
Abstract
Background
Neuronatin (NNAT) was recently identified as a novel mediator of estrogen receptor-positive (ER+) breast cancer cell proliferation and migration, which correlated with decreased tumorigenic potential and prolonged patient survival. However, despite these observations, the molecular and pathophysiological role(s) of NNAT in ER + breast cancer remains unclear. Based on high protein homology with phospholamban, we hypothesized that NNAT mediates the homeostasis of intracellular calcium [Ca2+]i levels and endoplasmic reticulum (EndoR) function, which is frequently disrupted in ER + breast cancer and other malignancies.
Methods
To evaluate the role of NNAT on [Ca2+]i homeostasis, we used a combination of bioinformatics, gene expression and promoter activity assays, CRISPR gene manipulation, pharmacological tools and confocal imaging to characterize the association between ROS, NNAT and calcium signaling.
Results
Our data indicate that NNAT localizes predominantly to EndoR and lysosome, and genetic manipulation of NNAT levels demonstrated that NNAT modulates [Ca2+]i influx and maintains Ca2+ homeostasis. Pharmacological inhibition of calcium channels revealed that NNAT regulates [Ca2+]i levels in breast cancer cells through the interaction with ORAI but not the TRPC signaling cascade. Furthermore, NNAT is transcriptionally regulated by NRF1, PPARα, and PPARγ and is strongly upregulated by oxidative stress via the ROS and PPAR signaling cascades.
Conclusion
Collectively, these data suggest that NNAT expression is mediated by oxidative stress and acts as a regulator of Ca2+ homeostasis to impact ER + breast cancer proliferation, thus providing a molecular link between the longstanding observation that is accumulating ROS and altered Ca2+ signaling are key oncogenic drivers of cancer.
Similar content being viewed by others


Background
Metabolic reprogramming that coincides with oxidative stress and remodeling of intracellular calcium (Ca2+) signaling are hallmarks of cancer [1]. Under normal physiological conditions, free intracellular levels of Ca2+ are tightly maintained at ~ 100-150nM until Ca2+ is released downgradient from intracellular stores (e.g., endoplasmic reticulum (EndoR), Golgi, or lysosome) or across the plasma membrane to reach intracellular levels of > 1 μm [2]. Upon triggered release by a variety of stimuli, Ca2+ acts as a second messenger to diverse physiological pathways ranging from cell growth and motility to apoptotic cell death, depending on cell state and context [3]. Due to the rewiring of intracellular Ca2+ handling pathways, tumor cells may inhibit or otherwise adjust their intracellular Ca2+ signaling and concomitant increase in reactive oxygen species (ROS) [4], which contribute to immortalized growth, motility, and survival under conditions that would otherwise trigger cell senescence or death [2]. However, despite the mounting evidence that Ca2+ and ROS are critical hallmarks of cancer, the underlying molecular mechanisms that tumor cells exploit to rewire Ca2+ and ROS signaling for disease pathogenesis remain largely unknown.
The EndoR is a major store of intracellular Ca2+ that is maintained by the activity of sarco/endoplasmic reticulum Ca2+-ATPase (SERCA2) to transport Ca2+ from the cytoplasm into the EndoR [5]. SERCA2 activity is regulated by phospholamban (PLN) binding to maintain the EndoR Ca2+ gradient [6]. Neuronatin (NNAT) is a small proteolipid (9kD) with high sequence homology to phospholamban [7, 8], which was also recently identified as a novel modifier of estrogen receptor-positive (ER+) breast cancer incidence and survival [9]. Although the role of NNAT in Ca2+ homeostasis in breast cancer is unknown, several unrelated studies have collectively implicated elevated NNAT coincided with increased resting level of Ca2+ [10, 11], which was attributed to inhibition of SERCA2 activity [12]. Thus, based on these observations [10, 12, 13] and the homology of NNAT with PLN [7, 8], we hypothesized that NNAT potentially modulates intracellular Ca2+ levels via interaction with SERCA2. However, it remains yet to be determined whether NNAT modulates other regulators of intracellular Ca2+ homeostasis, including store-operated or ion Ca2+ channels.
In addition to control of intracellular Ca2+ homeostasis, NNAT has been implicated in ROS signaling [14]. Ca2+-ROS relationship is a key factor of cell survival during apoptosis and may play a significant importance in cancer cell proliferation and tumorigenesis. For example, it is well established that oxidative stress initiates apoptotic processes through the store-operated channels and mitochondrial-EndoR calcium crosstalk [4, 15, 16]. On the other hand, ROS has been shown to regulate a number of TRPC family members [17]. Peroxisome proliferation is a receptor-mediated process that is activated by the peroxisome proliferator activated receptor (PPAR) and is commonly classified as a tumor promoter by altering gene expression and phenotypically mimicking steroid hormone receptor ligands, such as estrogen [18, 19]. Peroxisome proliferation is produced by H2O2 and may be one of the main factors mediating an imbalance between the production and degradation of reactive oxygen species in cancer cells. NNAT was recently linked to PPAR levels in adipocytes [20] and was separately colocalized to the peroxisome in the pituitary cells [21]; however, the cancer-specific role of NNAT in peroxisome mediated oxidative stress has not been evaluated.
Here, we provide the first pathophysiological insight into NNAT in Ca2+ and ROS signaling in breast cancer. Our data indicate that ROS-mediated NNAT expression strongly regulates cell cycle mechanisms and may control unchecked cell division in cancer. We explore the potential involvement of NNAT in Ca2+-ROS relationship as a major mechanism in cell apoptosis and cancer cell survival. Using fluorescent imaging, we reveal the correlation between NNAT expression and Ca2+ levels in cytoplasm and EndoR storage. In addition, pharmacological inhibitors of key players in breast cancer cells Ca2+ signaling [22], ORAI and TRPC, were used to identify a potential Ca2+ pathway in which NNAT modulates Ca2+ levels. Collectively, the findings of this study suggest that NNAT couples EndoR Ca2+ and ROS signaling with suppression of ER + breast cancer cell proliferation.
