
Abstract
Background
It is known that increased gastrin concentration is negatively correlated with cardiovascular mortality, and plasma gastrin levels are increased in patients after myocardial infarction (MI). However, whether gastrin can play a protective role in MI remains unknown.
Methods
Adult C57BL/6 mice were subjected to ligation of the left anterior descending coronary artery (LAD) and subcutaneous infusion of gastrin (120 μg/Kg body weight/day, 100 μL in the pump) for 28 days after MI. Plasma gastrin concentrations were measured through an ELISA detection kit. Mice were analyzed by echocardiography after surgery. CD31 and VEGF expression were quantified using immunofluorescence staining or/and western blot to assess the angiogenesis in peri-infarct myocardium. Capillary-like tube formation and cell migration assays were performed to detect gastrin-induced angiogenesis.
Results
We found that gastrin administration significantly ameliorated MI-induced cardiac dysfunction and reduced fibrosis at 28 days in post-MI hearts. Additionally, gastrin treatment significantly decreased cardiomyocyte apoptosis and increased angiogenesis in the infarct border zone without influencing cardiomyocyte proliferation. In vitro results revealed that gastrin up-regulated the PI3K/Akt/vascular endothelial growth factor (VEGF) signaling pathway and promoted migration and tube formation of human coronary artery endothelial cells (HCAECs). Cholecystokinin 2 receptor (CCK2R) mediated the protective effect of gastrin since the CCK2R blocker CI988 attenuated the gastrin-mediated angiogenesis and cardiac function protection.
Conclusion
Our data revealed that gastrin promoted angiogenesis and improved cardiac function in post-MI mice, highlighting its potential as a therapeutic target candidate.
Similar content being viewed by others


Background
Myocardial infarction (MI) is the leading cause of mortality for cardiovascular disease. Despite several therapeutic strategies that successfully restored the epicardial coronary flow, such as thrombolysis, surgical bypass grafting, and percutaneous coronary intervention (PCI), some patients remain ineligible for those therapies since microvasculature dysfunction promotes inefficient reperfusion of the entire myocardium (Laan et al. 2009). Hence, de novo formation of microvessel, also called angiogenesis, can rescue ischemic myocardium at the early stage after MI occurrence and is also essential for long-term cardiac remodeling to prevent the transition to heart failure (Cochain et al. 2013).
Gastrin is a peptide hormone secreted mainly by G-cells in the stomach after a meal (Varro and Ardill 2003). The physiological function of gastrin in regulating gastric acid secretion is currently well established. However, there is much more to discover concerning the biology of gastrin than is indicated. A study has shown that gastrin stimulated islet-cell growth and insulin secretion, which is proposed as a potential drug therapy for diabetes mellitus (Rehfeld 2019). Besides, gastrin plays an essential role in maintaining normal blood pressure (Chen et al. 2013) and protecting the kidney function against hypertensive injury (Gu et al. 2021). Diabetes and hypertension are important causes of coronary atherosclerotic heart disease. A previous study demonstrated that increased gastrin levels were negatively correlated with cardiovascular mortality risk (Goetze et al. 2016). Clinical data also revealed that serum gastrin levels were increased in patients with MI (Lapidus 1985; Tansey et al. 1981) and were associated with lower serum levels of cardiac troponin I in patients with unstable angina pectoris undergoing PCI (Yang et al. 2018). However, it is not clear if the increase in serum gastrin level is the result of MI or an attempt of the body to protect itself against cardiomyocyte damage after MI occurred. Additionally, gastrin is expressed in the heart and blood vessels (Gersl et al. 1981; Grossini et al. 2011). Two different studies reported that gastrin promoted a transient increase of Ca2+ (Grossini et al. 2013) and up-regulated nitric oxide production in porcine coronary artery endothelial cells (Grossini et al. 2012). Moreover, another study showed that gastrin increased coronary blood flow and myocardial contractility, dose-dependently, through intracoronary infusion in anesthetized pigs (Grossini et al. 2011). Besides, a work conducted by Yang et al. proposed that gastrin exerted a protective role against myocardial ischemia/reperfusion injury (IRI) in rats (Yang et al. 2018). Therefore, it is possible that gastrin may exert a protective effect against myocardial infarction.
An in vitro study revealed that gastrin induced chemotactic effects on endothelial cell migration and increased the tubulogenesis levels in endothelial cells, suggesting a potential pro-angiogenic role for this peptide (Clarke et al. 2006). Indeed, several studies found that gastrin induced pro-angiogenic effects in tumors (Bertrand et al. 2010; Lefranc et al. 2004). However, whether gastrin promotes cardiac angiogenesis after MI is still unclear. In this study, we aimed to investigate whether gastrin can improve the cardiac function after MI by increasing angiogenesis in the infarcted myocardium and to further explore its relative mechanism.
