissues, including 13 brain tissues (amygdala, anterior cingulate cortex BA24, caudate basal ganglia, cerebellar hemispheres, cerebellum, cerebral cortex, anterior cerebral cortex BA9, hippocampus, hypothalamus, volar nucleus basal ganglia, Choroidal nucleus basalis ganglia, cerebral spinal cord cervical c-1, brain substantia nigra), whole blood, and 3 vascular tissues (aorta arteries, tibial arteries, and coronary arteries). These tissues were chosen due to their relevance to the pathophysiology and LDSC-SEG results of migraine [11, 17, 18]. The JTI method allowed us to identify genetic variants associated with migraine in multiple tissues, providing insight into the complex genetic basis of this neurological disorder.
Transcriptome-wide association study
S-PrediXcan is an approach used to predict gene expression levels based on genetic data, specifically single nucleotide polymorphisms (SNPs), and a reference panel of gene expression data [19, 20]. It estimates gene expression weights by training a linear prediction model using a reference sample that includes both gene expression and SNP genotype data. In our application of S-PrediXcan, we used migraine GWAS summary statistics as the study set. We utilized expression weights for 17 tissues with S-PrediXcan expression weights from the JTI model, and LD information from the 1000 Genomes Project Phase 3. To address the issue of multiple testing, we employed Bonferroni multiple testing correction to adjust the significance threshold (p value). Genes with p value lower than the Bonferroni-corrected threshold were considered potentially significant in relation to migraine.
TWAS fine mapping
To identify relationships between different characteristics within specific genetic regions, we used a method known as TWAS fine mapping, specifically employing the FOCUS method [9, 10]. This method helps us estimate the likelihood that a particular genetic feature is involved in causing the trait of interest. It does this by combining data from GWAS, which look at the entire genome, with expression quantitative trait loci (eQTL) analysis, which examines how genetic variations influence gene expression [10]. A key metric in this process is the posterior inclusion probability (PIP), which is the estimated probability that a particular genetic feature is involved in the trait of interest. In statistical terms, PIP is the marginal posterior probability that a variable (in this case, a genetic feature) should be included in the model. If the PIP is greater than 0.9, this suggests that we can be 90% confident that the genetic feature plays a role in the development and manifestation of the trait. In simpler terms, a high PIP indicates a strong likelihood that the gene is involved in the trait studying. The FOCUS method has been shown to improve the precision of identifying these causal genes and is more sensitive compared to other methods. This makes it a powerful tool for identifying genes associated with diseases.
TWAS-based gene set enrichment analysis
Following the identification of risk genes through TWAS analysis, we categorized them based on their Z-score. Genes with Z-score greater than 0 were classified as risk genes, suggesting their potential role in increasing the likelihood of developing migraines. Conversely, genes with Z-score less than 0 were considered protective factors against migraines. This categorization was conducted for genes obtained from TWAS analyses of the central nervous system (CNS), whole blood, and vascular tissues. To further investigate the roles of these risk and protective genes, we employed several analytical tools. We used the Enrichr online tool to conduct a Gene Ontology (GO) analysis, a Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis, and a Reactome database analysis [21]. These analyses aimed to explore the specific pathways and processes associated with the identified genes. Finally, to visualize the network of these pathways, we utilized the Metascape online tool [22]. By using these tools, we aim to provide a more comprehensive understanding of the genetic underpinnings of migraines.
Cell type specificity analysis
CoExp Web is an online tool that allows for the annotation of genes using co-expression networks based on brain transcriptomic data or transcriptomic data from other tissues [23]. We utilized this tool in our study to identify specific cell types that may be involved in the pathogenesis of migraine by using transcriptomic data from the brain and other tissues [24]. This approach allows us to gain a deeper understanding of the cell types that play a key role in the development and manifestation of the disorder.
