
Abstract
HMGB1, a nucleoprotein, is expressed in almost all eukaryotic cells. During cell activation and cell death, HMGB1 can function as an alarm protein (alarmin) or damage-associated molecular pattern (DAMP) and mediate early inflammatory and immune response when it is translocated to the extracellular space. The binding of extracellular HMGB1 to Toll-like receptors (TLRs), such as TLR2 and TLR4 transforms HMGB1 into a pro-inflammatory cytokine, contributing to the occurrence and development of autoimmune diseases. TLRs, which are members of a family of pattern recognition receptors, can bind to endogenous DAMPs and activate the innate immune response. Additionally, TLRs are key signaling molecules mediating the immune response and play a critical role in the host defense against pathogens and the maintenance of immune balance. HMGB1 and TLRs are reported to be upregulated in several autoimmune diseases, such as rheumatoid arthritis, systemic lupus erythematosus, type 1 diabetes mellitus, and autoimmune thyroid disease. The expression levels of HMGB1 and some TLRs are upregulated in tissues of patients with autoimmune diseases and animal models of autoimmune diseases. The suppression of HMGB1 and TLRs inhibits the progression of inflammation in animal models. Thus, HMGB1 and TLRs are indispensable biomarkers and important therapeutic targets for autoimmune diseases. This review provides comprehensive strategies for treating or preventing autoimmune diseases discovered in recent years.
Similar content being viewed by others
Introduction
High-mobility group (HMG) proteins, which are non-histone DNA-binding proteins, are expressed in eukaryotic cells and exhibit various biological functions (Dumitriu et al. 2005). HMGB1, which is the most abundant member of the HMG family (Ge et al. 2021), is abundantly expressed in almost all human cells. Additionally, HMGB1 can actively and passively shuttle between the nucleus and the cytoplasm of all cells (Kang et al. 2014). HMGB1 is mainly localized to the nucleus where it interacts with DNA and regulates chromosome structure, maintains the integrity of nucleosomes, and promotes gene transcription. During biological processes, such as apoptosis, necrosis, and cell scorching, HMGB1 is transferred to the extracellular space (Chen et al. 2022). In the extracellular environment, HMGB1 can function as a damage-associated molecular pattern (DAMP) or alarm protein (alarmin) and mediate early inflammatory and immune response (Kang et al. 2014). Previous studies have reported that HMGB1 functions as an important inflammatory cytokine in various autoimmune diseases. Especially in rheumatoid arthritis (RA) (Schierbeck et al. 2013), systemic lupus erythematosus (SLE) (Wirestam et al. 2015), and autoimmune thyroid disease (AITD) (Li et al. 2017). Toll-like receptors (TLRs) are members of a family of pattern recognition receptors and can express on various cells, such as monocytes, macrophages, dendritic cells, and B cells (Iwasaki and Medzhitov 2004). Furthermore, TLRS can function as pathogen-associated molecular models (PAMPs), bind to endogenous DAMPs and activate the innate immune response (Chuang and Ulevitch 2001). Some pathogenic microorganisms, such as bacteria, viruses and fungi, can activate TLR signaling pathway, trigger innate immune response and promote the release of inflammatory mediators (Ulevitch 2004). Generally, TLRs are key signaling molecules mediating the immune response and play a critical role in the host defense against pathogens and the maintenance of immune balance. Previous studies have demonstrated that TLRs are also involved in the occurrence and development of various autoimmune diseases, such as RA (Drexler and Foxwell 2010), SLE (Wen et al. 2023), type 1 diabetes mellitus (T1DM) (Dandona et al. 2013), and AITD (Peng et al. 2016). HMGB1 can bind to TLRs and other PRRs to activate the TLR signaling pathway and induce cytokine production and immune cell proliferation, promoting the production of pro-inflammatory cytokines and eliciting inflammatory responses. The potential of HMGB1 and TLRs as therapeutic targets has been previously examined.
This review summarized the structural, functional, and immunological characteristics of HMGB1 and TLRs and the key roles of HMGB1 and TLRs in development of autoimmune diseases. Additionally, recent advances in establishing HMGB1 and TLRs as therapeutic targets for autoimmune diseases have been discussed.
HMGB1: origin and structure
In 1973, the research group of Ernest Johns and Graham Goodwin successfully extracted an abundant non-histone nuclear protein from calf thymus chromatin. This protein was named "high-mobility group" protein (Goodwin et al. 1973) owing to its rapid migration in polyacrylamide gel electrophoresis without aggregation (Goodwin and Johns 1973). This was the first time that HMGB1 was discovered. In 2001, Michael Bustin reclassified these proteins into three superfamilies and renamed them HMGA, HMGB and HMGN (Bustin 2001). Among them, the high-mobility group box (HMGB) family is the most abundant protein family of HMGs (Goodwin and Johns 1978). HMGB is highly conserved and comprises four members (HMGB1, HMGB2, HMGB3, and HMGB4). In 2001 and 2003, the homologs of mammalian HMGB1 were identified in bacteria, drosophila, echinoderms, plants, and fish (Bustin 2001; Wu et al. 2003). HMGB1 mRNA is polyadenylated (Bustin et al. 1981). Meanwhile, the homology of HMGB1 protein sequence between mice and rats is 100%, while that between rodents and humans is 99% (Ferrari et al. 1994; Wen et al. 1989).
Human HMGB1, which comprises 215 amino acid residues, contains two DNA-binding domains (HMG A box domain [9–79 amino acids] and HMG B box domain [95–163 amino acids]), a C-terminal acidic tail (186–215 amino acids) (Bianchi et al. 1992) (Fig. 1), and a functionally important N-terminal region. The DNA-binding domain contains nuclear export signals (NESs) (Tang et al. 2014). The homeostasis of HMGB1 in the nucleus is dependent on two nuclear localization signals (NLSs) (NLS1 [28–44 amino acids] and NLS2 [179–185 amino acids]) (Bonaldi et al. 2003). The localization of HMGB1 is disrupted upon alterations in NES and NLS. HMGB1 can bind to various proteins. This binding is critical for the activity and function of HMGB1. For example, the residues 150–183 bind to RAGE to promote cell migration (Huttunen et al. 2002), while the residues 89–108 bind to the TLR4 structural domain to promote inflammation (Venereau et al. 2016). The extracellular B box exhibits pro-inflammatory activity, whereas the A box functions as an antagonist of HMGB1. The A box can bind to the B box and exert anti-inflammatory effects (Li et al. 2003). The structures of two HMG boxes of HMGB1 are similar to those of a typical DNA-binding domain, comprising three alpha helices arranged in an "L" shape and two loops (Hardman et al. 1995; Thomas and Travers 2001). Three lysine residues are encoded at positions 23, 45, and 106 of HMGB1 (Hoppe et al. 2006). Generally, the structure of HMGB1 is highly conserved among species.

The structure and function of HMGB1. HMGB1 contains two DNA binding domains (HMG A box domain [9–79 aa] and HMG B box domain [95–163 aa]), a C-terminal acidic tail (186–215 aa) and a N-terminal region. Residues 150–183 bind to RAGE to promote cell migration, while residues 89–108 in charge of activating cytokine and binding to the TLR4 structural domain to promote inflammation. Two nuclear localization signals: NLS1 (28–44 aa) and NLS2 (179–185 aa). HMGB1 encode three cysteines: Cys23, Cys45 and Cys106
Mechanism of HMGB1 release
In 1999, after treating mouse macrophages with endotoxin lipopolysaccharide (LPS), Wang et al. found that large amounts of HMGB1 were secreted into the extracellular space (Wang et al. 1999). This was the first study to report that HMGB1 is a secretory protein (Xue et al. 2021). Additionally, this study highlighted the role of extracellular HMGB1 in inflammation and infection. In 2003, the research group of Tiziana Bonaldi and Marco Bianchi, for the first time, demonstrated that the acetylation and deacetylation of NLS1 and NLS2 promote the rapid shuttling of HMGB1 from the nucleus to the cytoplasm and consequently promote the secretion of HMGB1 from activated monocytes (Bonaldi et al. 2003). Inflammatory stimuli promote the acetylation of lysine residues in HMGB1, preventing the nuclear entry of HMGB1 (Bonaldi et al. 2003), which is critical for its translocation from the nucleus to the cytoplasm (Gardella et al. 2002; Rendon-Mitchell et al. 2003). Additionally, the phosphorylation of serine residues in NLS1 and NLS2 promotes the translocation of HMGB1 from the nucleus to the cytoplasm (Bonaldi et al. 2003; Wang et al. 2019). HMGB1 reaches the external cellular environment through active secretion by cells of the innate immune system (Wang et al. 1999; Agnello et al. 2002) or passive release after necrotic cell death (Scaffidi et al. 2002). During apoptosis, necrosis, cell scorching cell death, and injury resulting from various stimuli, such as irradiation, hypoxia, and hyperthermia (Pisetsky 2014), soluble HMGB1 can be released into the extracellular space in large quantities by these dead or dying cells and elicit inflammatory responses in vitro and in vivo (Scaffidi et al. 2002). Several studies have demonstrated that endogenous stimuli, exogenous microbial products, and infections with various pathogens can induce active secretion of HMGB1 by immune cells, endothelial cells, epithelial cells, fibroblasts, or other cells (Magna and Pisetsky 2014; Andersson et al. 2018). The active release of HMGB1 from immune cells also involves two modalities. In the first modality, the activation of target cells by exogenous stimuli leads to the secretion of HMGB1 into the extracellular space (Bonaldi et al. 2003). The second modality involves the lack of a secretory signal peptide for HMGB1, which cannot be transported through the classical endoplasmic reticulum and Golgi apparatus secretion pathways (Gardella et al. 2002; Rendon-Mitchell et al. 2003). This HMGB1 is then packaged into intracellular vesicles (such as lysosomes), which fuse with the cytoplasmic membrane and release HMGB1 into the extracellular space (Pisetsky 2014). Extracellular HMGB1 can function as a DAMP to activate the innate immune system by recruiting inflammatory cells and smooth muscle cells and stimulate macrophages and endothelial cells to produce pro-inflammatory cytokines that promote the inflammatory response (Andersson et al. 2000; Fiuza et al. 2003). Additionally, as an immune molecule, extracellular HMGB1 can trigger the inflammatory response of immune cells and endothelial cells. Subsequently, immune cells and endothelial cells activated by HMGB1 can secrete HMGB1, resulting in the formation of a positive feedback loop (Kang et al. 2014). Thus, HMGB1 can maintain a long-term inflammatory state under a variety of stimulation conditions (Kang et al. 2014).