Materials and methods
MI mice model
C57/BL6 male mice (eight weeks old) were induced to myocardial infarction by the ligation of the left anterior descending (LAD) coronary artery as previously described (Li et al. 2020). Under isoflurane anesthesia, the LAD coronary arteries were ligated 2–3 mm far from the origin with a 7-0 silk suture. For the sham-operated control group, mice were exposed to all surgical procedures except the ligation of the LAD. Alzet osmotic minipumps (Model 1004, Durect Corporation, CA) were subcutaneously implanted in the mice dorsal region under isoflurane anesthesia. The minipumps were loaded with saline (control) or the chemicals and infused at a 2.64 μL/days rate for 28 days, according to the previous study (Huang et al. 2010). The infusate contained the saline vehicle (control, 2.64 μL/day, 100 μL in the pump), gastrin I (120 μg/Kg body weight/day, 100 μL in the pump, Cat. No. G1276, Sigma-Aldrich) (Gu et al. 2021; Rooman and Bouwens 2004), or/and the CCK2R inhibitor, CI988 (0.5 mg/Kg body weight/day, 100 μL in the pump, Cat. No. sc-205244A, Santa Cruz) (Xu et al. 1992). All procedures were approved by the Animal Use Subcommittee of Southwest Jiaotong University.
Echocardiography
Echocardiography was performed using an echocardiographic monitor (Vivid9, GE Healthcare, WI), and 2% isoflurane was used to keep the mice under sedation. Hearts were observed in the short-axis with M-mode on the parasternal long. Diastolic left ventricle internal diameter (LVIDd) and systolic left ventricle internal diameter (LVIDs) were measured to find cardiac morphologic and functional changes. The percentage of fractional shortening (%FS) was calculated as (LVIDd– LVIDs)/LVIDd × 100. Also, the percentage of left ventricle ejection fraction (%EF) was calculated as ((LVIDd)3 -(LVIDs)3) (LVIDd)3 × 100. All of the echocardiography measurements were performed blindly.
Plasma gastrin measurements
After an overnight fast, mice blood samples were collected from the orbital venous while under isoflurane anesthesia on the day of myocardial infarction (day 1), as well as on days 3, 5, 7, 9, 11, and 13. Blood samples were contained in EDTA-coated tubes then centrifuged at 1000 rpm for 5 min and plasma stored at −80 ℃ until assayed. Plasma gastrin concentration was determined by using a mice gastrin-17 ELISA detection kit (Cat. No. ml569213-J, mlbio, Shanghai) according to the manufacturer’s standard procedures.
Histology
Mice heart tissues were fixed in 4% paraformaldehyde solution overnight at 25 °C and then undergoing paraffin embed and section. In order to analyze the myocardial fibrotic area after the myocardial infarction procedure with or without drug infusion, paraffin sections were cut through the entire ventricle from apex to base into serial sections with 0.5-mm intervals. After that, Masson’s trichrome staining (Cat. No. G1345, Solarbio, Beijing) was performed according to the manufacturer’s protocol following the dewaxing procedure.
Apoptosis detection
An in situ apoptotic cell death detection kit based on the terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL, Cat. No. C1088, Beyotime, Shanghai, China) was used to detect apoptosis, according to the manufacturer’s standard procedures. Briefly, after the dewaxing procedure, cardiomyocytes were labeled with anti-cardiac troponin T antibody (cTnT, 1:100, Cat. No. MA5-12960, Invitrogen, MA) at 4 °C for 10 h (hrs), the nuclei were detected through 4’,6-diamidino-2-phenylindole (DAPI, Cat. No. C0065, Solarbio) staining. The TUNEL-positive cardiomyocytes were detected in 5 randomly selected nonadjacent images of peri-infarcted areas. The index of apoptosis was presented as the TUNEL-positive cardiomyocytes per 1 × 106 Nuclei.
Immunofluorescence staining
Paraffin-embedded hearts from all groups of mice were cut into 5 µm thick sections and then deparaffinized and dehydrated. After treatment with immunostaining blocking solution (Cat. No. P0102, Beyotime, Shanghai) for 1 h at room temperature, the sections were incubated with anti-cluster of differentiation 31 (CD31) primary antibody (1:100, Cat. No. GB11063-2, Servicebio), anti Ki67 antibody (1:100, Cat. No. 9129S, Cell Signaling Technology), and anti-cTnT antibody (1:100) for 10 h at 4 °C, subsequently washed three times with phosphate buffer saline and incubated with secondary antibodies conjugated with Alexa Fluor 488 or 555 (1:200, Cat. No. A-11001, A-21428, A-21422, Invitrogen, MA) for 2 h at 37 °C. DAPI was used for nuclear staining. The slides were mounted in an antifade mounting medium. Angiogenesis was detected by calculating the capillary density per field in the infarct border zone as previously reported. In order to quantify the number of Ki67+ (cell cycle marker) cardiomyocytes, the results were acquired from sections of the heart with at least five different fields and positions. In all cell-counting experiments, visual fields were randomized to reduce counting bias.
Tube formation assay
Human coronary artery endothelial cells (HCAECs, Pricells, Wuhan, China) were cultured in primary endothelial cell basal medium (RPMI1640, Cat. No. A4192301, Gibco) supplemented with 10% fetal bovine serum (FBS, Cat. No.10099–141, Gibco), in a humidified incubator at 37 °C with 95% air/5% CO2 atmosphere as we previously described (Fu et al. 2016). All groups of cells were starved in a basal medium containing 1% FBS for 12 h before performing the experiments. The 24-well plate, pipette tips, and matrix gels were pre-cooled before tube formation assay. 300 μL Matrigel matrix (Cat. No. 354234, Corning, USA) was added to a 24-well plate and put into an incubator for gel formation. Subsequent to gel solidification, 500 μL of HCAECs suspension (2 × 105/mL) was added to each well (Song et al. 2020). The plate was incubated at 37 °C. Tubule formation was observed using a Nikon inverted microscope (Tokyo, Japan) and determined by measuring branch number and length using the National Institutes of Health ImageJ software 6 h after treatment with gastrin (1 nM to 100 nM) or/and CI988 (100 nM). ImageJ software was used to analyze the tube lengths in 5 randomly selected visual fields.