Results
PWAS of migraine
In our study, we identified six genes (CISD2, ICA1L, STAT6, SUGP1, TREX1, and UFL1) through PWAS approach. This approach involved integrating proteomics data from the ROS/MAP with migraine GWAS data (Fig. 2A and Additional file 1: Table S3). The PWAS approach works by combining genetic data with protein abundance data to identify genes that may influence the risk of migraine by regulating protein abundance in the brain. The significance of these genes was determined using a Bonferroni multiple testing correction, with a stringent p value threshold set at 4.363E-5. We also identified two additional genes, HNRNPK and PACSIN3, in the PWAS by integrating proteomics data from the Banner Sun Health Research Institute with migraine GWAS data (Fig. 2B and Additional file 1: Table S4). Furthermore, we integrated the plasma proteomic dataset with migraine GWAS data for another PWAS, identifying five more genes (MRVI1, PAPPA, B3GNT8, XCL2, and EPHA10) as potential risk genes (Fig. 2C and Additional file 1: Table S5). These genes may influence the risk of migraine by regulating plasma protein abundance. In total, we identified eight candidate risk genes for migraine using brain pQTL, and five candidate risk genes using plasma pQTL. pQTLs are genetic locations that are associated with variations in protein levels. And, we did not observe a clear overlap or trend between the significant proteins identified by brain-based pQTL and those identified by plasma-based pQTL. CISD2, ICA1L, STAT6, and TREX1 all demonstrated significance at the Bonferroni multiple testing corrected p value threshold between two brain proteomics data (Table 1).

Manhattan plots for the migraine PWASs in the human brain and plasma proteomes. Manhattan plot for the PWAS integrating the migraine GWAS with the ROSMAP proteomes (n = 376) (A), Banner proteomes (n = 152) (B), and plasma proteomes (n = 152) (C). Each dot on the x-axis represents a gene, and the association strength on the y-axis represents the -log10(p) of PWAS. Proteome-wide significance level in the ROSMAP dataset was set at p < 4.363 × 10–5(adjusted by Bonferroni multiple testing correction method). Proteome-wide significance level was set at p < 4.41 × 10–5 (adjusted by the method of multiple testing correction is the Bonferroni adjustment.) for the Banner dataset. Proteome-wide significance level in the ROSMAP dataset was set at p < 3.71 × 10–5(adjusted by Bonferroni multiple testing correction method). Proteome-wide significant genes (ICA1L, TREX1, CISD2, and STAT6) in both brain proteomes are shown in red. Chr, chromosome
TWAS analysis
In the TWAS analysis using the JTI reference transcriptome interpolation model, we identified 95 genes associated with migraine (Additional file 1: Tables S6). Among these, 47 were found in CNS tissues, 75 were found in whole blood and vascular tissues, with 27 overlapping (Additional file 1: Tables S7-8, Additional file 2: Figures S1 and Table S1). Of these, 33 have been reported in previous studies, while the remaining 62 genes are newly identified risk genes (Additional file 1: Tables S9-10). Five genes, including ICA1L, TREX1, STAT6, UFL1, and B3GNT8, showed significant correlation with migraine in both the proteome and transcriptome (Table 1). In this study, FOCUS identified 33 genes with a strong causal association with migraine, of which 29 overlapped with the results identified by TWAS (Additional file 1: Table S11, Additional file 2: Figure S2). Of these 29 genes identified by both TWAS and FOCUS analyses, 10 were novel genes not previously reported in migraine-related GWAS studies (Additional file 1: Table S12; Table 2).
Gene set enrichment analysis based on TWAS results
Among the risk genes identified in CNS tissues, only the Fanconi anemia pathway was found to be significant. In the enrichment analysis of pathways associated with protective genes, we observed a significant enrichment in pathways pertinent to lipid and cholesterol transport and regulation, nuclease activity, STING-mediated immune responses, and cell apoptosis were significantly enriched (Fig. 3). However, no significant findings were observed in the pathway enrichment results of both risk and protective genes in whole blood and vascular tissues. (Additional file 1: S13-16; Additional file 2: Fig S3).