Materials and methods
Analysis of RNAseq data
RNAseq data from normal and ER+ (N = 699) cases from The Cancer Genome Atlas Breast Invasive Carcinoma (TCGA-BRCA) cohort were downloaded from the Broad GDAC Firehose (https://gdac.broadinstitute.org/). The top correlated genes with NNAT expression were determined by Spearman’s correlation (137 genes correlated; absolute correlation coefficient ≥ 0.5). Enrichment analysis of biological pathways was performed using the Ingenuity Pathway Analysis (IPA) tool. Enrichment analysis of gene ontologies (GO) and network connectivity were performed using the Search Tool for the Retrieval of Interacting Genes (STRING) database (https://string-db.org/).
RNAseq data were assessed for ER + breast cancer cell lines T47D and ZR75 that transgenically expressed NNAT (n = 3) or GFP (n = 3; control). Total RNA was extracted by Trizol followed by library preparation using Illumina’s TruSeq RNA library kit and sequencing on an Illumina HiSeq2500 (Illumina, Inc., San Diego, CA). The Trim Galore program (v0.4.1), only reads with a Phred quality score equal or higher than 20 were taken for analysis. The RSEM program function “rsem-prepare-reference” (v1.3.0) was used to extract the transcript sequences human genome (NCBI Build GRCh38.p2) followed by read alignment using the “rsem-calculate-expression” function. Differential expression analysis was performed using the Bioconductor package DESeq2 version 1.12.4 to compute log2 fold changes and false discovery rate-adjusted p values. Statistical significance of gene expression was determined as FDR < 0.05.
Cell culture
ER + breast cancer cell lines T47D and ZR75 (luminal B ER + cell lines), MCF10A normal mammary epithelial cells, and MDA-MB-231 triple-negative breast cancer cell lines originally obtained from ATCC (American Type Culture Collection) were cultured in RPMI1640 (Gibco) with 10% FBS. All cell lines were mycoplasma-negative and were cultured for < 6 months.
NNAT promoter activity
T47D or ZR75 cell lines were seeded in 24-well plates (100k cells/well). 24 h post seeding, each well was transfected using Lipofectamine 2000 or Transit-BrCa with 250 ng of the Dual Luminescence NNAT promoter plasmid (GeneCopoeia) and one of the following plasmids: pLX304 (empty vector control), NRF1, E2F1, E2F4 (250 ng of each), and Peroxisome Proliferator Activated Receptor (PPAR)/Retinoid X Receptor heterodimers: PPARα + RXR, PPARγ + RXR (125 ng of each). 24 h post transfection, media from each well was collected, and luminescence activity was measured based on the manufacturer’s protocol (GeneCopeia).
NNAT expression during oxidative stress and clofibrate treatment
T47D, ZR75, MCF10A, and MDA-MB-231 cell lines were seeded in 6 well plates at a seeding density of 500k cells/well. 24 h post seeding, each well was treated with or without hydrogen peroxide (H2O2) (specific concentrations of 100–500 µM were used on the different cell types based on how much H2O2 induced cell death) for 7 h. Similarly, each well was treated with Clofibrate for 24 h. After that, cells were collected in 1 mL of Trizol, and the total RNA was extracted. The RNA was subsequently made into cDNA for qPCR analysis of CDKN1A, CDKN2B, NNAT, and NRF1 mRNA expression (Supplemental Table 1).
Characterizing the role of the EndoR localization signal of NNAT protein on ER + breast cancer cell proliferation
The amino acid sequence of NNAT was analyzed through the Eukaryotic Linear Motif (ELM) database, which revealed a consensus EndoR localization sequence (AA75-78). To determine if the EndoR localization signal was required for NNAT function, a lentiviral construct was generated to express NNAT lacking the EndoR sequence (dER). To assess the cell growth effects, 50k cells/well of T47D or ZR75 cell lines were transduced with lentiviral expression constructs expressing the wild-type NNAT or dER NNAT variant in 24-well plates (n = 9 wells per cell line). At 48-hours post seeding, cells were counted with a Countess™ II FL Automated Cell Counter (Life Technologies).
Characterizing the subcellular localization of NNAT with confocal imaging
Cells were observed under a laser scanning confocal microscope (Nikon A1-R) for dual staining of NNAT and subcellular markers in still images and z-stacks for colocalization. T47D or ZR75 NNAT-GFP tagged cells were seeded on glass bottom culture dishes (20k cells/well; MatTek dishes). At 24 h post seeding, cells were incubated with 2 µl of 1:10,000 dilution of Hoechst 33,342 (ThermoFisher) to stain the nucleus and one of the following organelle markers for the EndoR, ER-Tracker™ Red (Invitrogen), Mitochondria, MitoTracker® Red CMXRos (Invitrogen), Golgi apparatus, CellLight™ Golgi-RFP, BacMam 2.0 (Invitrogen), or Lysosome, Cell Navigator™ Lysosome Staining Kit (ATT Bioquest), in Hank’s Balanced Salt Solution with calcium and magnesium (Gibco) for 30 min.
Confocal imaging of NNAT-dependent regulation of calcium levels in ER + breast cancer cells
T47D and ZR75 cells overexpressing GFP, NNAT, pLentiCRISPRv2 empty vector control (CRISPR control) or NNAT CRISPR Guide RNA3 (GenScript) (n = 3 plates per group; 20k cells/well) were seeded on glass bottom culture dishes. 24 h after cell seeding, Ca2+ imaging was performed with laser scanning confocal microscope (Nikon A1-R). CRISPR knockout was validated via Western blot on NNAT-FLAG cell lines. Basal intracellular calcium [Ca2+]i calculations were made based on methods previously described [23]. Briefly, cells were incubated with the fluorescent Ca2+ indicator Cal-590™ AM (AAT Bioquest) for 1 h. The cultured media was replaced with a 2 mM Ca2+ HEPES buffer solution. Basal fluorescence was recorded for at least 1 min to ensure the signal was stable, at which point 10mM of ionomycin (Sigma) was applied to detect maximum uptake of Ca2+. Once the fluorescence reached a max, 10 mM of MnCl2 (Sigma) / 2.5 mM of EDTA (AMRESCO) was applied to induce a fluorescent minimum.