Cell migration assay
Transwell migration assay was performed as previously described. Briefly, cells were slowly put into the upper well of the transwell migration chamber (Cat. No. MCEP24H48, Millipore, Germany). After 24 h of treatment with gastrin (100 nM) or/and CY294002 (1000 nM, Cat. No. S1737, Beyotime, Shanghai, China) (Liu et al. 2016), the non-migrating cells were removed with a cotton swab, and the migrating cells on the underside of the membrane were stained with crystal violet. Cell migration was also analyzed using a scratch wound migration assay. Briefly, HCAECs were seeded into a 12-well plate in basal medium containing 10% FBS for 48 h. Culture media were then changed to basal medium containing 1% FBS for 12 h. Two parallel scratches were made by using a 10 µL pipette tip. Subsequently, HCAECs were treated with gastrin (100 nM) or/and CY294002 (1000 nM) for 8 h. The scratch was observed using a Nikon inverted microscope, as mentioned before. In order to calculating cell migration distance, HCAECs that migrated into the scratched area from the edge area were photographed and then measured in each image to yield an average value compared with the time zero point.
Western blot
Protein was extracted from HCAECs after treatment with gastrin (1 nM to 100 nM), or/and CI988 (100 nM), or/and LY294002 (1000 nM) for 24 h. Western blotting was carried out as we previously described. Cells and heart tissue lysates containing 100 μg of protein were separated by SDS-PAGE and then electrophoretically transferred onto NC membranes (Bio-Rad, CA). After treatment with skimmed milk to blocking non-specific bands for 3 h, the blots were probed with the anti-phospho protein kinase B (Akt) antibody (1:800, Cat. No. 4060 T, Cell Signaling Technology), anti-Akt antibody (1:800, Cat. No. 9272S, Cell Signaling Technology), anti-phospho phosphatidylinositol 3 kinase (PI3K) antibody (1:800, Cat. No. 17366S, Cell Signaling Technology), anti-PI3K antibody (1:800, Cat. No. 4257S, Cell Signaling Technology), anti-VEGFA antibody (1:1000, Cat. No. 66828-1-Ig, Proteintech, Wuhan, China), anti-CD31 primary antibody (1:500, Cat. No.11265-1-AP, Proteintech), anti-Caspase 3 antibody (1:800, Cat. No. 19677-1-AP, Proteintech), anti-cleaved Caspase 3 antibody (1:500, Cat. No. 9661S, Cell Signaling Technology), and GAPDH (1:500, Cat. No. 60004-1-Ig, Proteintech) at 4 °C overnight. Membranes were washed three times with western washing buffer (TBST) and incubated with the corresponding secondary antibodies (Li-Cor, IRDye 800CW, 1:10000) for 2 h at room temperature. After that, NC membranes were washed three times using TBST again. Bound complexes were detected using the Odyssey Infrared Imaging System (Li-Cor Biosciences). Images were analyzed using the Odyssey Application Software to obtain the integrated intensities.
Statistical analysis
Data were expressed as means ± standard deviation. The SPSS 20.0 software was used for general statistical analysis. Comparison within groups was made by repeated-measures ANOVA (or paired t-test when only two groups were compared), and the differences between groups were evaluated by factorial ANOVA with the Holm-Sidak post hoc test. Log-rank (Mantel-Cox) test was used for statistical survival analysis. A two-sided P-value less than 0.05 was considered statistically significant. Histograms were plotted using the GraphPad Prism 5.01 software.
Results
The plasma level of gastrin is increased in mice after MI
We induced MI by permanent ligation of the LAD in mice. To evaluate the changes in gastrin levels after MI, we detected the plasma concentration of gastrin in mice every two days for 13 days. Plasma concentration of gastrin was slightly increased in days 3 and 5, compared to the sham-operated group, in post-MI mice, but there were no significant differences. However, after 7 days, the plasma concentration of gastrin was significantly increased in the post-MI compared sham-operated group (Fig. 1). These data revealed that the plasma level of gastrin was increased in mice after MI.

Comparison of plasma concentrations of gastrin at different days (day 1, 3, 5, 7, 11, and 13) in the myocardial infarction (MI) mice and in sham-operated mice. Data are expressed as the mean ± SD. n = 8. *P < 0.05, vs. sham-operated mice
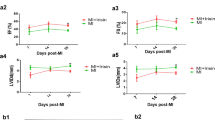
Gastrin improves cardiac function and survival rate in post-MI mice
Ventricular function was evaluated in all groups to assess the gastrin effects on cardiac hemodynamics. After MI injury, mice were submitted to subcutaneous infusion of gastrin for 28 days. Cardiac function was measured by echocardiography at 7, 14, and 28 days after myocardial infarction (Fig. 2a1–a5). Our results suggested that compared with sham-operated mice, post-MI mice had increased ventricular dilation and decreased cardiac function. At 7 and 14 days, gastrin slightly increased LVEF and LVFS and slightly decreased LVIDd and LVIDs, but there were no statistical differences compared with the MI group. However, on day 28 post-MI, gastrin improved LVEF and LVFS compared with the control MI mice. Besides, gastrin-treated mice had statistically smaller left ventricle size than MI mice, as indicated by the significant reduction of LVIDd and LVIDs. Meanwhile, survival rates of gastrin-treated mice were lower than the control mice subjected to MI (Fig. 2b). These data demonstrated that gastrin improved cardiac function in post-MI hearts.