GO-KEGG-Reactome pathway enrichment analyses. A Pathway enrichment analysis results of genes identified by TWAS analysis in CNS tissues, exhibiting negative Z-score
Specific cell type annotation
We used the 95 significant genes from the TWAS results as input for the Co-Exp Web analysis. This analysis assigned weight values to the input gene set and enriched them into corresponding modules. We focused on three identified genes, ICA1L, STAT6, and UFL1, which were shared by both PWAS and TWAS analyses, and examined their specific cell type enrichment in different brain regions. The TWAS results for ICA1L in whole blood and vascular tissues were significant, but the specific cell types in which it was expressed in vascular tissue were unclear. In the CNS, ICA1L was enriched in the Ependymal-External module (p-value = 1.128e-07) and the Neuron Interneuron-External module (p-value = 0.002184) in the shell nucleus, with a module membership (MM) value of 0.8662 for the latter module. ICA1L was also enriched in the amygdala module of the brain, with an MM value of 0.8282. In this module, the meaningful cell types included cortical neurons (p-value = 1.238e-44) and cerebral neurons (p-value = 2.2e-09). In the Co-Exp analysis, STAT6 had an MM value of 0.8343 in the spinal cord module and was specifically expressed in microglia (p-value 1.661e-69). It also had higher specificity in microglia in the brain hypothalamus module (p-value 8.64e-120). The gene UFL1 was more clearly clustered in the hippocampal and cerebrospinal modules of the brain, with MM values of 0.9178 and 0.9092, respectively. In the hippocampus, UFL1 was specifically expressed in oligodendrocytes (p-value = 0.02051), while in the cerebrospinal cord, it was mainly expressed in cortical neurons (p-value = 3.457e-07). The specific cell types for the remaining genes can be found in the supplementary material (Additional file 1: Table S17 and Additional file 2: Table S2).
Discussion
Migraine is a complex neurological disorder, with both CNS and vascular mechanisms playing significant roles in its pathophysiology [25]. Despite the widespread prevalence of migraine, the contemporary diagnostic and therapeutic approaches warrant further refinement and advancement. In this study, we conducted an integrative analysis of PWAS, TWAS, and FOCUS using data derived from the brain, vascular tissues, and plasma.
We identified eight candidate risk genes for migraine in the ROS/MAP and Banner datasets, and five candidate risk genes for migraine in the plasma pQTL through PWAS. However, we did not observe a clear overlap between the significant proteins identified by brain-based pQTL and those identified by plasma-based pQTL. This lack of overlap could be attributed to several factors. Firstly, the distinct biological environments of the brain and plasma could contribute to this observation. Proteins that play a significant role in the cellular environment of the brain may not have the same prominence in plasma, and vice versa. This discrepancy could be due to differences in protein expression, secretion, degradation, or function between the two tissues. Secondly, the limited power of pQTL, due to the relatively smaller sample size, could have also contributed to this lack of overlap. This limitation underscores the need for further studies with larger sample sizes and more comprehensive proteomic data to provide clearer insights into these observations. Despite the lack of a clear trend, we believe that our findings still provide valuable insights into the potential risk genes for migraine. Each set of proteins identified could be contributing to different aspects of the disease mechanism, reflecting the complex and multifactorial nature of migraine. Additionally, after Bonferroni multiple testing correction, we found 95 risk genes significantly associated with migraine. According to FOCUS analysis, 23 of these genes have a strong causal association with migraine within a 90% confidence interval. Through our analysis of two different brain proteomes using PWAS and TWAS of brain and vascular transcriptomes, we identified three potential causal genes for migraine (STAT6, ICA1L, and TREX1). However, in our TWAS analysis, regarding genes (CACNA1A, ATP1A2, and SCN1A) associated with monogenic forms of complex migraine, such as Familial Hemiplegic Migraine (FHM), we did not find overlap gene with our identified risk genes [18]. We identified five genes, including ICA1L, TREX1, STAT6, UFL1, and B3GNT8, which were revealed by both PWAS and TWAS. This shows that the results of this study are consistent at the level of translation and transcription. While, four of these (ICA1L, TREX1, STAT6, and UFL1) have been previously reported in association with migraine [26,27,28,29]. The discovery of new gene (B3GNT8) demonstrates the feasibility of the PWAS study methodology and also benefits from the more novel transcriptomic data we used.
Interestingly, ICA1L, STAT6, and UFL1 were further supported by FOCUS (with pip = 0.954,1, and 1, respectively). We also observed that the expression of STAT6 in whole blood is significantly associated with an increased risk of migraine, while its expression in CNS and vascular tissues is significantly associated with a decreased risk of migraine. This suggests that the effect of STAT6 on the risk of developing migraine may be tissue-specific. These results suggest that the identified genes may play a role in the regulation of the pathogenesis of migraine and may be potential targets for further research. Our findings underscore the complexity of the genetic basis of migraine and highlight the potential of integrative bioinformatics methods in revealing this complexity.