To evaluate EndoR Ca2+, cells were incubated with Mag-Fluo-4 AM (AAT Bioquest) for 1 h. Media was then replaced with a 0 mM Ca2+ HEPES buffer solution. Basal fluorescence was recorded for at least 1 min to ensure the signal was stable, at which point 2 µM of thapsigargin (Sigma) was applied to the cells to induce EndoR Ca2+ release. For the inhibition of store-operated calcium entry (SOCE) related calcium channels, cells pretreated with or without 6 μm of pyr6 (ORAI1/3 inhibitor, Sigma) or 5 μm of pyr3 (TRPC3 inhibitor, Sigma) for 5 min. The selectivity between ORAI and TRPC for the pyrazole (pyr) compounds was based on the previous reports [24,25,26]. The selectivity of the available pharmacological tools for ORAI1 and ORAI3 is limited, and we cannot discriminate between those two CRAC channels known to be expressed in estrogen receptor-positive (ER+) breast cancer cell lines [27, 28]. The area under the curve was analyzed to measure the total amount of Ca2+ being released from the EndoR. Images were processed with ImageJ (v.1.51u, NIH).
Statistical analysis
All statistical analyses were performed using Sigma Plot 11.0 software. Data are presented as ± standard error of the mean (SEM). Data were tested for normality (Shapiro-Wilk) and equal variance (Levene’s homogeneity test). Paired t-test was used to detect the statistical difference between two variables for the same subject. For more than two groups of variables for the same subject, the analysis of variance (ANOVA) was used with corresponding Tukey or Dunnett multiple-comparisons adjustments.
Results
NNAT expression correlates with ROS and PPAR signal-transduction pathway in TCGA-BRCA ER + cohort
Elevated NNAT protein in ER + breast tumor biopsies was previously associated with decreased tumorigenic potential and prolonged patient survival [9], yet the underlying regulators of NNAT expression remain elusive. To identify pathways related to NNAT expression, we used publicly available expression data from the ER + breast cancer cohort (N = 699 patients) of TCGA. Ingenuity Pathway Analysis (IPA) analysis of 137 correlated genes (R ≥ 0.5) revealed a significant enrichment (p < 10− 18) of ROS and PPAR signaling pathways (Fig. 1A and Supplemental Fig. 1). Likewise, STRING network analysis revealed significant connectivity and enrichment (p < 1.0− 16) of ROS and PPAR pathways in TCGA-BRCA ER + patients with elevated NNAT expression (Supplemental Fig. 2). Finally, STRING analysis of transcriptomics data following exogenous overexpression of NNAT in ER + breast cancer cell models, T47D and ZR75, revealed enrichment of oxidative stress and apoptosis pathways (Fig. 1B; Table 1), in addition to the inhibition of cell cycle pathways reported Plasterer et al. [9]. Combined with our previous observations that NNAT suppresses ER + breast cancer cell proliferation [9], these data collectively suggested that NNAT integrates cell cycling with oxidative stress and peroxisome proliferation signaling.

NNAT expression correlated with ROS and PPAR signal-transduction pathway in TCGA-BRCA ER + cohort and ER + breast cancer cells. A Gene expression that correlates with NNAT expression in breast cancer patients. Analysis of NNAT mRNA expression in TCGA-BRCA from ER + tumors (n = 699). Data are presented as log expression values and mean values are statistical different between groups (2.2 × 10− 16), as tested by ANOVA. B The most significant modules of oxidative stress genes selected from protein–protein interaction network upregulated by NNAT overexpression in both ZR75 and T75D cell lines
NNAT expression is regulated by ROS and PPAR signaling that coincides with decreased breast cancer proliferation
In silico analysis of the NNAT promoter revealed consensus binding sites for transcriptional regulators of the cell cycle (E2F1, E2F4), oxidative stress (NRF1), and peroxisome proliferation (PPAR), prompting further investigation of transcriptional regulation of NNAT by dual-luciferase promoter assay. Briefly, the ER + breast cancer cell lines, T47D and ZR75, were co-transfected with the NNAT promoter-reporter construct, and plasmids that constitutively expressed E2F1, E2F4, NRF1, PPARα/RXR, PPARγ/RXR, or control (pLX304). At 24 h post-transfection, NNAT promoter activity was significantly elevated by E2F4, NRF1, PPARα/RXR, and PPARγ/RXR compared to the empty vector control (p < 0.001), whereas E2F1 showed no effects (Fig. 2A). Likewise, NNAT expression was significantly elevated in MCF10A, MDA-MB-231, T47D, and ZR75 cells treated with the PPAR agonist, clofibrate, or under oxidative stress conditions (H2O2 100 μm), which also coincided with the elevation of the cell cycle inhibitors, CDKN1A and CDKN2B (Fig. 2B-C). Collectively, these data confirmed that NNAT is transcriptionally elevated in ER + breast cancer cells undergoing oxidative stress and peroxisome proliferation.