Gastrin ameliorates cardiac function and survival rate in myocardial infarction mice. a1 Representative M-mode images of hearts with the sham operation or myocardial infarction 28 days after the saline vehicle and gastrin infusion. a2–5 Represent left ventricle ejection fraction (EF), fractional shortening (FS), diastolic left ventricle internal diameter (LVIDd) and systolic left ventricle internal diameter (LVIDs) after MI. n = 10. *P < 0.05, vs. MI mice; #P < 0.05, vs. MI + Gastrin. b Gastrin improves Kaplan–Meier survival rate of mice subjected to MI. n = 10. Log-rank test P < 0.05
Gastrin decreases cardiomyocyte fibrosis and apoptosis in post-MI hearts
Masson’s trichrome staining was performed to evaluated fibrosis 4 weeks post-MI. The result indicated that gastrin-treated mice had decreased fibrosis level in the peri-infarct border zone compared with the control MI group (Fig. 3a1–a3). Cardiomyocytes are susceptible to apoptosis during MI (Matsumoto et al. 2019). Gastrin has been reported to protect cells from apoptosis (Ramamoorthy et al. 2004) and promote cell proliferation (Duckworth et al. 2013). Next, we evaluated cardiomyocyte apoptosis and proliferation levels in the hearts. Apoptosis was detected in the peri-infarct border zone collected two days post-MI with TUNEL staining (Li et al. 2020). Cardiomyocytes were identified by cTnT staining. We found that the gastrin-treated group had decreased TUNEL-positive cardiomyocytes (Fig. 3b1–b2). Moreover, western blot data revealed that the expression of cleaved caspase-3 was also significantly decreased in gastrin-treated MI mice than control MI mice. These data suggested that gastrin protected the myocardium partially by reducing cardiomyocyte apoptosis. Cardiomyocyte proliferation was determined by Ki67 staining. Our study found that the rate of Ki67 positive cardiomyocytes was comparable between the gastrin-treated MI mice and the control mice (Fig. 3c1–c2), indicating that gastrin does not influence cardiomyocyte proliferation ability in the post-MI heart.

Gastrin decreases cardiomyocyte apoptosis and fibrosis after myocardial infarction. a Representative images (a1) and quantification (a2) of the fibrotic area in infarct border zone by Masson trichrome staining at 28 days post-MI. b Representative images (b1) and quantification (b2) of terminal deoxynucleotidyl transferase dUTP nick-end labeling–positive (TUNEL+) cardiomyocytes in infarct border zone at 2 days after MI. TUNEL+ nuclei are stained green, cardiac troponin T antibody (cTnT) is red, and 4'-6-diamidino-2-phenylindole (DAPI) is blue. b3 Western blot was used to measure the protein levels of Caspase 3 in the infarct border zone at 2 days after MI. Results are expressed as the ratio of cleaved caspased 3 to total caspase 3. c, Representative images (c1) and quantification (c2) of cardiomyocyte proliferation 28 days after myocardial infarction, through Ki67 immunostaining. Cardiac troponin T antibody (cTnT) is green, Ki67 is red, and 4'-6-diamidino-2-phenylindole (DAPI) is blue. a and c Scale bar = 50 µm. b Scale bar = 20 µm. n = 8. *P < 0.05, vs. MI mice. P = NS means no significant difference
Gastrin promotes angiogenesis after myocardial infarction
Previous studies have shown that gastrin induced chemotactic effects on endothelial cell migration and increased the tubulogenesis levels of the endothelial cells (Clarke et al. 2006). Insufficient post-MI angiogenesis has been regarded as a non-negligible event that promotes heart failure development. In contrast, stimulating angiogenesis ameliorates cardiac remodeling after myocardial infarction (Laan et al. 2009). CD31 plays an important role in endothelial cell intercellular junctions and is widely used as a marker for angiogenesis quantification (Feng et al. 2020; Ho et al. 2012). We, therefore, assessed the de novo formation of microvessel in peri-infarct myocardium at 14 days post-MI through CD31 staining and expression. Both immunofluorescence staining and Western blot showed that CD31 expression in the peri-infarct border zone were increased in gastrin-treated mice (Fig. 4a1–a2). The in vitro experiment also revealed that gastrin (1 nM–100 nM) promoted tube formation of HCAECs in a concentration-dependent manner (Fig. 4b1–b2). These results suggested that gastrin improves angiogenesis in the post-MI heart.