ICA1L, a gene implicated in neuronal signaling, exhibits enriched expression in ependymal cells of the putamen and neurons of the cerebral cortex. This enrichment suggests a potential role for ICA1L in the transmission of information within the trigeminal vasculature. Previous research has established a correlation between elevated ICA1L expression and a decreased risk of Alzheimer's disease, stroke, and small vessel strokes [30,31,32]. Furthermore, ICA1L has been identified as a shared risk gene between migraine and coronary artery disease (CAD) [26]. The pathophysiological mechanisms of migraine, Alzheimer's disease (AD), and small vessel disease (SVD) may all contribute to the development of white matter damage and cognitive deficits. Vascular dysfunction represents a shared mechanism among these diseases, particularly evident in the context of migraine. Moreover, neuroinflammation, a common factor in the development of both AD and migraine, underscores the potential overlapping pathophysiology among these conditions. Therefore, it can be postulated that ICA1L plays a convergent role in the initiation and progression of migraine, AD, and SVD. This shared genetic influence underscores the interconnected nature of these seemingly disparate conditions and highlights the need for further exploration into the multifaceted roles of genes like ICA1L. This implies that treatments for these diseases might also aid in migraine management. UFL1, a protein-encoding gene, regulates humoral immune processes and endoplasmic reticulum stress, potentially altering vascular morphology and inflammation [25, 33]. Earlier research suggests that UFL1 plays a role in histone H4 acylation and ATM activation [34]. Our cell-specific analysis shows UFL1 enrichment in oligodendrocytes and neurons, involved in neuroexcitatory signal regulation and protein modifications in migraine [35, 36]. This suggests UFL1 as a potential target gene for antihistamines targeting the H4 receptor for migraine prevention and treatment. Evidence suggests that both immune responses and neuroinflammation, observable in peripheral blood, contribute to the pathogenesis of migraine [36, 37]. The STAT6 gene, in particular, may play a pivotal role in this context. It is hypothesized that STAT6 may contribute to the activation of the trigeminal vascular system, a process that can trigger an inflammatory response and sustain the state of migraine. This inflammation, potentially manifesting in plasma, could lower the thresholds of injury receptors, leading to heightened sensitization in both central and peripheral regions [25]. Further supporting this hypothesis, STAT6 shows enriched expression in microglia and macrophages, which are immune cells present in the central nervous system (CNS) and arterial tissues [38, 39]. The activation of these cells can lead to an inflammatory response, thereby potentially exacerbating migraine [40]. TREX1, a gene encoding a DNA exonuclease. TREX1-deficient brain cells exhibit neuroinflammatory and neurotoxic effects, a critical factor in the pathophysiology of migraine. This signaling pathway contributes to inflammatory responses and the persistence of headaches, key characteristics of migraine [41, 42]. Therefore, TREX1 may play a significant role in the initiation and maintenance of migraine episodes, potentially through the modulation of neuroinflammatory processes. B3GNT8 is highly enriched in the esophagus and vagina and is associated with gastrointestinal symptoms such as nausea and vomiting in migraine patients [43]. Given the higher prevalence of migraines in women and the influence of estrogen levels on migraine incidence [44], B3GNT8 emerges as a key candidate gene for the regulation of migraine-associated gastrointestinal symptoms.. B3GNT8 is a key candidate gene for regulating migraine-associated gastrointestinal symptoms and hormonal modulation for migraine prevention.
Our research reveals that identified genes play a complex role in migraine development, impacting lipid homeostasis, immune response, cell clearance, and nucleotide metabolism. Enriched pathways related to lipid transport and regulation, particularly cholesterol transport and efflux, suggest a key role for lipid balance in migraine development. This aligns with recent research linking lipid metabolism to migraines, indicating potential for lipid-lowering treatments [45]. We observed enrichment in the STING-mediated immune response pathway, suggesting the genes could regulate immune responses, potentially controlling inflammation-related migraine symptoms. The apoptotic cell clearance pathway was also enriched, indicating a role for the genes in preventing secondary necrosis and inflammation, potentially alleviating migraine symptoms. Finally, enrichment in pathways related to 3'-5' exonuclease activity and nucleobase-containing compound catabolic process suggests involvement in DNA repair and nucleotide metabolism, crucial for genomic stability and cellular balance, disruptions of which could contribute to migraines.