TLRs: structure and function
TLRs, which are transmembrane receptors mainly expressed by innate immune cells, are part of a family of pattern recognition receptors (PRRs) utilized by the innate immune system (Luchner et al. 2021). In humans, TLR1, TLR2, TLR4, TLR5, and TLR6 are expressed on the cell surface, while TLR3, TLR7, TLR8, and TLR9 are expressed on the endosomal membrane (Dvornikova et al. 2020). TLRs, an evolutionarily conserved type I transmembrane protein superfamily, comprise a leucine-rich repeat extracellular domain that can interact with PAMPs or DAMPs and a cytoplasmic Toll/interleukin (IL)-1 receptor (TIR) domain that is involved in downstream signal transduction and eliciting inflammatory responses (Botos et al. 2011; Lim and Staudt 2013). Additionally, TLRs are synthesized in the endoplasmic reticulum, processed in the Golgi apparatus, and transported to the plasma membrane based on the localization of PAMPs they recognize (Ullah et al. 2016; Gay et al. 2014). TLRs can recognize various PAMPs, including DAMPs, gram-negative and gram-positive bacteria, viruses, flagellin, and nucleic acids, according to their localization on the cell surface or within the cells (Marongiu et al. 2019). Upon binding to ligands, TLRs undergo homodimerization or heterodimerization and recruit adaptor proteins containing the TIR structural domain to activate the downstream signaling pathways (Luchner et al. 2021). For example, TLRs can activate interferon (IFN)-β (TRIF) signaling through the recruitment of the adapter molecule myeloid differentiation primary response differentiation gene 88 (MyD88) or MyD88-independent TIR domain-containing adapter molecules (Satoh and Akira 2016). This is followed by the activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling and mitogen-activated protein kinase (Owen et al. 2020), which promotes the expression of pro-inflammatory cytokines, such as tumor necrosis factor-α (TNF-α) or Interleukin-6 (IL-6) (Kawasaki and Kawai 2014). Additionally, TLRs can activate type I IFN responses mediated by IFN regulatory factors (IRFs), such as IRF3 and IRF7, which play a major role in adaptive immunity (Moynagh 2005; Zhao et al. 2015). Thus, TLRs recognize several danger signals to activate innate immune responses against infection and injury.
HMGB1 and TLRs
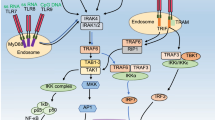
HMGB1 can interact with TLRs (TLR2, TLR4, and TLR9) to activate TLR signaling. Upon stimulation by the ligand, TLRs recruit downstream junction molecules, including TIR domain-containing adapter-inducing IFN-β (TRIF) and MyD88, which subsequently activate the NF-κB and IRF signaling pathways, promote the production of cytokines and chemokines involved in immune responses, and induce inflammation (Beutler 2004). TLR4 signaling is essential for HMGB1-induced cytokine production. Macrophages lacking TLR4 cannot release pro-inflammatory cytokines in response to HMGB1 (Li et al. 2020; Zhang et al. 2021). The intramolecular amino acids C23, C45, and C106 of HMGB1 are required for TLR4 binding and activation (Yang et al. 2012). The TLR4 structural domain can bind to the residues 89–108 of the HMGB1 B box to promote inflammation. The knockdown of TLR4 in vitro or in vivo mitigates HMGB1-induced tissue damage (Laird et al. 2014), inflammation (Zong et al. 2013), and immune response. In addition to TLR4, HMGB1 also requires TLR2 to promote tissue damage and inflammatory responses (Herzog et al. 2014). However, TLR9 mediates immune responses against nucleotides induced by the HMGB1-DNA complex. As HMGB1 interacts with multiple TLRs, it is a key protein for innate immunity. The specific pathways of HMGB1-induced TLR signaling are described below (Fig. 2).

HMGB1 and TLRs signaling pathway. HMGB1 can combined with cell surface TLRs (TLR1, TLR2, TLR4, TLR5, TLR6), while the nucleic acid of HMGB1 can cross the cell membrane and bind to the endosomal TLRs (TLR3, TLR7, TLR8, TLR9). TLRs recruit TIR domain-containing adaptor proteins like MyD88 and TRIF, which activate NF-κB signaling and IRFs. This initiates the expression of pro-inflammatory cytokines such as IL-1β, IL-6, TNF-α, IFN-γ or the type I interferons IFN-α and IFN-β. The figure distinguishes by color the adaptor proteins MyD88 and TRIF that bind to different TLRs. TRIF: TIR domain-containing adaptor protein inducing IFNβ, MyD88: molecule myeloid differentiation primary response differentiation gene 88, NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells, IRF: Interferon Regulatory Factor, IFN: Interferon, TNF-α: tumor necrosis factor-alpha, IL: Interleukin
Autoimmune diseases
Rheumatoid arthritis
Rheumatoid Arthritis (RA) is an autoimmune disease that is closely related to HMGB1. HMGB1 is reported to be an important factor in the development of RA (Li et al. 2011; Oktayoglu et al. 2013). RA is characterized by chronic synovial inflammation, articular cartilage erosion and damage, synovial cell proliferation, and bone destruction (Chen et al. 2013). Several studies examining the correlation between RA and HMGB1 or TLRs have demonstrated that HMGB1 and TLRs play an indispensable role in the progression of RA as pro-inflammatory cytokines (Kokkola et al. 2002; Taniguchi et al. 2003; Elshabrawy et al. 2017).
In 2002, the research group of Kokkola first reported that HMGB1 was upregulated in the nucleus, cytoplasm, and extracellular environment of the synovial tissue (ST) and synovial cells in patients with RA and experimental arthritis rat models (Kokkola et al. 2002). In 2003, Taniguchi et al. reported that the serum and ST levels of HMGB1 in patients with RA are higher than those in healthy human controls. Stimulation with HMGB1 promotes the release of pro-inflammatory cytokines, such as TNF-α, IL-1β, and IL-6 from synovial fluid macrophages. Meanwhile, TNF-α stimulation upregulated HMGB1 expression and shifted the localization of HMGB1 from the nucleus to the cytoplasm, which further confirmed the key role of HMGB1 in the pathogenesis of RA and suggested the presence of a pro-inflammatory circuit between HMGB1 and TNF-α (Taniguchi et al. 2003). Pullerits et al. demonstrated that the injection of recombinant HMGB1 protein into mice induced arthritis (Pullerits et al. 2003). Other studies have demonstrated that the suppression of HMGB1 expression or treatment with therapeutic agents (including polyclonal and monoclonal anti-HMGB1 antibodies, the A-frame structural domain of recombinant HMGB1, soluble RAGE, and corticosteroids) can inhibit the progression of RA (Palmblad et al. 2007; Af Klint et al. 2005; Hofmann et al. 2002; Wouwer et al. 2006; Zetterström et al. 2008; Hamada et al. 2008). Intra-articular corticosteroid injection has been shown to suppress the extracellular effect of HMGB1 in RA patients (Af Klint et al. 2005). In experimental RA animal models, the suppression of HMGB1 expression alleviates cartilage and bone damage (Ostberg et al. 2010; Schierbeck et al. 2011). Additionally, HMGB1 can promote the activation and polarization of Th17 cells by upregulating TLR2 and Th17 cell-related cytokines in patients with RA. This suggests that the increase of Th17 cells may partly contribute to HMGB1-mediated immune dysregulation in RA (Shi et al. 2012; He et al. 2012). Samarpita et al. (2020) reported that in the adjuvant-induced arthritis rat model, TAK-242(an inhibitor of TLR4) downregulated the serum levels of IL-6 and significantly alleviated inflammatory symptoms in the joint tissues on day 21 of treatment. Lu et al. (2021) demonstrated that celastrol treatment inhibited the release of inflammatory cytokines, such as TNF-α, IL-6, and IL-1β in the collagen-induced arthritis (CIA) animal model and suppressed RA-induced cellular autophagy by inhibiting the activation of the TLR2/HMGB1 signaling pathway.