Gastrin induces angiogenesis after myocardial infarction. a1 Immunofluorescence staining with anti-cluster of differentiation 31 (CD31) antibody showed the microvessel densities in the infarcted border zone 14 days after MI. DAPI is blue, and CD31 is red. Scale bar = 50 µm. a2 Western blot was used to measure the protein levels of CD31 in the infarcted border zone of the heart 14 days after MI. Results are expressed as the ratio of CD31 to GAPDH. n = 8. *P < 0.05, vs. MI mice. b Human coronary artery endothelial cells (HCAECs) were incubated with gastrin at the indicated concentrations. Representative images (b1) and quantification (b2) of tube formation assay with HCAECs. Scale bar = 50 µm. n = 4. *P < 0.05, vs. Control
Gastrin induces angiogenesis via the PI3K/Akt/VEGF signaling pathway
Vascular endothelial growth factor (VEGF), a key factor known to stimulate angiogenesis, attenuates cardiac dysfunction after MI in animal models (Cui et al. 2018; Rasanen et al. 2021). Previous studies reported that gastrin induced angiogenesis via stimulating VEGF signaling pathway (Bertrand et al. 2010). Liu et al. found that gastrin attenuated kidney ischemia/reperfusion injury through PI3K/Akt mediated anti-apoptosis signaling pathway (Liu et al. 2020). PI3K/Akt signaling also plays a critical role in angiogenesis (Liang et al. 2019; Sun et al. 2014). Therefore, we measured the phosphorylation status of PI3K, Akt, and VEGFA protein levels. As shown in Fig. 5a–c, after 24 h of treatment, gastrin (1 nM–100 nM) increased PI3K and Akt (Ser473) phosphorylation and VEGFA expression in a concentration-dependent manner in HCAECs but had no effect on total PI3K and Akt. Besides, gastrin (100 nM) treatment increased Akt phosphorylation and VEGFA protein level, which can be blocked by PI3K inhibitor, LY294002 (Fig. 5d–e). Endothelial cell migration is essential for angiogenesis. In our transwell migration study, gastrin (100 nM) treatment significantly boosted HCAECs migration, which was partially inhibited by LY294002 (Fig. 5f1–f2). Besides, the scratch wound healing result also revealed that HCAECs migration was statistically increased by gastrin (100 nM), which was partially repressed by LY294002 (Fig. 5g1–g2). These results suggested that the gastrin-induced angiogenesis, at least partly, was dependent on PI3K/Akt/VEGF signal pathway.

PI3K/Akt/VEGF signaling pathway is involved in the pro-angiogenic effect of gastrin. Endothelial cells were incubated with gastrin at the indicated concentrations for 24 h. VEGFA (a), phospho-PI3K (b), and phospho-Akt (c) results were normalized by GAPDH, total PI3K, and Akt, respectively. Endothelial cells were treated with gastrin (100 nM, 24 h) in the presence or absence of a PI3K inhibitor, LY294003 (1000 nM). Phospho-Akt (d) and VEGFA (e) were normalized by GAPDH and total Akt, respectively. n = 4. *P < 0.05, vs. Control group; #P < 0.05, vs gastrin-treated group. f Representative images and quantification of endothelial cell migration detected by transwell migration assay. g, Representative images and quantification of endothelial cell migration detected by scratch wound-healing assay. f and g scale bar = 50 µm. n = 4. *P < 0.05, vs. Control group; #P < 0.05, vs. gastrin-treated group. VEGF, vascular endothelial growth factor; PI3K, phosphatidylinositol 3 kinase; Akt, protein kinase B; DU, density units; p, phospho
Gastrin, via CCK2R, exerts a protective role against myocardial infarction
Gastrin and cholecystokinin (CCK) share the same receptor (Guilloteau et al. 2006). Cholecystokinin receptor (CCKR) has 2 subtypes, type1 and type 2. The expression of CCK2R is much more than CCK1R and has higher affinity for gastrin than CCK1R in the coronary arteries (Dockray et al. 2005). In the in vitro study, we found that the gastrin-mediated upregulation of VEGFA was blocked by CI988 (100 nM), a specific CCK2R inhibitor (Fig. 6a). The pro-angiogenic effect of gastrin was also inhibited in the presence of CI988 (Fig. 6b). Moreover, gastrin up-regulated CD31 expression in the border zone (Fig. 6c1–c2), and the improved cardiac function (Fig. 6d1–d2) was also abolished in the presence of CI988. These data indicated that CCK2R mediated the protective role of gastrin against MI.