The datasets used in this study encompass a diverse range of proteins, which are crucial for understanding the potential biological mechanisms underlying migraine. These proteins were linked to genetic variation through pQTL analysis, which investigates the influence of genetic variants on protein abundance, measured in the dorsolateral prefrontal cortex (dPFC) region of the brain. However, our study has several limitations. First, the sample size of the migraine GWAS dataset and pQTL data was limited, which may have affected the robustness of our findings. As more migraine GWAS and pQTL data become available in the future, we anticipate that the power and significance of PWAS in understanding diseases like migraine will become more evident. Second, our study was limited by its focus on European populations, which may have influenced the detection of some gene transcriptomic and proteomic expression effects. This limits the generalizability of our findings, and further studies with more diverse populations are needed to validate our results. Third, we only analyzed 17 tissues deemed relevant to migraine, potentially overlooking associations with migraine in other tissues. This includes the possibility that some transcripts are expressed in the brain but not in the blood. Finally, the clinical relevance of our findings requires further validation. The lack of clinical data to correlate with our molecular findings is a significant limitation of this study. Future research should aim to explain how these genes modulate and influence the pathophysiological processes of migraine through scientific experiments. Despite these limitations, by combining these datasets, we were able to identify multiple proteins potentially involved in the development of migraine and gain insights into their potential mechanisms of action.
Conclusions
In conclusion, by integrating proteomic and transcriptomic data from PWAS and TWAS, we have identified causal genes for migraine, including some that have not been reported in previous TWAS analyses, providing new insights compared to previous TWAS analyses. Our findings shed light on the transcriptomic changes and potential pathogenic mechanisms of these genes in the context of migraine. This makes them promising candidates for future studies aimed at understanding the pathogenesis of migraine and developing effective treatments for this debilitating condition.
Availability of data and materials
The Migraine GWAS dataset provided by Hautakangas et al. can be obtained by contacting the International Headache Genetics Consortium (IHGC) [11]. The interpolation model for the JTI reference transcriptome used in TWAS can be downloaded at https://zenodo.org/record/3842289[8]. The ROSMAP and Banner datasets, including weights and pQTL data, supporting the results of this study can be accessed at https://www.synapse.org/#!Synapse:syn23627957 and the plasma dataset can be downloaded from http://nilanjanchatterjeelab.org/pwas/[12,13,14,15]. These resources can be used to further investigate the mechanisms behind GWAS-identified risk variants for migraine.
This study used R version 4.2.1 for data collation and analysis. The S-PrediXcan package (https://github.com/hakyimlab/MetaXcan) was used to analyze TWAS [19, 20], while TWAS fine mapping was conducted using the FOCUS package (https://github.com/bogdanlab/focus/) [9, 10]. Gene enrichment analysis was performed with the Enrichr tool (https://maayanlab.cloud/Enrichr/) [21]. The pathway network was built with https://metascape.org/ [22]. The FUSION package was utilized for proteome-wide association studies (PWAS), and cell type-specific analyses were performed using the CoExpNets online tool (https://rytenlab.com/coexp/Run) [23].