In RA, TLR1 is expressed on fibroblasts (Ospelt and Gay 2010). Previous studies have demonstrated that TLR2 and TLR4 are upregulated in the macrophages and fibroblasts in the inner layer of ST of patients with RA (Huang et al. 2007; Iwahashi et al. 2004). In vitro studies have confirmed that the TLR2 and TLR4 expression levels were upregulated in peripheral blood monocytes, ST macrophages, and fibroblasts of patients with RA (Radstake et al. 2004), suggesting that TLR2 and TLR4 are associated with the pathogenesis of RA. IL-17 upregulates the TLR2, TLR3, and TLR4 expression levels in ST fibroblasts of patients with RA (Lee et al. 2014). The expression of TLR2 in ST fibroblasts of patients with RA is regulated by TNF, IL-1, LPS, and miR-19 (Iwahashi et al. 2004; Ospelt et al. 2008; Seibl et al. 2003; Jung et al. 2007; Meng et al. 2010; Sacre et al. 2007; Kim et al. 2007). Treatment with macrophage colony-stimulating factor and IL-10 upregulated the TLR2 expression levels in monocytes and ST macrophages of patients with RA (Iwahashi et al. 2004). In ST, articular fibroblasts exhibit upregulated levels of TLR3 during the early stages of RA (Ospelt et al. 2008). One study demonstrated that TLR3 expression is similar in the monocytes of patients with RA and healthy controls (Roelofs et al. 2009). IFNα upregulates TLR3 expression in myeloid cells and ST fibroblasts of patients with RA (Roelofs et al. 2009; Zhu et al. 2011), suggesting a close correlation between TLR3 and IFN-α in ST of patients with RA. TLR5 expression is reported to be upregulated in the inner layer fibroblasts and macrophages of patients with RA (Kim et al. 2014, 2013; Chamberlain et al. 2012), TNF and IL-17 upregulate the TLR5 expression levels in the peripheral blood mononuclear cells and differentiated macrophages of patients with RA in vitro. Meanwhile, multiple single factors upregulated TLR5 levels in ST fibroblasts of patients with RA. TLR5 expression was positively correlated with RA disease activity (DAS28) and TNF levels. Additionally, TLR5 expression in patients treated with antirheumatic drugs (DMARDs) and anti-TNF antibodies was significantly downregulated when compared with that in patients treated with DMARDs alone (Chamberlain et al. 2012). Pro-inflammatory cytokines, such as IL-1β, IL-17, IL-6, and IL-8 can significantly upregulate TLR5 expression in fibroblasts of patients with RA (Chamberlain et al. 2012). Previous studies have demonstrated that TLR7 and TLR8 are mainly expressed in ST-lining/sub-lining macrophages and ST-lining fibroblasts of patients with RA (Chamberlain et al. 2013). IL-17 and IL-8 can upregulate TLR7 expression. LPS and IL-1 upregulated the TLR8 levels in the monocytes and macrophages of patients with RA. TLR7 expression in monocytes of patients with RA is correlated with DAS28 and TNF levels, whereas TLR8 expression in the myeloid cells was not correlated with the DAS28 or TNF levels. TLR9 expression is upregulated in ST fibroblasts of patients with RA and can be induced by hypoxia (Hu et al. 2014). Additionally, the peripheral blood mononuclear cell and SF macrophage expression levels of TLR9 in patients with active RA were significantly higher than those in healthy control (Jongbloed et al. 2006). TLR9 (rs187084) single-nucleotide polymorphism (SNP) is correlated with RA susceptibility in the Turkish population (Etem et al. 2011). TNF suppression can significantly downregulate the expression levels of TLR1, TLR4, and TLR6 in the whole blood of CIA rats (Clanchy et al. 2021). TLR3 agonists can relieve arthritis by inhibiting synovial cell proliferation and inflammatory response (Yarilina et al. 2007). TLR4 antagonists alleviate joint inflammation in CIA rats by downregulating the IL-1 levels (Abdollahi-Roodsaz et al. 2007). The topical application of TLR5 agonists exacerbates joint inflammation and bone erosion (Kim et al. 2013; Chamberlain et al. 2012). Anti-TLR5 antibody treatment can reduce M1 macrophage and Th17 cell polarization (Kim et al. 2014). Hegewald et al. (2020), for the first time, demonstrated that enhanced osteoclast maturation is mediated by TLR7/8 signaling, which is activated by miR-574-5p. This is a novel mechanism for the involvement of small extracellular vesicles and miRNAs in RA pathogenesis, suggesting that miR-574-5p inhibitors can exert protective effects against osteoclast-mediated bone destruction in RA and inhibit bone resorption. The administration of TLR9 agonists relieves joint inflammation by promoting IL-12 and IFN-γ production and inhibiting synovial neutrophil infiltration (Wu et al. 2007). Data from a machine learning model revealed the correlation between TLR9 polymorphism (rs352139) and treatment response in patients with RA who were treated with TNF-α inhibitors (Kim et al. 2021).
Systemic lupus erythematosus
Systemic lupus erythematosus (SLE), an autoimmune disease closely associated with HMGB1 and TLRs, is characterized by autoantibody production and systemic inflammation involving multiple organ systems (Tamirou et al. 2018). Previous studies have demonstrated that HMGB1 levels are upregulated in the blood of patients with SLE and are correlated with disease activity (Jiang and Pisetsky 2008; Ma et al. 2012; Li et al. 2010). The circulating immune complexes in patients with SLE also contain HMGB1, which is required to activate the immune response. In skin lesions of patients with SLE, the expression of HMGB1, which is expressed in the cytoplasmic and extracellular spaces, is upregulated in the epidermis and dermis and is correlated with the levels of IL-1β and TNF (Ardoin and Pisetsky 2008; Popovic et al. 2005). Ultraviolet radiation increases the translocation of HMGB1 into the cytoplasm and extracellular spaces in the skin of patients with SLE (Barkauskaite et al. 2007), suggesting that photosensitivity can induce SLE through HMGB1. The HMGB1-DNA complex can induce the production of anti-DNA antibodies in mice under the condition of DNA inactivation alone. The production of this antibody is dependent on TLR2 (Urbonaviciute et al. 2008).
TLRs are also involved in the pathogenesis of SLE. The peripheral blood mononuclear cell expression levels of TLR2 and TLR4 in patients with SLE are higher than those in the healthy control group (Komatsuda et al. 2008; Lee et al. 2016). In the lupus-prone model, TLR2 or TLR4 knockdown downregulated the antinuclear antibody levels and alleviated disease symptoms (Lartigue et al. 2009). TLR4 ligand endotoxin promotes the progression of lupus nephritis and autoantibody production (Cavallo and Granholm 1990a, b; Liu et al. 2006). TLR7 overexpression promoted autoantibody production (Pisitkun et al. 2006; Subramanian et al. 2006). In contrast, in the absence of TLR7, the levels of circulating autoantibodies and inflammatory cytokines, such as IL-6 and INF-α were significantly downregulated and the disease symptoms were alleviated (Christensen et al. 2006; Lee et al. 2008; Kono et al. 2009). TLR7 is closely correlated with the expression of IFN-α and IFN-β in patients with SLE (Paradowska-Gorycka et al. 2021). TLR8 overexpression is associated with the progression of SLE glomerulonephritis (Kimura et al. 2014). Estrogen treatment stimulates STAT1-dependent transcriptional activation of TLR8 (Young et al. 2014, 2017), suggesting that estrogen application may contribute to the progression of SLE. Some studies found that in the lupus-prone model, TLR9 knockdown did not improve the disease status and promoted disease progression (Christensen et al. 2006; Nickerson et al. 2010; Jackson et al. 2014), suggesting that TLR9 may exert a positive regulatory effect on the development of SLE. In the absence of TLR9, autoimmune disease is exacerbated, resulting in heightened activation of lymphocytes and plasmacytoid DC, as well as elevated levels of serum IgG and IFN-α (Christensen et al. 2006), suggesting the activation of TLR9 may be potential therapeutic strategies for SLE. A recent study has identified a mechanism through which TLR9 hampers the progression of SLE. The researchers observed that TLR9P915H (TLR9 point mutant—lacking MyD88), in contrast to TLR9K51E, exhibited a more pronounced inhibition of disease progression compared to TLR9WT, and TLR9P915H mice exhibited less disease than TLR9K51E mice (TLR9 point mutant—lacking either ligand) (Leibler et al. 2022). These findings suggest that the anti-inflammatory response is encoded by a signal dependent on the TLR9 ligand, rather than MyD88. However, the study also reveals an unexpected pro-inflammatory effect of TLR9-MyD88 signaling, despite TLR9's primary role being thought to prevent SLE (Leibler et al. 2022). The study found that selective elimination of TLR9-MyD88 (TLR9P915H) signaling reduced disease compared to WT mice.
Ji et al. reported that HMGB1 expression was significantly upregulated in the bone marrow of patients with SLE. The clinical signs of lupus nephritis were alleviated and the survival time was prolonged in MRL/LPR mice administered with the safe HMGB1 inhibitor ethyl pyruvate (EP) for 8 weeks (Ji et al. 2019), suggesting that EP is a potential therapeutic for SLE. The benzylamine derivative FC-99 inhibited the TLR ligand-induced activation of pro-inflammatory cytokines, such as IL-12 and CXCL10, alleviated albuminuria, and upregulated immunoglobulin levels associated with renal disease and lupus-like syndrome in vitro and in vitro (Gao et al. 2017).