NNAT mRNA expression is regulated by oxidative stress and activation of PPAR signaling cascade. A Luminescence activity of NNAT promoter activity co-transfected with E2F1, E2F4, NRF1, PPARα + RXR, PPARγ + RXR, or pLX304 control plasmid in ER + breast cancer cell lines. Data presented as mean percentage of GLuc/SEAP ratio normalized to pLX304 ± SEM (n = 9 per group; paired t-test, ***p < 0.001 vs. pLX304 control). B Evaluation of mRNA expression of NNAT, tumor suppressors genes CDKN1A and CDKN2B, and the transcription factor NRF1 responsible for cellular growth, during the exposure to H2O2. MCF10A, MDA-MB-231, T47D, and ZR75 cell lines untreated or treated with H2O2 (n = 3 per group; paired t-test, *p < 0.05, **p < 0.01, ***p < 0.001 vs. control). C NNAT mRNA expression of MCF10A, MDA-MB-231, T47D, and ZR75 cells treated with or without PPAR agonist, Clofibrate (n = 3 per group; paired t-test, *p < 0.05, **p < 0.01 vs. untreated control)
NNAT colocalizes to the endoplasmic reticulum and lysosome in ER + breast cancer
Prediction of NNAT functional sites using the Eukaryotic Linear Motif database (http://elm.eu.org/search.html) revealed a transmembrane domain and an endoplasmic reticulum (EndoR) retention sequence, suggesting that NNAT is localized to the EndoR of breast cancer cells. To test this hypothesis, the subcellular localization of NNAT-GFP in T47D and ZR75 cells was assessed by fluorescent confocal imaging using organelle-specific dyes for the EndoR, lysosome, Golgi apparatus, and mitochondria. As shown in Fig. 3A, NNAT protein (green) was largely colocalized with the EndoR and lysosome (yellow/merged images) in both T47D and ZR75 cell lines. In contrast, NNAT expression was not colocalized with markers for the Golgi apparatus or mitochondria (Supplemental Fig. 3). Based on the observation that NNAT expression correlated with oxidative stress pathways in ER + breast cancer patients, these data suggest that NNAT might mediate EndoR function(s) in the cellular response to ROS that impacts the progression of ER + breast cancer.

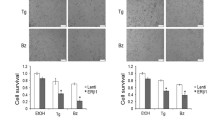
NNAT regulates intracellular calcium in ER + breast cancer cells through EndoR calcium storage. A Confocal fluorescent imaging revealed NNAT (green) colocalization with EndoR and lysosome (red and yellow merged images). B Basal Ca2+ concentration in T47D and ZR75 CRISPR knockout of NNAT cell lines (NNAT CRISPR) (n = 30 per group, paired t-test, p < 0.001 vs. control). C EndoR Ca2+ release following knockout of NNAT (n ≥ 16 per group; paired t-test, **p < 0.01 vs. respective CRISPR control). D Basal Ca2+ concentration in T47D and ZR75 cell lines overexpressing NNAT (n = 30 per group, paired t-test, p < 0.001 vs. control). E Change in proliferative capacity in T47D and ZR75 breast cancer cell lines overexpressed NNAT deletion construct (wild-type, NNAT, and dER). (n = 9 per group; one-way ANOVA tests (Tukey post hoc test) *p < 0.05, ***p < 0.001 vs. GFP).
NNAT regulates intracellular calcium in ER + breast cancer cells through EndoR calcium storage
Intracellular Ca2+ homeostasis is altered in cancer cells and involved in tumor initiation, angiogenesis, progression and metastasis [29]. To assess the impact of NNAT on Ca2+ homeostasis in breast cancer cells, intracellular free Ca2+ levels detected by Cal-590™ AM staining was measured by confocal microscopy in T47D and ZR75 cells lacking NNAT expression (Supplemental Fig. 4). Compared with non-targeting controls, depletion of endogenous NNAT by CRISPR resulted in a significant decrease in intracellular free Ca2+ levels (Fig. 3B). Likewise, the loss of NNAT significantly decreased Ca2+ in the cytoplasm and EndoR, as estimated by the presence of the SERCA inhibitor, thapsigargin (Fig. 3C). In comparison, stable overexpression of NNAT in T47D and ZR75 cells significantly increased basal cytoplasmic Ca2+ concentration (Fig. 3D), which coincided with significantly decreased tumor cell proliferation compared with control cells (Fig. 3E). Notably, proliferation of T47D and ZR75 cells was not suppressed by overexpression of NNAT lacking the putative EndoR retention signal (Fig. 3E and Supplemental Fig. 4), suggesting that the tumor suppressive role(s) of NNAT function through the EndoR. Collectively, these data, combined with our previous observations that elevated NNAT correlated with better survival of ER + breast cancer patients [9], suggest that NNAT is a key regulator of Ca2+ homeostasis that impacts ER + breast cancer cell progression.
ORAI but not TRPC3 inhibition reduces NNAT-mediated elevation in intracellular Ca2+ concentration in breast cancer cells.
Intracellular Ca2+ levels are restored by (SOCE) upon EndoR depletion, and recent studies have shown that SOCE function is required for ER + breast cancer growth and progression [30]. SOCE is mediated via STIM1 and native pore-forming unit ORAI, and we confirmed the presence of both ORAI1 and ORAI3 in ZR751 and T47D lines ( [27, 28].; https://doi.org/10.6084/m9.figshare.22779170.v1). To test the role of SOCE in NNAT-mediated regulation of intracellular Ca2+ levels, ER + breast cancer cells overexpressing NNAT or empty vector control were treated with selective blockers of ORAI (pyr6) or TRPC (pyr3) channels, followed by measurement of intracellular Ca2+ by fluorescent confocal microscopy. The NNAT overexpression significantly increases Ca2+ release from the EndoR estimated by thapsigargin application (Fig. 4A). Compared with the empty vector controls, NNAT overexpression significantly elevated intracellular Ca2+ levels in both ER + breast cancer cell lines, which was reduced by blockade of ORAI channels (Fig. 4B) but not TRPC channels (Fig. 4C). Compared with the empty vector controls, NNAT overexpression significantly elevated intracellular Ca2+ levels in both ER + breast cancer cell lines, which was reduced by blockade of ORAI channels (Fig. 4B) but not TRPC channels (Fig. 4C). Notably, although inhibition of ORAI channels significantly reduced intracellular Ca2+ in the presence of NNAT overexpression, this reduction did not approach the level of ORAI channel blockade in the parental cells (Fig. 4B). Collectively, these data suggest that NNAT modulates homeostatic levels of intracellular Ca2+ by shifting the equilibrium of Ca2+ at steady state between intracellular stores and the extracellular space.