Abbreviations
- SNP:
-
Single nucleotide polymorphism
- GWAS:
-
Genome-wide association study
- PWAS:
-
Proteome-wide association study
- TWAS:
-
Transcriptome-wide association study
- JTI:
-
Joint-tissue imputation
- PIP:
-
Posterior inclusion probability
- CNS:
-
Central nervous system
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- eQTL:
-
Expression quantitative trait loci
- pQTL:
-
Protein quantitative trait loci
- MM:
-
Module membership
References
Robbins MS (2021) Diagnosis and management of headache: a review. Jama 325:1874–1885. https://doi.org/10.1001/jama.2021.1640
Charles A (2018) The pathophysiology of migraine: implications for clinical management. Lancet Neurol 17:174–182. https://doi.org/10.1016/s1474-4422(17)30435-0
Biondi DM (2006) Is migraine a neuropathic pain syndrome? Curr Pain Headache Rep 10:167–178. https://doi.org/10.1007/s11916-006-0042-y
Choquet H, Yin J, Jacobson AS, Horton BH, Hoffmann TJ, Jorgenson E et al (2021) New and sex-specific migraine susceptibility loci identified from a multiethnic genome-wide meta-analysis. Commun Biol 4:864. https://doi.org/10.1038/s42003-021-02356-y
Gormley P, Anttila V, Winsvold BS, Palta P, Esko T, Pers TH et al (2016) Meta-analysis of 375,000 individuals identifies 38 susceptibility loci for migraine. Nat Genet 48:856–866. https://doi.org/10.1038/ng.3598
Wainberg M, Sinnott-Armstrong N, Mancuso N, Barbeira AN, Knowles DA, Golan D et al (2019) Opportunities and challenges for transcriptome-wide association studies. Nat Genet 51:592–599. https://doi.org/10.1038/s41588-019-0385-z
Brandes N, Linial N, Linial M (2020) PWAS: proteome-wide association study-linking genes and phenotypes by functional variation in proteins. Genome Biol 21:173. https://doi.org/10.1186/s13059-020-02089-x
Zhou D, Jiang Y, Zhong X, Cox NJ, Liu C, Gamazon ER (2020) A unified framework for joint-tissue transcriptome-wide association and Mendelian randomization analysis. Nat Genet 52:1239–1246. https://doi.org/10.1038/s41588-020-0706-2
Wu C, Pan W (2020) A powerful fine-mapping method for transcriptome-wide association studies. Hum Genet 139:199–213. https://doi.org/10.1007/s00439-019-02098-2
Mancuso N, Freund MK, Johnson R, Shi H, Kichaev G, Gusev A et al (2019) Probabilistic fine-mapping of transcriptome-wide association studies. Nat Genet 51:675–682. https://doi.org/10.1038/s41588-019-0367-1
Hautakangas H, Winsvold BS, Ruotsalainen SE, Bjornsdottir G, Harder AVE, Kogelman LJA et al (2022) Genome-wide analysis of 102,084 migraine cases identifies 123 risk loci and subtype-specific risk alleles. Nat Genet 54:152–160. https://doi.org/10.1038/s41588-021-00990-0
Bennett DA, Buchman AS, Boyle PA, Barnes LL, Wilson RS, Schneider JA (2018) Religious orders study and rush memory and aging project. J Alzheimers Dis 64:S161-s189. https://doi.org/10.3233/jad-179939
Beach TG, Adler CH, Sue LI, Serrano G, Shill HA, Walker DG et al (2015) Arizona study of aging and neurodegenerative disorders and brain and body donation program. Neuropathology 35:354–389. https://doi.org/10.1111/neup.12189
Zhang J, Dutta D, Köttgen A, Tin A, Schlosser P, Grams ME et al (2022) Plasma proteome analyses in individuals of European and African ancestry identify cis-pQTLs and models for proteome-wide association studies. Nat Genet 54:593–602. https://doi.org/10.1038/s41588-022-01051-w
Wingo AP, Liu Y, Gerasimov ES, Gockley J, Logsdon BA, Duong DM et al (2021) Integrating human brain proteomes with genome-wide association data implicates new proteins in Alzheimer’s disease pathogenesis. Nat Genet 53:143–146. https://doi.org/10.1038/s41588-020-00773-z
Gusev A, Ko A, Shi H, Bhatia G, Chung W, Penninx BW et al (2016) Integrative approaches for large-scale transcriptome-wide association studies. Nat Genet 48:245–252. https://doi.org/10.1038/ng.3506
Yazar HO, Yazar T, Aygün A, Kaygisiz Ş, Kirbaş D (2020) Evaluation of simple inflammatory blood parameters in patients with migraine. Ir J Med Sci 189:677–683. https://doi.org/10.1007/s11845-019-02136-y
Sutherland HG, Griffiths LR (2017) Genetics of migraine: insights into the molecular basis of migraine disorders. Headache 57:537–569. https://doi.org/10.1111/head.13053