Type 1 diabetes mellitus
Type 1 diabetes mellitus (T1DM), an autoimmune disease, is characterized by the progressive destruction of pancreatic β-cells mediated by immune cells. During autoimmune responses, HMGB1 can be passively released from damaged pancreatic cells and actively secreted by islet-infiltrating immune cells. The suppression of HMGB1 significantly inhibited the progression of diabetes in mice (Han et al. 2008). The serum HMGB1 levels were significantly upregulated in patients with T1DM and animal models (Han et al. 2008; Zhang et al. 2009; Wu et al. 2016). In T1DM, extracellular HMGB1 promotes autoimmune response by activating TLR4 and destabilizing and disrupting the function of regulatory T cells (Zhang et al. 2020). The upregulation of circulating HMGB1 in patients with T1DM leads to the instability of T regulatory cells, suggesting that blocking HMGB1 may be an effective therapeutic approach for T1DM (Zhang et al. 2020).
The peripheral blood mononuclear cell surface expression levels of TLR2 and TLR4 in patients with T1DM were significantly higher than those in the healthy control group. Additionally, the expression levels of TLRs, MyD88, TRIF, and downstream proteins were significantly upregulated in patients with T1DM. Furthermore, the secretory levels of IL-1 and TNF-α from the peripheral blood mononuclear cells were significantly upregulated in patients with T1DM and positively correlated with the TLR2 and TLR4 expression levels (Devaraj et al. 2008). Previous studies have reported that the inhibition of TLR2 or TLR4 in diabetes suppresses the inflammatory response (Lin et al. 2012; Ma et al. 2014; Jialal et al. 2014).
Continuous intake of glycyrrhizin acid after the onset of diabetes significantly downregulates retinal HMGB1 expression (Abu El-Asrar et al. 2014). Resveratrol, a dietary antioxidant, prevents morphological and functional ventricular remodeling and downregulates HMGB1 expression in T1DM rats (Delucchi et al. 2012). Sodium butyrate inhibited the expression of HMGB1 and NF-κB proteins in the pancreas and suppressed the progression of T1DM by inhibiting HMGB1 and downregulating the NF-κB-mediated inflammatory signaling pathways (Guo et al. 2018). Insulin infusion inhibits the expression of HMGB1 and TLRs in monocytes of patients with T1DM (Dandona et al. 2013). Further studies are needed to identify other drugs that can delay the progression of T1DM or its complications by inhibiting HMGB1 or TLRs.
Autoimmune thyroid disease
HMGB1 may also be involved in the pathogenesis of Autoimmune Thyroid Disease (AITD). AITD, including Hashimoto's thyroiditis (HT) and Graves' disease (GD), are organ-specific autoimmune diseases characterized by lymphocytic infiltration of the thyroid gland (Lee et al. 2015). HMGB1 expression is upregulated in the peripheral blood of patients with AITD (including patients with HT or GD) (Peng et al. 2016), the thyroid tissue and serum of NOD.H-2h4 mice (murine model of autoimmune thyroiditis) (Li et al. 2017), the thyroid tissue and serum of a rat model of thyroglobulin-induced experimental autoimmune thyroiditis (EAT) (Guo et al. 2021), and thyroid follicular epithelial cells (Guo et al. 2021). The upregulated HMGB1 expression was positively correlated with thyroglobulin antibody and thyroid peroxidase antibody levels in the peripheral blood of patients with AITD (Peng et al. 2016). Previous studies have demonstrated that the number of HMGB1-positive monocytes is upregulated in patients with GD and that HMGB1 expression is downregulated by approximately 50% upon treatment with antithyroid drug (Mobarrez et al. 2016). These findings suggest that HMGB1 may play an important role in the development of AITD and that it is associated with disease progression.
The expression levels of TLR2, TLR3, TLR9, and TLR10 in peripheral blood monocytes of AITD patients were up-regulated (Peng et al. 2016). Additionally, the expression levels of TLR2, TLR3, TLR9, and the downstream adaptor protein MyD88 were significantly upregulated in the thyroid tissue and serum of NOD.H-2h4 mice or EAT rats (Li et al. 2017; Guo et al. 2021; Harii et al. 2005), as well as in thyroid follicular epithelial cells (Zhang et al. 2022). This suggests that TLR2, TLR3, and TLR9 are involved in the development of AITD. HMGB1 expression was upregulated in the peripheral blood mononuclear cells of patients with AITD both at resting state and upon TLR9 stimulation (Peng et al. 2016). This indicates a positive correlation between HMGB1 and TLR9 expression. Thus, HMGB1 plays an important role in the development of chronic inflammation in AITD by activating the TLR9 pathway. TLR10 SNP is significantly associated with the pathogenesis of AITD (Cho et al. 2015). Additionally, TLR10 is suggested to function as a co-receptor for TLR2 (Guan et al. 2010). However, the role of TLR10 in thyroid cells must be examined further.
HMGB1 promotes the expression of early pro-inflammatory cytokines, such as TNF-α, IL-1β, and IL-6 (Li et al. 2017). The inhibition of HMGB1 using glycyrrhizin (GL) suppresses thyroid lymphocyte infiltration and inhibits the downstream proteins MyD88 and NF-κB and the expression of pro-inflammatory cytokines through the HMGB1-TLR2 signaling pathway (Li et al. 2017). These findings suggest that the GL-mediated inhibition of HMGB1 mitigates tissue damage by alleviating inflammatory response and that GL may inhibit the development of AITD by suppressing the HMGB1-TLR2 signaling pathway. Guo reported that Prunella vulgaris L. (PV)-mediated HMGB1 inhibition can suppress inflammatory response and cytokine production by inhibiting the expression of MyD88 (downstream protein of the HMGB1-TLR9 signaling pathway), decreasing the proportion of Th1, Th2, and Th17 cells in splenocytes, and reversing the enhanced production of pro-inflammatory cytokines, such as TNF-α, IL-6, and IL-1β in vivo and in vitro (Guo et al. 2021). This suggests that PV can delay the progression of AITD by inhibiting the HMGB1-TLR9 signaling pathway.
HMGB1, TLR2, TLR3, TLR9, and TLR10 may aggravate tissue damage by promoting the release of pro-inflammatory cytokines and play a major role in the development of AITD. Further studies are needed to elucidate the specific roles of TLR3 and TLR10. GL can target the HMGB1-TLR2 signaling pathway, while PV can inhibit the HMGB1-TLR9 signaling pathway and downstream proteins to delay the progression of AITD. Further studies are needed to identify targeted therapeutic drugs.
Conclusions and perspectives
HMGB1 is an early-warning protein that induces immune responses alone or in combination with TLRs. Clinical and experimental studies have demonstrated that HMGB1 and TLR-dependent pro-inflammatory mechanisms play a key role in the pathogenesis of autoimmune diseases. Thus, HMGB1 and TLRs are important biomarkers. Clinical trials on several drugs targeting HMGB1 and TLRs are currently ongoing. Targeting HMGB1 and TLRs for therapeutic purposes has shown promise, but there are several limitations and challenges that need to be considered. These include potential side effects and difficulties in developing effective drugs. One limitation is the pleiotropic nature of HMGB1 and TLRs signaling. Both HMGB1 and TLRs have diverse roles and functions in the immune system, inflammation, and various diseases. Modulating their activities may disrupt the delicate balance and potentially lead to adverse effects, including immune dysregulation, increased susceptibility to infections, or abnormal immune activation. Another challenge is the complexity of the HMGB1 and TLRs signaling networks. These pathways involve intricate molecular interactions and crosstalk with other signaling pathways. Designing drugs that specifically target HMGB1 or individual TLRs without affecting other related molecules requires a deep understanding of the underlying mechanisms. Moreover, there is a need for comprehensive preclinical and clinical studies to validate the safety and efficacy of HMGB1 and TLRs targeted therapies. In conclusion, while targeting HMGB1 and TLRs holds great therapeutic potential, it is crucial to address the limitations and challenges associated with their modulation.
Recent studies have demonstrated that HMGB1 and TLRs are closely related to inflammasomes, pyroptosis, and ferroptosis. The specific downstream pathways of HMGB1 and TLRs that lead to autoimmune and inflammatory diseases have also been gradually discovered. However, future studies must focus on distinguishing the origin of the positive feedback loop for HMGB1 (is it as inconclusive as the chicken or egg problem?), elucidating the mechanism through which HMGB1 enters the cells and binds to intracellular TLRs, and evaluating the therapeutic potential of HMGB1 and TLR antagonists in autoimmune disease as targeting HMGB1 and TLRs has been demonstrated to exert potent therapeutic effects on inflammatory and autoimmune diseases.
Availability of data and materials
Not applicable.