ORAI but not TRPC3 inhibition reduces NNAT-mediated elevation in intracellular Ca2+concentration in breast cancer cells. The overexpression of NNAT promotes significantly higher Ca2+ release from EndoR of ZR75 breast cancer cells. (n = 30 per group; paired t-test, ***p < 0.001 vs. control GFP) (A). Intracellular Ca2+ levels in standard and overexpressing NNAT T47D and ZR75 cell lines in the presence or absence of ORAI (pyr6) (B) or TRPC3 (pyr3) (C) pyrazole compound inhibitors (n = 30 per group; Two-way ANOVA tests (factors: NNAT and inhibitor; Tukey post hoc test) ***p < 0.001)
Discussion
We previously demonstrated that NNAT suppresses cell proliferation and migration, which correlates with decreased tumorigenic potential and prolonged survival in ER + breast cancer patients [9]. Elsewhere, NNAT has been reported to function as an intracellular Ca2+ regulator [10, 23, 31], leading to the hypothesis that NNAT suppresses ER + breast cancer by altering Ca2+ homeostasis in response to extracellular stimuli, such as ROS. Further, ROS and Ca2 homeostasis have long been implicated in tumorigenesis and progression across many cancers [4], yet the underlying mechanisms remain incompletely understood. To our knowledge, this study was the first to provide a mechanistic link between ROS and elevated NNAT expression and the downstream effects of NNAT on the regulation of intracellular Ca2+ through the EndoR that coincided with decreased proliferation of ER + breast cancer cells.
NNAT is elevated in ER + breast cancer by ROS and PPAR signaling
The role of ROS in breast cancer is based on intracellular levels; at low levels, ROS can promote proliferation in cancer by stimulating cyclin D1 expression, triggering ERK phosphorylation and MAPK activation, which are all associated with carcinogenesis and cancer cell survival [32,33,34,35]. The accumulation of ROS may induce irreparable damages subsequently resulting in tumor cell apoptosis and is used in some cancer therapy strategies [36]. ER + breast cancer patient RNAseq data from TCGA revealed an association of ROS and PPAR signaling with NNAT expression and in silico analysis of the NNAT promotor revealed consensus binding sites for NRF1 and PPARα/PPARγ, as well as the cell cycle transcription factor, E2F4. Co-transfection of the NNAT promoter-reporter constructs with vectors expressing NRF1, PPARα, PPARγ, and E2F4 increased NNAT promoter activity, suggesting that NNAT expression is transcriptionally regulated ROS and PPAR signaling in the context of cell cycling.
Exposure of ER + breast cancer cells to the ROS inducer, H2O2, or PPAR agonist, clofibrate, increased NNAT abundance, confirming that ROS and PPAR signaling activate NNAT expression. PPAR signaling is also known to elevate ROS production and decrease cancer progression [37,38,39,40], suggesting that NNAT expression might integrate ER + breast cancer response to both stimuli with cell cycling. Further supporting this hypothesis was the observation that NNAT elevation by ROS coincided with the upregulation of the cell cycle inhibitors, CDKN1A and CDKN2B. Additionally, E2F4 is a negative regulator of the cell cycle by increasing the expression of genes that inhibit the cell cycle [41]. Collectively, the observations that NNAT is transcriptionally regulated by E2F4 and that overexpression of NNAT decreased ER + breast cancer cell proliferation suggest that cell cycle inhibition in response to ROS or PPAR signaling is potentially sensed and reinforced by NNAT. This hypothesis also fits with our previous observations that NNAT overexpression decreased the expression of genes related to cell cycle progression and activated genes related to cell cycle inhibition [9]. Increasing ROS levels has been shown to directly trigger the release of lysosomal calcium, leading to lysosomal autophagy [42]. NNAT is localized in lysosome and its expression is regulated by ROS production. These data combined lead to the hypothesis that NNAT may be a mediator of lysosomal autophagy through increased ROS production.
Role of NNAT in regulating Ca2+ homeostasis in ER + breast cancer cells
Confocal fluorescent microscopy revealed that NNAT co-localized to the EndoR and modulation of NNAT expression modify intracellular Ca2+ homeostasis in ER + breast cancer cells, fitting with reports that NNAT regulates intracellular Ca2+ in other physiological and pathophysiological settings [10, 31]. NNAT has been hypothesized to regulate SERCA2 on the EndoR membrane based on homology to phospholamban and sarcolipin [7, 43]. SERCA2 is important regulator of a EndoR Ca2+ during apoptosis and integral in maintaining Ca2+ levels to support the protein synthesis and folding machinery of the ER [44, 45]. Cancer cells were shown to inhibit Ca2+-ROS dependent apoptosis and inactivation of SERCA may be one of the mechanisms underlying this process [46]. To test the relevance of NNAT and EndoR Ca2+ storage, the SERCA2 inhibitor, thapsigargin [47], was used to induce EndoR Ca2+ release into the cytosol and indirectly measure the total amount of Ca2+ in the EndoR, which revealed that NNAT levels correspond with changes in EndoR Ca2+ storage. Combined with the observations that NNAT colocalized to the EndoR and required the EndoR localization sequence for antiproliferative effects, the data collectively suggest that NNAT suppresses ER + breast cancer by altering EndoR Ca2+ storage.