Pasman JA, Verweij KJH, Gerring Z, Stringer S, Sanchez-Roige S, Treur JL et al (2018) GWAS of lifetime cannabis use reveals new risk loci, genetic overlap with psychiatric traits, and a causal influence of schizophrenia. Nat Neurosci 21:1161–1170. https://doi.org/10.1038/s41593-018-0206-1
Barbeira AN, Dickinson SP, Bonazzola R, Zheng J, Wheeler HE, Torres JM et al (2018) Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nat Commun 9:1825. https://doi.org/10.1038/s41467-018-03621-1
Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z et al (2016) Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44:W90-97. https://doi.org/10.1093/nar/gkw377
Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O et al (2019) Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun 10:1523. https://doi.org/10.1038/s41467-019-09234-6
García-Ruiz S, Gil-Martínez AL, Cisterna A, Jurado-Ruiz F, Reynolds RH, Cookson MR et al (2021) CoExp: a web tool for the exploitation of co-expression networks. Front Genet 12:630187. https://doi.org/10.3389/fgene.2021.630187
Botía JA, Vandrovcova J, Forabosco P, Guelfi S, D’Sa K, Hardy J et al (2017) An additional k-means clustering step improves the biological features of WGCNA gene co-expression networks. BMC Syst Biol 11:47. https://doi.org/10.1186/s12918-017-0420-6
Ashina M (2020) Migraine. N Engl J Med 383:1866–1876. https://doi.org/10.1056/NEJMra1915327
Sutherland HG, Albury CL, Griffiths LR (2019) Advances in genetics of migraine. J Headache Pain 20:72. https://doi.org/10.1186/s10194-019-1017-9
Winsvold BS, Nelson CP, Malik R, Gormley P, Anttila V, Vander Heiden J et al (2015) Genetic analysis for a shared biological basis between migraine and coronary artery disease. Neurol Genet 1:e10. https://doi.org/10.1212/nxg.0000000000000010
Meng W, Adams MJ, Hebert HL, Deary IJ, McIntosh AM, Smith BH (2018) A genome-wide association study finds genetic associations with broadly-defined headache in UK Biobank (N=223,773). EBioMedicine 28:180–186. https://doi.org/10.1016/j.ebiom.2018.01.023
Zhao H, Eising E, de Vries B, Vijfhuizen LS, Anttila V, Winsvold BS et al (2016) Gene-based pleiotropy across migraine with aura and migraine without aura patient groups. Cephalalgia 36:648–657. https://doi.org/10.1177/0333102415591497
Zhang C, Qin F, Li X, Du X, Li T (2022) Identification of novel proteins for lacunar stroke by integrating genome-wide association data and human brain proteomes. BMC Med 20:211. https://doi.org/10.1186/s12916-022-02408-y
Cullell N, Gallego-Fábrega C, Cárcel-Márquez J, Muiño E, Llucià-Carol L, Lledós M, et al (2022) ICA1L Is Associated with Small Vessel Disease: A Proteome-Wide Association Study in Small Vessel Stroke and Intracerebral Haemorrhage. Int J Mol Sci; 23. https://doi.org/10.3390/ijms23063161.
Ou YN, Yang YX, Deng YT, Zhang C, Hu H, Wu BS et al (2021) Identification of novel drug targets for Alzheimer’s disease by integrating genetics and proteomes from brain and blood. Mol Psychiatry 26:6065–6073. https://doi.org/10.1038/s41380-021-01251-6
Liang JR, Lingeman E, Luong T, Ahmed S, Muhar M, Nguyen T et al (2020) A genome-wide ER-phagy screen highlights key roles of mitochondrial metabolism and ER-resident UFMylation. Cell 180:1160-1177.e1120. https://doi.org/10.1016/j.cell.2020.02.017
Qin B, Yu J, Nowsheen S, Wang M, Tu X, Liu T et al (2019) UFL1 promotes histone H4 ufmylation and ATM activation. Nat Commun 10:1242. https://doi.org/10.1038/s41467-019-09175-0
Eising E, de Leeuw C, Min JL, Anttila V, Verheijen MH, Terwindt GM et al (2016) Involvement of astrocyte and oligodendrocyte gene sets in migraine. Cephalalgia 36:640–647. https://doi.org/10.1177/0333102415618614
Renthal W (2018) Localization of migraine susceptibility genes in human brain by single-cell RNA sequencing. Cephalalgia 38:1976–1983. https://doi.org/10.1177/0333102418762476
Aczél T, Körtési T, Kun J, Urbán P, Bauer W, Herczeg R et al (2021) Identification of disease- and headache-specific mediators and pathways in migraine using blood transcriptomic and metabolomic analysis. J Headache Pain 22:117. https://doi.org/10.1186/s10194-021-01285-9
Cai W, Dai X, Chen J, Zhao J, Xu M, Zhang L, et al (2019) STAT6/Arg1 promotes microglia/macrophage efferocytosis and inflammation resolution in stroke mice. JCI Insight; 4. https://doi.org:https://doi.org/10.1172/jci.insight.131355.