Abbreviations
- DAMP:
-
Damage-associated molecular pattern
- TLRs:
-
Toll-like receptors
- HMG:
-
High-mobility group
- RA:
-
Rheumatoid arthritis
- SLE:
-
Systemic lupus erythematosus
- AITD:
-
Autoimmune thyroid disease
- PAMPs:
-
Pathogen-associated molecular models
- HMGB:
-
High-mobility group box
- NESs:
-
Nuclear export signals
- NLSs:
-
Nuclear localization signals
- LPS:
-
Lipopolysaccharide
- PRRs:
-
Pattern recognition receptors
- TRIF:
-
TIR domain-containing adaptor protein inducing IFNβ
- TNF-α:
-
Tumor necrosis factor-α
- NF-κB:
-
Nuclear factor kappa-light-chain-enhancer of activated B cells
- IL:
-
Interleukin
- MyD88:
-
Molecule myeloid differentiation primary response differentiation gene 88
- IRFs:
-
IFN regulatory factors (IRFs)
- ST:
-
Synovial tissue (ST)
- CIA:
-
Collagen-induced arthritis
- SNP:
-
Single-nucleotide polymorphism
- EP:
-
Ethyl pyruvate
- HT:
-
Hashimoto's thyroiditis
- GD:
-
Graves' disease
- EAT:
-
Experimental autoimmune thyroiditis
- GL:
-
Glycyrrhizin
- PV:
-
Prunella vulgaris L.
References
Abdollahi-Roodsaz S, Joosten LAB, Roelofs MF, Radstake TRDJ, Matera G, Popa C, et al. Inhibition of Toll-like receptor 4 breaks the inflammatory loop in autoimmune destructive arthritis. Arthritis Rheum. 2007;56:2957–67.
Abu El-Asrar AM, Siddiquei MM, Nawaz MI, Geboes K, Mohammad G. The proinflammatory cytokine high-mobility group box-1 mediates retinal neuropathy induced by diabetes. Mediat Inflamm. 2014;2014: 746415.
Af Klint E, Grundtman C, Engström M, Catrina AI, Makrygiannakis D, Klareskog L, et al. Intraarticular glucocorticoid treatment reduces inflammation in synovial cell infiltrations more efficiently than in synovial blood vessels. Arthritis Rheum. 2005;52:3880–9.
Agnello D, Wang H, Yang H, Tracey KJ, Ghezzi P. HMGB-1, a DNA-binding protein with cytokine activity, induces brain TNF and IL-6 production, and mediates anorexia and taste aversion. Cytokine. 2002;18:231–6.
Andersson U, Wang H, Palmblad K, Aveberger AC, Bloom O, Erlandsson-Harris H, et al. High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. J Exp Med. 2000;192:565–70.
Andersson U, Yang H, Harris H. Extracellular HMGB1 as a therapeutic target in inflammatory diseases. Expert Opin Ther Targets. 2018;22:263–77.
Ardoin SP, Pisetsky DS. Developments in the scientific understanding of lupus. Arthritis Res Ther. 2008;10:218.
Barkauskaite V, Ek M, Popovic K, Harris HE, Wahren-Herlenius M, Nyberg F. Translocation of the novel cytokine HMGB1 to the cytoplasm and extracellular space coincides with the peak of clinical activity in experimentally UV-induced lesions of cutaneous lupus erythematosus. Lupus. 2007;16:794–802.
Beutler B. Inferences, questions and possibilities in Toll-like receptor signalling. Nature. 2004;430:257–63.
Bianchi ME, Falciola L, Ferrari S, Lilley DM. The DNA binding site of HMG1 protein is composed of two similar segments (HMG boxes), both of which have counterparts in other eukaryotic regulatory proteins. EMBO J. 1992. https://doi.org/10.1002/j.1460-2075.1992.tb05144.x.
Bonaldi T, Talamo F, Scaffidi P, Ferrera D, Porto A, Bachi A, et al. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 2003;22:5551–60.
Botos I, Segal DM, Davies DR. The structural biology of Toll-like receptors. Structure. 2011;19:447–59.
Bustin M. Revised nomenclature for high mobility group (HMG) chromosomal proteins. Trends Biochem Sci. 2001;26:152–3.
Bustin M, Neihart NK, Fagan JB. mRNA of chromosomal proteins HMG-1 and HMG-2 are polyadenylated. Biochem Biophys Res Commun. 1981;101:893–7.
Cavallo T, Granholm NA. Lipopolysaccharide from gram-negative bacteria enhances polyclonal B cell activation and exacerbates nephritis in MRL/lpr mice. Clin Exp Immunol. 1990a;82:515–21.
Cavallo T, Granholm NA. Bacterial lipopolysaccharide transforms mesangial into proliferative lupus nephritis without interfering with processing of pathogenic immune complexes in NZB/W mice. Am J Pathol. 1990b;137:971–8.
Chamberlain ND, Vila OM, Volin MV, Volkov S, Pope RM, Swedler W, et al. TLR5, a novel and unidentified inflammatory mediator in rheumatoid arthritis that correlates with disease activity score and joint TNF-α levels. J Immunol. 2012;189:475–83.
Chamberlain ND, Kim S, Vila OM, Volin MV, Volkov S, Pope RM, et al. Ligation of TLR7 by rheumatoid arthritis synovial fluid single strand RNA induces transcription of TNFα in monocytes. Ann Rheum Dis. 2013;72:418–26.
Chen Y, Sun W, Gao R, Su Y, Umehara H, Dong L, et al. The role of high mobility group box chromosomal protein 1 in rheumatoid arthritis. Rheumatology. 2013;52:1739–47.
Chen R, Kang R, Tang D. The mechanism of HMGB1 secretion and release. Exp Mol Med. 2022;54:91–102.
Cho WK, Jang J-P, Choi E-J, Jeon YJ, Jung IA, Kim S-H, et al. Association of Toll-like receptor 10 polymorphisms with autoimmune thyroid disease in Korean children. Thyroid. 2015;25:250–5.
Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity. 2006;25:417–28.
Chuang T, Ulevitch RJ. Identification of hTLR10: a novel human Toll-like receptor preferentially expressed in immune cells. Biochim Biophys Acta. 2001;1518:157–61.
Clanchy FIL, Borghese F, Bystrom J, Balog A, Penn H, Hull DN, et al. TLR expression profiles are a function of disease status in rheumatoid arthritis and experimental arthritis. J Autoimmun. 2021;118: 102597.
Dandona P, Ghanim H, Green K, Sia CL, Abuaysheh S, Kuhadiya N, et al. Insulin infusion suppresses while glucose infusion induces Toll-like receptors and high-mobility group-B1 protein expression in mononuclear cells of type 1 diabetes patients. Am J Physiol Endocrinol Metab. 2013;304:E810-818.
Delucchi F, Berni R, Frati C, Cavalli S, Graiani G, Sala R, et al. Resveratrol treatment reduces cardiac progenitor cell dysfunction and prevents morpho-functional ventricular remodeling in type-1 diabetic rats. PLoS ONE. 2012;7: e39836.
Devaraj S, Dasu MR, Rockwood J, Winter W, Griffen SC, Jialal I. Increased toll-like receptor (TLR) 2 and TLR4 expression in monocytes from patients with type 1 diabetes: further evidence of a proinflammatory state. J Clin Endocrinol Metab. 2008;93:578–83.
Drexler SK, Foxwell BM. The role of toll-like receptors in chronic inflammation. Int J Biochem Cell Biol. 2010;42:506–18.
Dumitriu IE, Baruah P, Manfredi AA, Bianchi ME, Rovere-Querini P. HMGB1: guiding immunity from within. Trends Immunol. 2005;26:381–7.
Dvornikova KA, Bystrova EY, Platonova ON, Churilov LP. Polymorphism of toll-like receptor genes and autoimmune endocrine diseases. Autoimmun Rev. 2020;19: 102496.
Elshabrawy HA, Essani AE, Szekanecz Z, Fox DA, Shahrara S. TLRs, future potential therapeutic targets for RA. Autoimmun Rev. 2017;16:103–13.
Etem EO, Elyas H, Ozgocmen S, Yıldırım A, Godekmerdan A. The investigation of toll-like receptor 3, 9 and 10 gene polymorphisms in Turkish rheumatoid arthritis patients. Rheumatol Int. 2011;31:1369–74.
Ferrari S, Ronfani L, Calogero S, Bianchi ME. The mouse gene coding for high mobility group 1 protein (HMG1). J Biol Chem. 1994;269:28803–8.
Fiuza C, Bustin M, Talwar S, Tropea M, Gerstenberger E, Shelhamer JH, et al. Inflammation-promoting activity of HMGB1 on human microvascular endothelial cells. Blood. 2003;101:2652–60.
Gao S, Gong Y, Ji J, Yuan L, Han L, Guo Y, et al. A new benzenediamine derivative modulates Toll-like receptors-induced myeloid dendritic cells activation and ameliorates lupus-like syndrome in MRLlpr/lpr mice. Eur J Pharmacol. 2017;803:94–102.
Gardella S, Andrei C, Ferrera D, Lotti LV, Torrisi MR, Bianchi ME, et al. The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep. 2002;3:995–1001.
Gay NJ, Symmons MF, Gangloff M, Bryant CE. Assembly and localization of Toll-like receptor signalling complexes. Nat Rev Immunol. 2014;14:546–58.
Ge Y, Huang M, Yao Y-M. The effect and regulatory mechanism of high mobility group box-1 protein on immune cells in inflammatory diseases. Cells. 2021;10:1044.
Goodwin GH, Johns EW. Isolation and characterisation of two calf-thymus chromatin non-histone proteins with high contents of acidic and basic amino acids. Eur J Biochem. 1973;40:215–9.
Goodwin GH, Sanders C, Johns EW. A new group of chromatin-associated proteins with a high content of acidic and basic amino acids. Eur J Biochem. 1973;38:14–9.