The SOCE channels are required to replenish EndoR Ca2+ levels after store depletion [48], which is mediate by activation of STIM1 proteins in the EndoR membrane that translocate to the cell membrane and activate channels comprised of either ORAI or TRPC proteins [49,50,51,52,53,54]. ORAI channels are a well-characterized regulators of the proliferation and migration of many basal breast cancer cells [28]. ORAI1 is responsible SOCE-regulated calcium entry in a variety of cell types, and ER + breast cancer cells used in this study were reported to show normal or decreased ORAI1 expression [27]. In addition, recent data suggest that ORAI3 plays crucial role in calcium regulation of ER + breast cancer cells in particular [27, 28], and is capable of complexing with ORAI1 to form heteromultimeric channel [55]. The differential expression of ORAI isoforms directly related to the redox sensitivity, since ORAI3 is lacking redox sensor [56]. TRPC channels are known to be activated by ROS [57], and TRPC3 was proposed to interact with STIM1/ORAI1 complexes promoting Ca2+ entry [58]. We hypothesize that NNAT might increase intracellular Ca2+ levels and EndoR Ca2+ storage by activating SOCE function. To test this hypothesis, we used specific inhibitors of ORAI or TRPC channels, of which only ORAI inhibition modulate Ca2+ homeostasis. Notably, ORAI channels have been shown to mediate migration, metastasis, and proliferation in breast cancer cells [59, 60], suggesting that SOCE is critical to breast cancer progression. Overall, we propose that ROS activate NNAT expression, and high levels of NNAT regulates equilibrium of EndoR Ca2+ through SERCA function, which shifts the steady state between intracellular stores and the extracellular space.
Conclusions
Collectively, our data suggest that NNAT expression is associated with ROS and PPAR signaling in ER + breast cancer patients from the TCGA cohort. NNAT was localized in the endoplasmic reticulum and lysosomes of ER + breast cancer cells. Overexpression of NNAT in ER + breast cancer cell lines suppresses proliferation and leads to elevated basal calcium levels and calcium release induced by thapsigargin, while NNAT CRISPRKO reduces intracellular calcium levels. NNAT increases intracellular Ca2+ levels and EndoR Ca2+ storage by activating SOCE function through ORAI channels.
Data Availability
The data during the current study are available from the corresponding author on reasonable request.
Abbreviations
- [Ca2+]i :
-
Intracellular calcium
- APC/C:
-
Motif anaphase- promoting complex or cyclosome
- E2F1:
-
Transcription factor E2F1
- EndoR:
-
Endoplasmic reticulum
- ER+:
-
Estrogen receptor
- GFP:
-
Green fluorescent protein
- GO:
-
Gene Ontology
- NNAT:
-
Neuronatin
- NRF1:
-
Nuclear respiratory factor 1
- ORAI1:
-
Calcium release-activated calcium channel protein 1
- PPARγ:
-
Peroxisome proliferator-activated receptor gamma type
- ROS:
-
Reactive oxygen species
- SERCA2:
-
Sarco/endoplasmic reticulum Ca2+-ATPase
- SOCE:
-
Store-operated calcium entry
- STAT5:
-
Signal transducer and activator of transcription 5
- STIM1:
-
Stromal interaction molecule 1
- STRING:
-
Search Tool for the Retrieval of Interacting Genes
- TCGA-BRCA:
-
The Cancer Genome Atlas Breast Invasive Carcinoma
- TCGA:
-
The Cancer Genome Atlas
- TRPC3:
-
Short transient receptor potential channel 3
References
Hecht F, et al. The role of oxidative stress on breast cancer development and therapy. Tumour Biol. 2016;37:4281–91.
Roderick HL, Cook SJ. Ca2 + signalling checkpoints in cancer: remodelling Ca2 + for cancer cell proliferation and survival. Nat Rev Cancer. 2008;8:361–75.
Clapham DE. Calcium signaling. Cell. 2007;131:1047–58.
Hempel N, Trebak M. Crosstalk between calcium and reactive oxygen species signaling in cancer. Cell Calcium. 2017;63:70–96.
Brini M, Carafoli E. Calcium pumps in health and disease. Physiol Rev. 2009;89:1341–78.
Sharma J, et al. Neuronatin-mediated aberrant calcium signaling and endoplasmic reticulum stress underlie neuropathology in Lafora disease. J Biol Chem. 2013;288:9482–90.
Dou D, Joseph R. Cloning of human neuronatin gene and its localization to chromosome-20q 11.2–12: the deduced protein is a novel “proteolipid’. Brain Res. 1996;723:8–22.
Dou D, Joseph R. Structure and organization of the human neuronatin gene. Genomics. 1996;33:292–7.
Plasterer C, et al. Neuronatin is a modifier of estrogen receptor-positive breast cancer incidence and outcome. Breast Cancer Res Treat. 2019;177:77–91.
Joe MK, et al. Crucial roles of neuronatin in insulin secretion and high glucose-induced apoptosis in pancreatic beta-cells. Cell Signal. 2008;20:907–15.
Lin HH, et al. Neuronatin promotes neural lineage in ESCs via ca(2+) signaling. Stem Cells. 2010;28:1950–60.
Oyang EL, Davidson BC, Lee W, Poon MM. Functional characterization of the dendritically localized mRNA neuronatin in hippocampal neurons. PLoS ONE. 2011;6:e24879.
Siu IM, et al. Coexpression of neuronatin splice forms promotes medulloblastoma growth. Neuro Oncol. 2008;10:716–24.
Li X, Thomason PA, Withers DJ, Scott J. Bio-informatics analysis of a gene co-expression module in adipose tissue containing the diet-responsive gene Nnat. BMC Syst Biol. 2010;4:175.
Gorlach A, Bertram K, Hudecova S, Krizanova O. Calcium and ROS: a mutual interplay. Redox Biol. 2015;6:260–71.
Redza-Dutordoir M, Averill-Bates DA. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim Biophys Acta. 2016;1863:2977–92.
Roedding AS, et al. Effect of oxidative stress on TRPM2 and TRPC3 channels in B lymphoblast cells in bipolar disorder. Bipolar Disord. 2012;14:151–61.
Mehendale HM. PPAR-alpha: a key to the mechanism of hepatoprotection by clofibrate. Toxicol Sci. 2000;57:187–90.
Corton JC, Anderson SP, Stauber A. Central role of peroxisome proliferator-activated receptors in the actions of peroxisome proliferators. Annu Rev Pharmacol Toxicol. 2000;40:491–518.
Yang W et al. (2019) PPAR gamma/Nnat/NF-kappaB Axis Involved in Promoting Effects of Adiponectin on Preadipocyte Differentiation. Mediators Inflamm 2019: 5618023.