CCK2R role in the protective effects of gastrin against myocardial infarction. Endothelial cells were treated with gastrin (100 nM) with or without the CCK2R inhibitor, CI988 (100 nM). a Western blot was used to detect the protein levels of vascular endothelial growth factor (VEGFA). Results are expressed as the ratio of VEGFA to GAPDH. n = 4. *P < 0.05, vs. Control; #P < 0.05, vs. gastrin-treated group. b Representative images and quantification of endothelial cell tube formation assay in vitro. Scale bar = 50 µm. n = 4. *P < 0.05, vs. Control; #P < 0.05, vs gastrin-treated group. Myocardial infarction mice were infused with gastrin or/and CI988. c1 Immunofluorescence staining with anti-cluster of differentiation 31 (CD31) antibody showed the microvessel densities in the infarcted border zone 14 days after MI. DAPI is blue, and CD31 is red. Scale bar = 50 µm. c2 Western blot was used to detect the protein levels of CD31 in the peri-infarct area of the heart 14 days after MI. Results are expressed as the ratio of CD31 to GAPDH. n = 8. *P < 0.05, vs. MI group; #P < 0.05, vs gastrin-treated MI mice. d1 Representative M-mode images of hearts with MI 28 days after gastrin or/and CI988 infusion. d2–3 Left ventricle ejection fraction (EF), fractional shortening (FS) 7, 14, and 28 days after MI. n = 8. *P < 0.05, vs. MI + Gastrin + CI988
Discussion
Studies on several gastrointestinal hormones have demonstrated that there may exist a gastro-heart axis that protects cardiovascular function. For instance, glucagon-like peptide 1 (GLP-1) has been reported to protect myocardial ischemia/reperfusion injury (Andrikou et al. 2019; Bonaventura et al. 2019). A study also found that ghrelin may be essential in ameliorating the efficacy of mesenchymal stem cell-based therapy for ischemic heart disease (Han et al. 2015). Besides, cholecystokinin was proposed to improve cardiac function in endotoxic shock rats (Saia et al. 2013). Gastrin, as one of the main gastrointestinal hormones, together with cholecystokinin, was among the first gastrointestinal hormones discovered. The two peptides were structurally related and activated the same receptor (the CCK2 receptor) (Zeng et al. 2020). Serum gastrin level markedly increased after a meal, about 20-fold more than cholecystokinin (Rehfeld et al. 2007). A previous study reported that the serum concentration of gastrin was significantly increased after myocardial infarction (Tansey et al. 1981). Besides, high serum gastrin level was reported to be an independent predictor of MI (Lapidus 1985). Similarly, our result also revealed that the plasma level of gastrin was increased in mice after MI. However, the pathophysiological significance of this phenomenon remains to be elucidated.
A former study found that increased gastrin concentration might represent an adaptive response to hypoxic conditions (Laval et al. 2015). Indeed, Yang et al. proposed that gastrin protects against myocardial IRI through activation of RISK (Reperfusion Injury Salvage Kinase) and SAFE (Survivor Activating Factor Enhancement) pathways (Yang et al. 2018). Also, gastrin was reported to protect the brain from ischemia-induced dysfunction in stroke-prone spontaneously hypertensive rats (Yasui and Kawasaki 1995). More recently, gastrin attenuated renal IRI through anti-apoptosis signaling (Liu et al. 2020). Our data showed that gastrin treatment for 28 days improved cardiac function in the post-MI hearts. Earlier works have shown that gastrin displays proliferative and antiapoptotic effects (Duckworth et al. 2013; Ramamoorthy et al. 2004). Our data revealed that gastrin ameliorated apoptosis but did not increase cardiomyocyte proliferation in the post-MI heart.
Pro-angiogenic therapy has the potential to rescue the ischemic myocardium at the early stages after MI and is also essential for long-term left ventricular remodeling to prevent the transition to heart failure (Li et al. 2021). However, angiogenesis after myocardial infarction is insufficient. Thus, stimulating angiogenesis is an essential strategy for MI patients. Angiogenesis is based on endothelial cell proliferation and migration to form new vessels through sprouting in the hypoxic or ischemic region (Carmeliet and Jain 2011). Our in vivo study revealed that CD31-positive microvessels in the infarct border zone were increased by gastrin treatment after LAD ligation for 14 days, and the in vitro assay showed that migration of HCAECs and tube formation were stimulated by gastrin, meaning that gastrin promoted angiogenesis in ischemic myocardium.
VEGF, a critical growth factor for vascular endothelial cells, is a critical factor that regulates angiogenesis and attenuates cardiac dysfunction after MI in animal models (Carmeliet and Jain 2011; Saif and Emanueli 2014). Although there are other related genes, including VEGF-B and VEGF-C, VEGF-A plays a dominant role in regulating angiogenesis (Apte et al. 2019). PI3K/Akt signaling plays an important role in angiogenesis and exerts a protective role in myocardial infarction (Peng et al. 2021; Ruan et al. 2020). Bertrand et al. found that gastrin-gly stimulated VEGF expression through the PI3K/Akt pathway (Bertrand et al. 2010). In porcine coronary artery endothelial cells, gastrin has been reported to stimulate NO generation through PI3K/Akt signaling pathway (Grossini et al. 2012). We, therefore, performed a western-blot experiment using lysates from HCAECs and found that gastrin increased PI3K and Akt phosphorylation and VEGFA expression in a concentration-dependent manner, and this process was partly repressed by PI3K inhibitor LY294002. Similarly, the increased HCAECs migration stimulated by gastrin was also partly blocked by LY294002. These results indicated that the PI3K/Akt/VEGF signal pathway was involved in the pro-angiogenic effect of gastrin in post-MI mice.
The gastrin peptide family has two subtype receptors, namely, CCK1R and CCK2R. The expression of CCK2R is greater than CCK1R in the coronary arteries and has a higher affinity for gastrin than CCK1R (Grossini et al. 2013). Gastrin has been reported to up-regulate NO production in the artery, and the intracoronary infusion of gastrin significantly increased coronary blood flow through stimulating CCK2R in pigs (Grossini et al. 2011). In this study, we showed that CCK2R inhibitor C1988 abolished the pro-angiogenic function of gastrin. Therefore, the potential cardioprotective role of gastrin in post-MI was probably mediated by the CCK2R.
Conclusion
We demonstrated that increased plasma gastrin level played a protective role against cardiac dysfunction after myocardial infarction. Gastrin protected the post-MI heart from dysfunction by reducing cardiomyocyte apoptosis and promoting angiogenesis. The pro-angiogenic effect of gastrin was associated with CCK2R mediated PI3K/Akt/VEGF signal pathway activation.