Goodwin GH, Johns EW. Are the high mobility group non-histone chromosomal proteins associated with ‘active’ chromatin? Biochimica Et Biophysica Acta Nucleic Acids and Protein Synthesis. 1978;519:279–84.
Guan Y, Ranoa DRE, Jiang S, Mutha SK, Li X, Baudry J, et al. Human TLRs 10 and 1 share common mechanisms of innate immune sensing but not signaling. J Immunol. 2010;184:5094–103.
Guo Y, Xiao Z, Wang Y, Yao W, Liao S, Yu B, et al. Sodium butyrate ameliorates streptozotocin-induced type 1 diabetes in mice by inhibiting the hmgb1 expression. Front Endocrinol (lausanne). 2018;9:630.
Guo Q, Qu H, Zhang H, Zhong X. Prunella vulgaris L. attenuates experimental autoimmune thyroiditis by inhibiting HMGB1/TLR9 signaling. Drug Des Devel Ther. 2021;15:4559–74.
Hamada T, Torikai M, Kuwazuru A, Tanaka M, Horai N, Fukuda T, et al. Extracellular high mobility group box chromosomal protein 1 is a coupling factor for hypoxia and inflammation in arthritis. Arthr Rheum. 2008;58:2675–85.
Han J, Zhong J, Wei W, Wang Y, Huang Y, Yang P, et al. Extracellular high-mobility group box 1 acts as an innate immune mediator to enhance autoimmune progression and diabetes onset in NOD mice. Diabetes. 2008;57:2118–27.
Hardman CH, Broadhurst RW, Raine AR, Grasser KD, Thomas JO, Laue ED. Structure of the A-domain of HMG1 and its interaction with DNA as studied by heteronuclear three- and four-dimensional NMR spectroscopy. Biochemistry. 1995;34:16596–607.
Harii N, Lewis CJ, Vasko V, McCall K, Benavides-Peralta U, Sun X, et al. Thyrocytes express a functional toll-like receptor 3: overexpression can be induced by viral infection and reversed by phenylmethimazole and is associated with Hashimoto’s autoimmune thyroiditis. Mol Endocrinol. 2005;19:1231–50.
He Z, Shotorbani SS, Jiao Z, Su Z, Tong J, Liu Y, et al. HMGB1 promotes the differentiation of Th17 via up-regulating TLR2 and IL-23 of CD14+ monocytes from patients with rheumatoid arthritis. Scand J Immunol. 2012;76:483–90.
Hegewald AB, Breitwieser K, Ottinger SM, Mobarrez F, Korotkova M, Rethi B, et al. Extracellular miR-574-5p induces osteoclast differentiation via TLR 7/8 in rheumatoid arthritis. Front Immunol. 2020;11: 585282.
Herzog C, Lorenz A, Gillmann H-J, Chowdhury A, Larmann J, Harendza T, et al. Thrombomodulin’s lectin-like domain reduces myocardial damage by interfering with HMGB1-mediated TLR2 signalling. Cardiovasc Res. 2014;101:400–10.
Hofmann MA, Drury S, Hudson BI, Gleason MR, Qu W, Lu Y, et al. RAGE and arthritis: the G82S polymorphism amplifies the inflammatory response. Genes Immun. 2002;3:123–35.
Hoppe G, Talcott KE, Bhattacharya SK, Crabb JW, Sears JE. Molecular basis for the redox control of nuclear transport of the structural chromatin protein Hmgb1. Exp Cell Res. 2006;312:3526–38.
Hu F, Mu R, Zhu J, Shi L, Li Y, Liu X, et al. Hypoxia and hypoxia-inducible factor-1α provoke toll-like receptor signalling-induced inflammation in rheumatoid arthritis. Ann Rheum Dis. 2014;73:928–36.
Huang Q, Ma Y, Adebayo A, Pope RM. Increased macrophage activation mediated through toll-like receptors in rheumatoid arthritis. Arthr Rheum. 2007;56:2192–201.
Huttunen HJ, Fages C, Kuja-Panula J, Ridley AJ, Rauvala H. Receptor for advanced glycation end products-binding COOH-terminal motif of amphoterin inhibits invasive migration and metastasis. Cancer Res. 2002;62:4805–11.
Iwahashi M, Yamamura M, Aita T, Okamoto A, Ueno A, Ogawa N, et al. Expression of Toll-like receptor 2 on CD16+ blood monocytes and synovial tissue macrophages in rheumatoid arthritis. Arthr Rheum. 2004;50:1457–67.
Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–95.
Jackson SW, Scharping NE, Kolhatkar NS, Khim S, Schwartz MA, Li Q-Z, et al. Opposing impact of B cell-intrinsic TLR7 and TLR9 signals on autoantibody repertoire and systemic inflammation. J Immunol. 2014;192:4525–32.
Ji J, Fu T, Dong C, Zhu W, Yang J, Kong X, et al. Targeting HMGB1 by ethyl pyruvate ameliorates systemic lupus erythematosus and reverses the senescent phenotype of bone marrow-mesenchymal stem cells. Aging. 2019;11:4338–53.
Jialal I, Major AM, Devaraj S. Global Toll-like receptor 4 knockout results in decreased renal inflammation, fibrosis and podocytopathy. J Diabetes Complicat. 2014;28:755–61.
Jiang W, Pisetsky DS. Expression of high mobility group protein 1 in the sera of patients and mice with systemic lupus erythematosus. Ann Rheum Dis. 2008;67:727–8.
Jongbloed SL, Lebre MC, Fraser AR, Gracie JA, Sturrock RD, Tak PP, et al. Enumeration and phenotypical analysis of distinct dendritic cell subsets in psoriatic arthritis and rheumatoid arthritis. Arthr Res Ther. 2006;8:R15.
Jung YO, Cho M-L, Kang C-M, Jhun J-Y, Park J-S, Oh H-J, et al. Toll-like receptor 2 and 4 combination engagement upregulate IL-15 synergistically in human rheumatoid synovial fibroblasts. Immunol Lett. 2007;109:21–7.
Kang R, Chen R, Zhang Q, Hou W, Wu S, Cao L, et al. HMGB1 in health and disease. Mol Aspects Med. 2014;40:1–116.
Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol. 2014;5:461.
Kim K-W, Cho M-L, Lee S-H, Oh H-J, Kang C-M, Ju JH, et al. Human rheumatoid synovial fibroblasts promote osteoclastogenic activity by activating RANKL via TLR-2 and TLR-4 activation. Immunol Lett. 2007;110:54–64.
Kim S-J, Chen Z, Chamberlain ND, Volin MV, Swedler W, Volkov S, et al. Angiogenesis in rheumatoid arthritis is fostered directly by toll-like receptor 5 ligation and indirectly through interleukin-17 induction. Arthr Rheum. 2013;65:2024–36.
Kim S-J, Chen Z, Chamberlain ND, Essani AB, Volin MV, Amin MA, et al. Ligation of TLR5 promotes myeloid cell infiltration and differentiation into mature osteoclasts in rheumatoid arthritis and experimental arthritis. J Immunol. 2014;193:3902–13.
Kim W, Kim TH, Oh SJ, Kim HJ, Kim JH, Kim H-A, et al. Association of TLR 9 gene polymorphisms with remission in patients with rheumatoid arthritis receiving TNF-α inhibitors and development of machine learning models. Sci Rep. 2021;11:20169.
Kimura J, Ichii O, Miyazono K, Nakamura T, Horino T, Otsuka-Kanazawa S, et al. Overexpression of Toll-like receptor 8 correlates with the progression of podocyte injury in murine autoimmune glomerulonephritis. Sci Rep. 2014;4:7290.
Kokkola R, Sundberg E, Ulfgren A-K, Palmblad K, Li J, Wang H, et al. High mobility group box chromosomal protein 1: a novel proinflammatory mediator in synovitis. Arthr Rheum. 2002;46:2598–603.
Komatsuda A, Wakui H, Iwamoto K, Ozawa M, Togashi M, Masai R, et al. Up-regulated expression of Toll-like receptors mRNAs in peripheral blood mononuclear cells from patients with systemic lupus erythematosus. Clin Exp Immunol. 2008;152:482–7.
Kono DH, Haraldsson MK, Lawson BR, Pollard KM, Koh YT, Du X, et al. Endosomal TLR signaling is required for anti-nucleic acid and rheumatoid factor autoantibodies in lupus. Proc Natl Acad Sci U S A. 2009;106:12061–6.
Laird MD, Shields JS, Sukumari-Ramesh S, Kimbler DE, Fessler RD, Shakir B, et al. High mobility group box protein-1 promotes cerebral edema after traumatic brain injury via activation of toll-like receptor 4. Glia. 2014;62:26–38.
Lartigue A, Colliou N, Calbo S, François A, Jacquot S, Arnoult C, et al. Critical role of TLR2 and TLR4 in autoantibody production and glomerulonephritis in lpr mutation-induced mouse lupus. J Immunol. 2009;183:6207–16.
Lee PY, Kumagai Y, Li Y, Takeuchi O, Yoshida H, Weinstein J, et al. TLR7-dependent and FcgammaR-independent production of type I interferon in experimental mouse lupus. J Exp Med. 2008;205:2995–3006.
Lee S-Y, Yoon B-Y, Kim J-I, Heo Y-M, Woo Y-J, Park S-H, et al. Interleukin-17 increases the expression of Toll-like receptor 3 via the STAT3 pathway in rheumatoid arthritis fibroblast-like synoviocytes. Immunology. 2014;141:353–61.