Kanno N, et al. Expression studies of neuronatin in prenatal and postnatal rat pituitary. Cell Tissue Res. 2016;364:273–88.
Azimi I, Roberts-Thomson SJ, Monteith GR. Calcium influx pathways in breast cancer: opportunities for pharmacological intervention. Br J Pharmacol. 2014;171:945–60.
Lee KO, et al. Improved intracellular delivery of glucocerebrosidase mediated by the HIV-1 TAT protein transduction domain. Biochem Biophys Res Commun. 2005;337:701–7.
Schleifer H, et al. Novel pyrazole compounds for pharmacological discrimination between receptor-operated and store-operated ca(2+) entry pathways. Br J Pharmacol. 2012;167:1712–22.
Mamenko M, et al. Defective store-operated calcium entry causes partial nephrogenic diabetes insipidus. J Am Soc Nephrol. 2016;27:2035–48.
Dhande IS, et al. Stim1 polymorphism disrupts Immune Signaling and creates renal Injury in Hypertension. J Am Heart Assoc. 2020;9:e014142.
Motiani RK, Abdullaev IF, Trebak M. A novel native store-operated calcium channel encoded by Orai3: selective requirement of Orai3 versus Orai1 in estrogen receptor-positive versus estrogen receptor-negative breast cancer cells. J Biol Chem. 2010;285:19173–83.
Azimi I et al. (2019) ORAI1 and ORAI3 in breast Cancer Molecular Subtypes and the identification of ORAI3 as a Hypoxia Sensitive Gene and a Regulator of Hypoxia responses. Cancers (Basel) 11.
Cui C, Merritt R, Fu L, Pan Z. Targeting calcium signaling in cancer therapy. Acta Pharm Sin B. 2017;7:3–17.
Motiani RK, et al. Orai3 is an estrogen receptor alpha-regulated ca(2)(+) channel that promotes tumorigenesis. FASEB J. 2013;27:63–75.
Yang J, et al. Metformin induces ER stress-dependent apoptosis through miR-708-5p/NNAT pathway in prostate cancer. Oncogenesis. 2015;4:e158.
Wang P, et al. Chloride intracellular channel 1 regulates colon cancer cell migration and invasion through ROS/ERK pathway. World J Gastroenterol. 2014;20:2071–8.
Zhao W, Lu M, Zhang Q. Chloride intracellular channel 1 regulates migration and invasion in gastric cancer by triggering the ROS-mediated p38 MAPK signaling pathway. Mol Med Rep. 2015;12:8041–7.
Ranjan P, et al. Redox-dependent expression of cyclin D1 and cell proliferation by Nox1 in mouse lung epithelial cells. Antioxid Redox Signal. 2006;8:1447–59.
Shimura T, et al. Mitochondrial reactive oxygen species perturb AKT/cyclin D1 cell cycle signaling via oxidative inactivation of PP2A in lowdose irradiated human fibroblasts. Oncotarget. 2016;7:3559–70.
Liou GY, Storz P. Reactive oxygen species in cancer. Free Radic Res. 2010;44:479–96.
Kole L, Sarkar M, Deb A, Giri B. Pioglitazone, an anti-diabetic drug requires sustained MAPK activation for its anti-tumor activity in MCF7 breast cancer cells, independent of PPAR-gamma pathway. Pharmacol Rep. 2016;68:144–54.
Moon HS, et al. Alpha-eleostearic acid suppresses proliferation of MCF-7 breast cancer cells via activation of PPARgamma and inhibition of ERK 1 / 2. Cancer Sci. 2010;101:396–402.
Mehta RG, Williamson E, Patel MK, Koeffler HP. A ligand of peroxisome proliferator-activated receptor gamma, retinoids, and prevention of preneoplastic mammary lesions. J Natl Cancer Inst. 2000;92:418–23.
Srivastava N, et al. Inhibition of cancer cell proliferation by PPARgamma is mediated by a metabolic switch that increases reactive oxygen species levels. Cell Metab. 2014;20:650–61.
Bertoli C, Skotheim JM, de Bruin RA. Control of cell cycle transcription during G1 and S phases. Nat Rev Mol Cell Biol. 2013;14:518–28.
Zhang X, Yu L, Xu H. Lysosome calcium in ROS regulation of autophagy. Autophagy. 2016;12:1954–5.
Autry JM, Thomas DD, Espinoza-Fonseca LM. Sarcolipin promotes uncoupling of the SERCA ca(2+) pump by inducing a structural rearrangement in the Energy-Transduction Domain. Biochemistry. 2016;55:6083–6.
Primeau JO, Armanious GP, Fisher ME, Young HS. The SarcoEndoplasmic Reticulum Calcium ATPase. Subcell Biochem. 2018;87:229–58.
Chemaly ER, Troncone L, Lebeche D. SERCA control of cell death and survival. Cell Calcium. 2018;69:46–61.
Ruiz-Meana M, Abellan A, Miro-Casas E, Agullo E, Garcia-Dorado D. Role of sarcoplasmic reticulum in mitochondrial permeability transition and cardiomyocyte death during reperfusion. Am J Physiol Heart Circ Physiol. 2009;297:H1281–1289.
Kijima Y, Ogunbunmi E, Fleischer S. Drug action of thapsigargin on the Ca2 + pump protein of sarcoplasmic reticulum. J Biol Chem. 1991;266:22912–8.
Jardin I, Rosado JA. STIM and calcium channel complexes in cancer. Biochim Biophys Acta. 2016;1863:1418–26.
Feske S, et al. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441:179–85.
Soboloff J, Spassova MA, Dziadek MA, Gill DL. Calcium signals mediated by STIM and Orai proteins–a new paradigm in inter-organelle communication. Biochim Biophys Acta. 2006;1763:1161–8.
Putney JW. Capacitative calcium entry: from concept to molecules. Immunol Rev. 2009;231:10–22.
Park CY, et al. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell. 2009;136:876–90.