Xu J, Chen Z, Yu F, Liu H, Ma C, Xie D et al (2020) IL-4/STAT6 signaling facilitates innate hematoma resolution and neurological recovery after hemorrhagic stroke in mice. Proc Natl Acad Sci U S A 117:32679–32690. https://doi.org/10.1073/pnas.2018497117
Rawji KS, Mishra MK, Michaels NJ, Rivest S, Stys PK, Yong VW (2016) Immunosenescence of microglia and macrophages: impact on the ageing central nervous system. Brain 139:653–661. https://doi.org/10.1093/brain/awv395
Silberstein SD (2004) Migraine. Lancet 363:381–391. https://doi.org/10.1016/s0140-6736(04)15440-8
Thomas CA, Tejwani L, Trujillo CA, Negraes PD, Herai RH, Mesci P et al (2017) Modeling of TREX1-dependent autoimmune disease using human stem cells highlights l1 accumulation as a source of neuroinflammation. Cell Stem Cell 21:319-331.e318. https://doi.org/10.1016/j.stem.2017.07.009
Schwedt TJ (2014) Chronic migraine. Bmj 348:g1416. https://doi.org/10.1136/bmj.g1416
Chai NC, Peterlin BL, Calhoun AH (2014) Migraine and estrogen. Curr Opin Neurol 27:315–324. https://doi.org/10.1097/wco.0000000000000091
Castor K, Dawlaty J, Arakaki X, Gross N, Woldeamanuel YW, Harrington MG et al (2021) Plasma lipolysis and changes in plasma and cerebrospinal fluid signaling lipids reveal abnormal lipid metabolism in chronic migraine. Front Mol Neurosci 14:691733. https://doi.org/10.3389/fnmol.2021.691733
Acknowledgements
We would like to thank the International Headache Genetics Consortium for providing us with the migraine GWAS summary data, The Religious Orders Study and Memory and Aging Project (ROSMAP) Study, and The Banner Sun Health Research Institute (Banner) study for providing the brain proteomic data, and The Atherosclerosis Risk in Communities Study (ARIC) for providing the plasma proteomic data.
Consortia
International Headache Genetics Consortium (IHGC)
Funding
This work was supported by the National Natural Science Foundation of China to Dr. Chang-he Shi [grant number 81974211, 82171247], the Scientific Research and Innovation Team of The First Affiliated Hospital of Zhengzhou University to Dr. Chang-he Shi [grant number ZYCXTD2023011].
Author information
Authors and Affiliations
Contributions
CHS and YGW designed and supervised the study, while SJL performed the majority of the analysis and drafted the manuscript. JJS provided mentorship, and all authors contributed to the study, interpreted the findings, and reviewed the manuscript. The final version of the manuscript was approved by all authors before publication.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All data analyzed during this study have been previously published.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
The data information used in this study and the analysis files generated, with the title of each table displayed in Additional file1: List.
Additional file 2:
Figure S1. Venn plots of the migraine significant genes. Figure S2. FOCUS plot for each gene in one region. Figure S3. Pathway network for the migraine significant genes with negative Z-score for 13 central nervous systems tissues (TWAS). Table S1. Overlap genes of migraine risk genes identified by TWAS. Table S2. Cell type annotation of three important genes.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, Sj., Shi, Jj., Mao, Cy. et al. Identifying causal genes for migraine by integrating the proteome and transcriptome. J Headache Pain 24, 111 (2023). https://doi.org/10.1186/s10194-023-01649-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s10194-023-01649-3