Lee HJ, Li CW, Hammerstad SS, Stefan M, Tomer Y. Immunogenetics of autoimmune thyroid diseases: a comprehensive review. J Autoimmun. 2015;64:82–90.
Lee YH, Choi SJ, Ji JD, Song GG. Association between toll-like receptor polymorphisms and systemic lupus erythematosus: a meta-analysis update. Lupus. 2016;25:593–601.
Leibler C, John S, Elsner RA, Thomas KB, Smita S, Joachim S, et al. Genetic dissection of TLR9 reveals complex regulatory and cryptic proinflammatory roles in mouse lupus. Nat Immunol. 2022;23:1457–69.
Li J, Kokkola R, Tabibzadeh S, Yang R, Ochani M, Qiang X, et al. Structural basis for the proinflammatory cytokine activity of high mobility group box 1. Mol Med. 2003;9:37–45.
Li J, Xie H, Wen T, Liu H, Zhu W, Chen X. Expression of high mobility group box chromosomal protein 1 and its modulating effects on downstream cytokines in systemic lupus erythematosus. J Rheumatol. 2010;37:766–75.
Li Z-C, Cheng G-Q, Hu K-Z, Li M-Q, Zang W-P, Dong Y-Q, et al. Correlation of synovial fluid HMGB-1 levels with radiographic severity of knee osteoarthritis. Clin Invest Med. 2011;34:E298.
Li C, Peng S, Liu X, Han C, Wang X, Jin T, et al. Glycyrrhizin, a direct HMGB1 antagonist, ameliorates inflammatory infiltration in a model of autoimmune thyroiditis via inhibition of TLR2-HMGB1 signaling. Thyroid. 2017;27:722–31.
Li W, Deng M, Loughran PA, Yang M, Lin M, Yang C, et al. LPS induces active HMGB1 release from hepatocytes into exosomes through the coordinated activities of TLR4 and caspase-11/GSDMD signaling. Front Immunol. 2020;11:229.
Lim K-H, Staudt LM. Toll-like receptor signaling. Cold Spring Harb Perspect Biol. 2013;5: a011247.
Lin M, Yiu WH, Wu HJ, Chan LYY, Leung JCK, Au WS, et al. Toll-like receptor 4 promotes tubular inflammation in diabetic nephropathy. J Am Soc Nephrol. 2012;23:86–102.
Liu B, Yang Y, Dai J, Medzhitov R, Freudenberg MA, Zhang PL, et al. TLR4 up-regulation at protein or gene level is pathogenic for lupus-like autoimmune disease. J Immunol. 2006;177:6880–8.
Lu X, Gong S, Wang X, Hu N, Pu D, Zhang J, et al. Celastrol exerts cardioprotective effect in rheumatoid arthritis by inhibiting TLR2/HMGB1 signaling pathway-mediated autophagy. Int Arch Allergy Immunol. 2021;182:1245–54.
Luchner M, Reinke S, Milicic A. TLR agonists as vaccine adjuvants targeting cancer and infectious diseases. Pharmaceutics. 2021;13:142.
Ma C, Jiao Y, Zhang J, Yang Q, Zhang Z, Shen Y, et al. Elevated plasma level of HMGB1 is associated with disease activity and combined alterations with IFN-α and TNF-α in systemic lupus erythematosus. Rheumatol Int. 2012;32:395–402.
Ma J, Chadban SJ, Zhao CY, Chen X, Kwan T, Panchapakesan U, et al. TLR4 activation promotes podocyte injury and interstitial fibrosis in diabetic nephropathy. PLoS ONE. 2014;9: e97985.
Magna M, Pisetsky DS. The role of HMGB1 in the pathogenesis of inflammatory and autoimmune diseases. Mol Med. 2014;20:138–46.
Marongiu L, Gornati L, Artuso I, Zanoni I, Granucci F. Below the surface: the inner lives of TLR4 and TLR9. J Leukoc Biol. 2019;106:147–60.
Meng L, Zhu W, Jiang C, He X, Hou W, Zheng F, et al. Toll-like receptor 3 upregulation in macrophages participates in the initiation and maintenance of pristane-induced arthritis in rats. Arthr Res Ther. 2010;12:R103.
Mobarrez F, Abraham-Nordling M, Aguilera-Gatica K, Friberg I, Antovic A, Pisetsky DS, et al. The expression of microvesicles in the blood of patients with Graves’ disease and its relationship to treatment. Clin Endocrinol. 2016;84:729–35.
Moynagh PN. TLR signalling and activation of IRFs: revisiting old friends from the NF-kappaB pathway. Trends Immunol. 2005;26:469–76.
Nickerson KM, Christensen SR, Shupe J, Kashgarian M, Kim D, Elkon K, et al. TLR9 regulates TLR7- and MyD88-dependent autoantibody production and disease in a murine model of lupus. J Immunol. 2010;184:1840–8.
Oktayoglu P, Em S, Tahtasiz M, Bozkurt M, Ucar D, Yazmalar L, et al. Elevated serum levels of high mobility group box protein 1 (HMGB1) in patients with ankylosing spondylitis and its association with disease activity and quality of life. Rheumatol Int. 2013;33:1327–31.
Ospelt C, Gay S. TLRs and chronic inflammation. Int J Biochem Cell Biol. 2010;42:495–505.
Ospelt C, Brentano F, Rengel Y, Stanczyk J, Kolling C, Tak PP, et al. Overexpression of toll-like receptors 3 and 4 in synovial tissue from patients with early rheumatoid arthritis: toll-like receptor expression in early and longstanding arthritis. Arthritis Rheum. 2008;58:3684–92.
Ostberg T, Kawane K, Nagata S, Yang H, Chavan S, Klevenvall L, et al. Protective targeting of high mobility group box chromosomal protein 1 in a spontaneous arthritis model. Arthr Rheum. 2010;62:2963–72.
Owen AM, Fults JB, Patil NK, Hernandez A, Bohannon JK. TLR agonists as mediators of trained immunity: mechanistic insight and immunotherapeutic potential to combat infection. Front Immunol. 2020;11: 622614.
Palmblad K, Sundberg E, Diez M, Söderling R, Aveberger A-C, Andersson U, et al. Morphological characterization of intra-articular HMGB1 expression during the course of collagen-induced arthritis. Arthr Res Ther. 2007;9:R35.
Paradowska-Gorycka A, Wajda A, Stypinska B, Walczuk E, Rzeszotarska E, Walczyk M, et al. Variety of endosomal TLRs and Interferons (IFN-α, IFN-β, IFN-γ) expression profiles in patients with SLE, Ssc and MCTD. Clin Exp Immunol. 2021;204:49–63.
Peng S, Li C, Wang X, Liu X, Han C, Jin T, et al. Increased Toll-Like receptors activity and TLR ligands in patients with autoimmune thyroid diseases. Front Immunol. 2016;7:578.
Pisetsky DS. The expression of HMGB1 on microparticles released during cell activation and cell death in vitro and in vivo. Mol Med. 2014;20:158–63.
Pisitkun P, Deane JA, Difilippantonio MJ, Tarasenko T, Satterthwaite AB, Bolland S. Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science. 2006;312:1669–72.
Popovic K, Ek M, Espinosa A, Padyukov L, Harris HE, Wahren-Herlenius M, et al. Increased expression of the novel proinflammatory cytokine high mobility group box chromosomal protein 1 in skin lesions of patients with lupus erythematosus. Arthr Rheum. 2005;52:3639–45.
Pullerits R, Jonsson I-M, Verdrengh M, Bokarewa M, Andersson U, Erlandsson-Harris H, et al. High mobility group box chromosomal protein 1, a DNA binding cytokine, induces arthritis. Arthr Rheum. 2003;48:1693–700.
Radstake TRDJ, Roelofs MF, Jenniskens YM, Oppers-Walgreen B, van Riel PLCM, Barrera P, et al. Expression of toll-like receptors 2 and 4 in rheumatoid synovial tissue and regulation by proinflammatory cytokines interleukin-12 and interleukin-18 via interferon-gamma. Arthr Rheum. 2004;50:3856–65.
Rendon-Mitchell B, Ochani M, Li J, Han J, Wang H, Yang H, et al. IFN-gamma induces high mobility group box 1 protein release partly through a TNF-dependent mechanism. J Immunol. 2003;170:3890–7.
Roelofs MF, Wenink MH, Brentano F, Abdollahi-Roodsaz S, Oppers-Walgreen B, Barrera P, et al. Type I interferons might form the link between Toll-like receptor (TLR) 3/7 and TLR4-mediated synovial inflammation in rheumatoid arthritis (RA). Ann Rheum Dis. 2009;68:1486–93.
Sacre SM, Andreakos E, Kiriakidis S, Amjadi P, Lundberg A, Giddins G, et al. The Toll-like receptor adaptor proteins MyD88 and Mal/TIRAP contribute to the inflammatory and destructive processes in a human model of rheumatoid arthritis. Am J Pathol. 2007;170:518–25.
Samarpita S, Kim JY, Rasool MK, Kim KS. Investigation of toll-like receptor (TLR) 4 inhibitor TAK-242 as a new potential anti-rheumatoid arthritis drug. Arthr Res Ther. 2020;22:16.