Zhou Y, et al. STIM1 gates the store-operated calcium channel ORAI1 in vitro. Nat Struct Mol Biol. 2010;17:112–6.
Zhou Y, et al. Initial activation of STIM1, the regulator of store-operated calcium entry. Nat Struct Mol Biol. 2013;20:973–81.
Holzmann C, et al. Differential Redox Regulation of ca(2)(+) signaling and viability in normal and malignant prostate cells. Biophys J. 2015;109:1410–9.
Bogeski I, et al. Differential redox regulation of ORAI ion channels: a mechanism to tune cellular calcium signaling. Sci Signal. 2010;3:ra24.
Staruschenko A, Ma R, Palygin O, Dryer SE. Ion channels and channelopathies in glomeruli. Physiol Rev. 2023;103:787–854.
Liao Y, et al. Functional interactions among Orai1, TRPCs, and STIM1 suggest a STIM-regulated heteromeric Orai/TRPC model for SOCE/Icrac channels. Proc Natl Acad Sci U S A. 2008;105:2895–900.
Yang S, Zhang JJ, Huang XY. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell. 2009;15:124–34.
Isa L, Jean G, Silvani A, Arosio P, Taccagni GL. Evaluation of iron stores in patients with alcoholic liver disease: role of red cell ferritin. Acta Haematol. 1988;80:85–8.
Sachini N, et al. Promyelocytic leukemia protein (PML) controls breast cancer cell proliferation by modulating Forkhead transcription factors. Mol Oncol. 2019;13:1369–87.
Pickard MR, et al. Dysregulated expression of Fau and MELK is associated with poor prognosis in breast cancer. Breast Cancer Res. 2009;11:R60.
Stanley RF, et al. A myeloid tumor suppressor role for NOL3. J Exp Med. 2017;214:753–71.
Tavazoie MF, et al. LXR/ApoE activation restricts Innate Immune suppression in Cancer. Cell. 2018;172:825–840e818.
Griess B, Tom E, Domann F, Teoh-Fitzgerald M. Extracellular superoxide dismutase and its role in cancer. Free Radic Biol Med. 2017;112:464–79.
Zhu L, et al. Pan-Cancer Analysis of the Mitophagy-Related protein PINK1 as a biomarker for the immunological and prognostic role. Front Oncol. 2020;10:569887.
Ellinger J, et al. Systematic expression analysis of mitochondrial complex I identifies NDUFS1 as a Biomarker in Clear-Cell Renal-Cell Carcinoma. Clin Genitourin Cancer. 2017;15:e551–62.
Uhlen M et al. (2017) A pathology atlas of the human cancer transcriptome. Science 357.
Alsubait A, Aldossary W, Rashid M, Algamdi A, Alrfaei BM. CYP1B1 gene: implications in glaucoma and cancer. J Cancer. 2020;11:4652–61.
Saelee P, Pongtheerat T, Sophonnithiprasert T. Reduced expression of GPX3 in breast Cancer patients in correlation with clinical significance. Glob Med Genet. 2020;7:87–91.
Du J, et al. PDK1 promotes tumor growth and metastasis in a spontaneous breast cancer model. Oncogene. 2016;35:3314–23.
Men X, et al. Transcriptome profiling identified differentially expressed genes and pathways associated with tamoxifen resistance in human breast cancer. Oncotarget. 2018;9:4074–89.
Moraes LA, Ampomah PB, Lim LHK. Annexin A1 in inflammation and breast cancer: a new axis in the tumor microenvironment. Cell Adh Migr. 2018;12:417–23.
Jankovic-Karasoulos T, et al. Elevated levels of tumour apolipoprotein D independently predict poor outcome in breast cancer patients. Histopathology. 2020;76:976–87.
Chen Y, et al. NCF1/2/4 are prognostic biomarkers related to the Immune infiltration of kidney renal clear cell carcinoma. Biomed Res Int. 2021;2021:5954036.
Dawson SJ, et al. BCL2 in breast cancer: a favourable prognostic marker across molecular subtypes and independent of adjuvant therapy received. Br J Cancer. 2010;103:668–75.
Shi X, Singh S, Lin E, Li H. Chimeric RNAs in cancer. Adv Clin Chem. 2021;100:1–35.
Zhang L, et al. Novel read-through fusion transcript Bcl2l2-Pabpn1 in glioblastoma cells. J Cell Mol Med. 2022;26:4686–97.
Tajnik M, Strazisar M, Volavsek M, Bostjancic E, Glavac D. BBC3 is down-regulated with increased tumor size independently of p53 expression in head and neck cancer. Cancer Biomark. 2012;11:197–208.
Acknowledgements
Authors thank Dr. Mykola Mamenko (Department of Physiology, Augusta University) for providing his expertise on ORAI and TRPC channels pharmacology and Dr. Melinda R. Dwinell (Department of Physiology, Medical College of Wisconsin) for critical help during the course of these studies.
Funding
Research was supported by National Institutes of Health Grants R01 DK129227, R01 DK126720 (to OP), MUSC COMETS PPG (to OP), and SC SmartState Centers of Excellence (to OP).
Author information
Authors and Affiliations
Contributions
Conception and design of the study (CP, MF), acquisition of data (CP, MF, OP), analysis and interpretation of data (CP, MF, OP, MS), drafting the article (MF, OP, MS). All authors approved final version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
10020_2023_673_MOESM2_ESM.docx
Supplementary Material 2 - Supplemental Figure 1. NNAT expression and genetic correlation in ER+ breast cancer TGCA RNAseq data.
10020_2023_673_MOESM4_ESM.docx
Supplementary Material 4 - Supplemental Figure 3. Confocal imaging for NNAT colocalization with Golgi apparatus and mitochondria.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Plasterer, C., Semenikhina, M., Tsaih, SW. et al. NNAT is a novel mediator of oxidative stress that suppresses ER + breast cancer. Mol Med 29, 87 (2023). https://doi.org/10.1186/s10020-023-00673-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s10020-023-00673-y