References
Andrikou E, et al. GLP-1 receptor agonists and cardiovascular outcome trials: an update. Hellenic J Cardiol. 2019;60:347–51.
Apte RS, Chen DS, Ferrara N. VEGF in signaling and disease: beyond discovery and development. Cell. 2019;176:1248–64.
Bertrand C, et al. A gastrin precursor, gastrin-gly, upregulates VEGF expression in colonic epithelial cells through an HIF-1-independent mechanism. Int J Cancer. 2010;126:2847–57.
Bonaventura A, Carbone S, Dixon DL, Abbate A, Montecucco F. Pharmacologic strategies to reduce cardiovascular disease in type 2 diabetes mellitus: focus on SGLT-2 inhibitors and GLP-1 receptor agonists. J Intern Med. 2019;286:16–31.
Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307.
Chen Y, et al. Gastrin and D1 dopamine receptor interact to induce natriuresis and diuresis. Hypertension. 2013;62:927–33.
Clarke PA, Dickson JH, Harris JC, Grabowska A, Watson SA. Gastrin enhances the angiogenic potential of endothelial cells via modulation of heparin-binding epidermal-like growth factor. Cancer Res. 2006;66:3504–12.
Cochain C, Channon KM, Silvestre JS. Angiogenesis in the infarcted myocardium. Antioxid Redox Signal. 2013;18:1100–13.
Cui G, et al. A novel Ca (2+) current blocker promotes angiogenesis and cardiac healing after experimental myocardial infarction in mice. Pharmacol Res. 2018;134:109–17.
Dockray G, Dimaline R, Varro A. Gastrin: old hormone, new functions. Pflugers Arch. 2005;449:344–55.
Duckworth CA, et al. Progastrin-induced secretion of insulin-like growth factor 2 from colonic myofibroblasts stimulates colonic epithelial proliferation in mice. Gastroenterology. 2013;145:197-208 e3.
Feng Q, et al. PTEN inhibitor improves vascular remodeling and cardiac function after myocardial infarction through PI3k/Akt/VEGF signaling pathway. Mol Med. 2020;26:111.
Fu J, et al. Irisin lowers blood pressure by improvement of endothelial dysfunction via AMPK-Akt-eNOS-NO pathway in the spontaneously hypertensive rat. J Am Heart Assoc. 2016. https://doi.org/10.1161/JAHA.116.003433.
Gersl V, Goiny M, Uvnas-Wallensten K. The occurrence of gastrin-17 in the heart. Acta Physiol Scand. 1981;111:307–10.
Goetze JP, Rehfeld JF, Alehagen U. Cholecystokinin in plasma predicts cardiovascular mortality in elderly females. Int J Cardiol. 2016;209:37–41.
Grossini E, et al. Intracoronary gastrin 17 increases cardiac perfusion and function through autonomic nervous system, CCK receptors, and nitric oxide in anesthetized pigs. J Appl Physiol. 2011;110:95–108.
Grossini E, et al. CCK receptors-related signaling involved in nitric oxide production caused by gastrin 17 in porcine coronary endothelial cells. Mol Cell Endocrinol. 2012;350:20–30.
Grossini E, et al. Calcium handling in porcine coronary endothelial cells by gastrin-17. J Mol Endocrinol. 2013;50:243–53.
Gu D, et al. Gastrin, via activation of PPARalpha, protects the kidney against hypertensive injury. Clin Sci (lond). 2021;135:409–27.
Guilloteau P, Le Meuth-Metzinger V, Morisset J, Zabielski R. Gastrin, cholecystokinin and gastrointestinal tract functions in mammals. Nutr Res Rev. 2006;19:254–83.
Han D, et al. Ghrelin improves functional survival of engrafted adipose-derived mesenchymal stem cells in ischemic heart through PI3K/Akt signaling pathway. Biomed Res Int. 2015;2015:858349.
Ho VC, Duan LJ, Cronin C, Liang BT, Fong GH. Elevated vascular endothelial growth factor receptor-2 abundance contributes to increased angiogenesis in vascular endothelial growth factor receptor-1-deficient mice. Circulation. 2012;126:741–52.
Huang XR, et al. Smad3 mediates cardiac inflammation and fibrosis in angiotensin II-induced hypertensive cardiac remodeling. Hypertension. 2010;55:1165–71.
Lapidus L. Ischaemic heart disease, stroke and total mortality in women–results from a prospective population study in Gothenburg. Sweden Acta Med Scand Suppl. 1985;705:1–42.
Laval M, Baldwin GS, Shulkes A, Marshall KM. Increased gastrin gene expression provides a physiological advantage to mice under hypoxic conditions. Am J Physiol Gastrointest Liver Physiol. 2015;308:G76-84.
Lefranc F, et al. Characterization of gastrin-induced proangiogenic effects in vivo in orthotopic U373 experimental human glioblastomas and in vitro in human umbilical vein endothelial cells. Clin Cancer Res. 2004;10:8250–65.
Li L, et al. The role of G protein-coupled receptor kinase 4 in cardiomyocyte injury after myocardial infarction. Eur Heart J. 2020;42:1415.
Li Y, et al. Injectable hydrogel with MSNs/microRNA-21-5p delivery enables both immunomodification and enhanced angiogenesis for myocardial infarction therapy in pigs. Sci Adv. 2021;7:eabd6740.