Satoh T, Akira S. Toll-like receptor signaling and its inducible proteins. Microbiol Spectr. 2016. https://doi.org/10.1128/microbiolspec.MCHD-0040-2016.
Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–5.
Schierbeck H, Lundbäck P, Palmblad K, Klevenvall L, Erlandsson-Harris H, Andersson U, et al. Monoclonal anti-HMGB1 (high mobility group box chromosomal protein 1) antibody protection in two experimental arthritis models. Mol Med. 2011;17:1039–44.
Schierbeck H, Pullerits R, Pruunsild C, Fischer M, Holzinger D, Laestadius Å, et al. HMGB1 levels are increased in patients with juvenile idiopathic arthritis, correlate with early onset of disease, and are independent of disease duration. J Rheumatol. 2013;40:1604–13.
Seibl R, Birchler T, Loeliger S, Hossle JP, Gay RE, Saurenmann T, et al. Expression and regulation of Toll-like receptor 2 in rheumatoid arthritis synovium. Am J Pathol. 2003;162:1221–7.
Shi Y, Sandoghchian Shotorbani S, Su Z, Liu Y, Tong J, Zheng D, et al. Enhanced HMGB1 expression may contribute to Th17 cells activation in rheumatoid arthritis. Clin Dev Immunol. 2012;2012: 295081.
Subramanian S, Tus K, Li Q-Z, Wang A, Tian X-H, Zhou J, et al. A Tlr7 translocation accelerates systemic autoimmunity in murine lupus. Proc Natl Acad Sci U S A. 2006;103:9970–5.
Tamirou F, Arnaud L, Talarico R, Scirè CA, Alexander T, Amoura Z, et al. Systemic lupus erythematosus: state of the art on clinical practice guidelines. RMD Open. 2018;4: e000793.
Tang D, Kang R, Van Houten B, Zeh HJ, Billiar TR, Lotze MT. High mobility group box 1 (HMGB1) phenotypic role revealed with stress. Mol Med. 2014;20:359–62.
Taniguchi N, Kawahara K, Yone K, Hashiguchi T, Yamakuchi M, Goto M, et al. High mobility group box chromosomal protein 1 plays a role in the pathogenesis of rheumatoid arthritis as a novel cytokine. Arthr Rheum. 2003;48:971–81.
Thomas JO, Travers AA. HMG1 and 2, and related ‘architectural’ DNA-binding proteins. Trends Biochem Sci. 2001;26:167–74.
Ulevitch RJ. Therapeutics targeting the innate immune system. Nat Rev Immunol. 2004;4:512–20.
Ullah MO, Sweet MJ, Mansell A, Kellie S, Kobe B. TRIF-dependent TLR signaling, its functions in host defense and inflammation, and its potential as a therapeutic target. J Leukoc Biol. 2016;100:27–45.
Urbonaviciute V, Fürnrohr BG, Meister S, Munoz L, Heyder P, De Marchis F, et al. Induction of inflammatory and immune responses by HMGB1-nucleosome complexes: implications for the pathogenesis of SLE. J Exp Med. 2008;205:3007–18.
Van de Wouwer M, Plaisance S, De Vriese A, Waelkens E, Collen D, Persson J, et al. The lectin-like domain of thrombomodulin interferes with complement activation and protects against arthritis. J Thromb Haemost. 2006;4:1813–24.
Venereau E, De Leo F, Mezzapelle R, Careccia G, Musco G, Bianchi ME. HMGB1 as biomarker and drug target. Pharmacol Res. 2016;111:534–44.
Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285:248–51.
Wang Y, Wang L, Gong Z. Regulation of acetylation in high mobility group protein B1 cytosol translocation. DNA Cell Biol. 2019;38:491–9.
Wen L, Huang JK, Johnson BH, Reeck GR. A human placental cDNA clone that encodes nonhistone chromosomal protein HMG-1. Nucleic Acids Res. 1989;17:1197–214.
Wen L, Zhang B, Wu X, Liu R, Fan H, Han L, et al. Toll-like receptors 7 and 9 regulate the proliferation and differentiation of B cells in systemic lupus erythematosus. Front Immunol. 2023;14:1093208.
Wirestam L, Schierbeck H, Skogh T, Gunnarsson I, Ottosson L, Erlandsson-Harris H, et al. Antibodies against High Mobility Group Box protein-1 (HMGB1) versus other anti-nuclear antibody fine-specificities and disease activity in systemic lupus erythematosus. Arthr Res Ther. 2015;17:338.
Wu H-J, Sawaya H, Binstadt B, Brickelmaier M, Blasius A, Gorelik L, et al. Inflammatory arthritis can be reined in by CpG-induced DC-NK cell cross talk. J Exp Med. 2007;204:1911–22.
Wu H, Li R, Wei Z-H, Zhang X-L, Chen J-Z, Dai Q, et al. Diabetes-induced oxidative stress in endothelial progenitor cells may be sustained by a positive feedback loop involving high mobility group box-1. Oxid Med Cell Longev. 2016;2016:1943918.
Wu Q, Zhang W, Pwee K-H, Kumar PP. Cloning and characterization of rice HMGB1 gene. Gene. 2003;312:103–9.
Xue J, Suarez JS, Minaai M, Li S, Gaudino G, Pass HI, et al. HMGB1 as a therapeutic target in disease. J Cell Physiol. 2021;236:3406–19.
Yang D, Postnikov YV, Li Y, Tewary P, de la Rosa G, Wei F, et al. High-mobility group nucleosome-binding protein 1 acts as an alarmin and is critical for lipopolysaccharide-induced immune responses. J Exp Med. 2012;209:157–71.
Yarilina A, DiCarlo E, Ivashkiv LB. Suppression of the effector phase of inflammatory arthritis by double-stranded RNA is mediated by type I IFNs. J Immunol. 2007;178:2204–11.
Young NA, Wu L-C, Burd CJ, Friedman AK, Kaffenberger BH, Rajaram MVS, et al. Estrogen modulation of endosome-associated toll-like receptor 8: an IFNα-independent mechanism of sex-bias in systemic lupus erythematosus. Clin Immunol. 2014;151:66–77.
Young NA, Valiente GR, Hampton JM, Wu L-C, Burd CJ, Willis WL, et al. Estrogen-regulated STAT1 activation promotes TLR8 expression to facilitate signaling via microRNA-21 in systemic lupus erythematosus. Clin Immunol. 2017;176:12–22.
Zetterström CK, Jiang W, Wähämaa H, Ostberg T, Aveberger A-C, Schierbeck H, et al. Pivotal advance: inhibition of HMGB1 nuclear translocation as a mechanism for the anti-rheumatic effects of gold sodium thiomalate. J Leukoc Biol. 2008;83:31–8.
Zhang S, Zhong J, Yang P, Gong F, Wang C-Y. HMGB1, an innate alarmin, in the pathogenesis of type 1 diabetes. Int J Clin Exp Pathol. 2009;3:24–38.
Zhang J, Chen L, Wang F, Zou Y, Li J, Luo J, et al. Extracellular HMGB1 exacerbates autoimmune progression and recurrence of type 1 diabetes by impairing regulatory T cell stability. Diabetologia. 2020;63:987–1001.
Zhang J, Li Q, Zou Y-R, Wu S-K, Lu X-H, Li G-S, et al. HMGB1-TLR4-IL-23-IL-17A axis accelerates renal ischemia-reperfusion injury via the recruitment and migration of neutrophils. Int Immunopharmacol. 2021;94: 107433.
Zhang Z, Liu J, Mao C, Zhang S, Wang X, Dong L. SJMHE1 protects against excessive iodine-induced pyroptosis in human thyroid follicular epithelial cells through a toll-like receptor 2-dependent pathway. Int J Med Sci. 2022;19:631–9.
Zhao G-N, Jiang D-S, Li H. Interferon regulatory factors: at the crossroads of immunity, metabolism, and disease. Biochim Biophys Acta. 2015;1852:365–78.
Zhu W, Meng L, Jiang C, He X, Hou W, Xu P, et al. Arthritis is associated with T-cell-induced upregulation of Toll-like receptor 3 on synovial fibroblasts. Arthritis Res Ther. 2011;13:R103.
Zong M, Bruton JD, Grundtman C, Yang H, Li JH, Alexanderson H, et al. TLR4 as receptor for HMGB1 induced muscle dysfunction in myositis. Ann Rheum Dis. 2013;72:1390–9.
Acknowledgements
We thank Bullet Edits Limited for the linguistic editing and proofreading of the manuscript.
Authors’ statement
The authors hereby confirm that neither the manuscript nor any part of it has been published or is being considered for publication elsewhere. We acknowledge that all authors participated sufficiently in the work and take public responsibility for its content.
Funding
This study was funded by National Natural Science Foundation of China (Grant No. 82170804) and China postdoctoral Science Foundation (Grant No. 2022YFC3602300).
Author information
Authors and Affiliations
Contributions
WXR is the first author of this study. XGS is the corresponding author supervising this work. WXR drafted the manuscript. LZ provided major technical support. YS and XCW assisted in the literature review. All authors contributed to the article and approved the submitted version.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ren, W., Zhao, L., Sun, Y. et al. HMGB1 and Toll-like receptors: potential therapeutic targets in autoimmune diseases. Mol Med 29, 117 (2023). https://doi.org/10.1186/s10020-023-00717-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s10020-023-00717-3