Liang X, et al. Overexpression of ERBB4 rejuvenates aged mesenchymal stem cells and enhances angiogenesis via PI3K/AKT and MAPK/ERK pathways. FASEB J. 2019;33:4559–70.
Liu C, et al. Gastrin attenuates renal ischemia/reperfusion injury by a PI3K/Akt/Bad-mediated anti-apoptosis signaling. Front Pharmacol. 2020;11:540479.
Liu HS, Zhang J, Guo JL, Lin CY, Wang ZW. Phosphoinositide 3-kinase inhibitor LY294002 ameliorates the severity of myosin-induced myocarditis in mice. Curr Res Transl Med. 2016;64:21–7.
Matsumoto K, et al. Blockade of NKG2D/NKG2D ligand interaction attenuated cardiac remodelling after myocardial infarction. Cardiovasc Res. 2019;115:765–75.
Peng C, et al. LRIG3 suppresses angiogenesis by regulating the PI3K/AKT/VEGFA signaling pathway in glioma. Front Oncol. 2021;11:621154.
Ramamoorthy S, Stepan V, Todisco A. Intracellular mechanisms mediating the anti-apoptotic action of gastrin. Biochem Biophys Res Commun. 2004;323:44–8.
Rasanen M, et al. VEGF-B promotes endocardium-derived coronary vessel development and cardiac regeneration. Circulation. 2021;143:65–77.
Rehfeld JF. Premises for cholecystokinin and gastrin peptides in diabetes therapy. Clin Med Insights Endocrinol Diabetes. 2019;12:1179551419883608.
Rehfeld JF, Friis-Hansen L, Goetze JP, Hansen TV. The biology of cholecystokinin and gastrin peptides. Curr Top Med Chem. 2007;7:1154–65.
Rooman I, Bouwens L. Combined gastrin and epidermal growth factor treatment induces islet regeneration and restores normoglycaemia in C57Bl6/J mice treated with alloxan. Diabetologia. 2004;47:259–65.
Ruan Y, et al. Grape seed proanthocyanidin extract ameliorates cardiac remodelling after myocardial infarction through PI3K/AKT pathway in mice. Front Pharmacol. 2020;11:585984.
Saia RS, et al. Cardiovascular and inflammatory response to cholecystokinin during endotoxemic shock. Shock. 2013;39:104–13.
Saif J, Emanueli C. miRNAs in post-ischaemic angiogenesis and vascular remodelling. Biochem Soc Trans. 2014;42:1629–36.
Song W, Liang Q, Cai M, Tian Z. HIF-1alpha-induced up-regulation of microRNA-126 contributes to the effectiveness of exercise training on myocardial angiogenesis in myocardial infarction rats. J Cell Mol Med. 2020;24:12970–9.
Sun YY, et al. Period 2 is essential to maintain early endothelial progenitor cell function in vitro and angiogenesis after myocardial infarction in mice. J Cell Mol Med. 2014;18:907–18.
Tansey MJ, Opie LH, Vinik A, Kennelly BM. Plasma pancreatic polypeptide and gastrin in the assessment of autonomic activity in acute myocardial infarction. Eur J Cardiol. 1981;12:243–59.
van der Laan AM, Piek JJ, van Royen N. Targeting angiogenesis to restore the microcirculation after reperfused MI. Nat Rev Cardiol. 2009;6:515–23.
Varro A, Ardill JE. Gastrin: an analytical review. Ann Clin Biochem. 2003;40:472–80.
Xu XJ, Wiesenfeld-Hallin Z, Hughes J, Horwell DC, Hokfelt T. CI988, a selective antagonist of cholecystokininB receptors, prevents morphine tolerance in the rat. Br J Pharmacol. 1992;105:591–6.
Yang X, et al. Gastrin protects against myocardial ischemia/reperfusion injury via activation of RISK (Reperfusion Injury Salvage Kinase) and SAFE (Survivor Activating Factor Enhancement) pathways. J Am Heart Assoc. 2018. https://doi.org/10.1161/JAHA.116.005171.
Yasui M, Kawasaki K. CCKB receptor activation protects CA1 neurons from ischemia-induced dysfunction in stroke-prone spontaneously hypertensive rats hippocampal slices. Neurosci Lett. 1995;191:99–102.
Zeng Q, Ou L, Wang W, Guo DY. Gastrin, cholecystokinin, signaling, and biological activities in cellular processes. Front Endocrinol (lausanne). 2020;11:112.
Acknowledgements
None.
Funding
This study was supported by the National Natural Science Foundation of China (81800239), Science and Technology Department of Sichuan Province Project (2021YJ0210).
Author information
Authors and Affiliations
Contributions
Data collection and analysis: JF, YT, ZZ, LT, RY and LC. Study designed and manuscript writing: JF, RY and LC. All authors approved the final submission. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All the animal care and experimental procedures were approved by the Animal Care and Use Committee of the Southwest Jiaotong University.
Consent for publication
All of the authors have consented to the publication of this research.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fu, J., Tang, Y., Zhang, Z. et al. Gastrin exerts a protective effect against myocardial infarction via promoting angiogenesis. Mol Med 27, 90 (2021). https://doi.org/10.1186/s10020-021-00352-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s10020-021-00352-w