
Abstract
Interleukin-5 is produced by a number of cell types, and is responsible for the maturation and release of eosinophils in the bone marrow. In humans, interleukin-5 is a very selective cytokine as a result of the restricted expression of the interleukin-5 receptor on eosinophils and basophils. Eosinophils are a prominent feature in the pulmonary inflammation that is associated with allergic airway diseases, suggesting that inhibition of interleukin-5 is a viable treatment. The present review addresses the data that relate interleukin-5 to pulmonary inflammation and function in animal models, and the use of neutralizing anti-interleukin-5 monoclonal antibodies for the treatment of asthma in humans.
Similar content being viewed by others


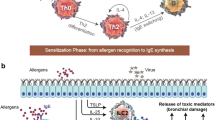
Introduction
Several allergic diseases, such as nasal rhinitis, nasal polyps, asthma, idiopathic eosinophilic syndromes, and atopic dermatitis, have prominent inflammatory components that are characterized by pronounced eosinophilic infiltration [1]. As a result, the role of chronic pulmonary inflammation in the pathophysiology of asthma has been studied extensively in human and in animal models. In asthma, pulmonary inflammation is characterized by edema, decreased mucociliary clearance, epithelial damage, increased neuronal responsiveness, and bronchoalveolar eosinophilia [1].
Eosinophils form in the bone marrow from myeloid precursors in response to cytokine activation, and are released into the circulation following an appropriate stimulus [2]. Once in the circulation they accumulate rapidly in tissue, where they synthesize and release lipid mediators that can cause edema, bronchoconstriction and chemotaxis, and secrete enzymes and proteins that can damage tissue [2]. The eosinophil is therefore an ideal target for selectively inhibiting the tissue damage that accompanies allergic diseases, without inducing the immunosuppressive consequences that can arise from systemic use of pleiotropic drugs such as steroids.
Interleukin-5 acts as a homodimer, and is essential for maturation of eosinophils in the bone marrow and their release into the blood [3,4,5,6]. In humans, interleukin-5 acts only on eosinophils and basophils, in which it causes maturation, growth, activation, and survival [7,8]. This specificity occurs because only those cells possess the interleukin-5 receptor. The functional high-affinity interleukin-5 receptor (250 pmol/l) is composed of two subunits: an α-subunit that is unique to interleukin-5, and a βc-subunit that is shared with interleukin-3 and granulocyte macrophage-colony stimulating factor (GM-CSF) [9,10].
In animals and in humans, inhibiting interleukin-5 with monoclonal antibodies can reduce blood and bronchoalveolar eosinophilia caused by allergic challenge or chronic disease [11,12,13,14]. Therefore, exclusively inhibiting the actions of interleukin-5 can suppress at least one of the alleged causes of asthma, namely tissue damage due to eosinophil accumulation during pulmonary inflammation.
Although a relationship exists between pulmonary eosinophilia and asthma in humans [15,16], the correlation in animal models between airway hyperreactivity and eosinophilia is less convincing [13,17,18]. However, selective inhibition of interleukin-5 by antibodies can block hyperreactivity in nonhuman primates [14]. Much of the same biology is evident in interleukin-5-knockout mice [19]. Although these mice can produce constitutive levels of eosinophils, they do not react to an allergic challenge with blood or lung eosinophilia or hyperreactivity, compared to normal controls. Of interest, interleukin-5-knockout mice do not develop an enhanced Mesocestoides corti infection after exposure, as measured by the worm burden [20].
Clinical trials with humanized antibodies against interleukin-5 have begun. In the current trials these therapeutics inhibit eosinophilia in asthmatic persons, but an effect on lung function has yet to be established [21,22]. Further trials designed to measure eosinophil accumulation and lung function in asthmatic persons are currently underway, and will help to define the role of interleukin-5 and eosinophils in general in this disease.
Genomics and biochemistry of the interleukin-5 system
There are clusters of T-helper (Th)2-type cytokine genes, including that which encodes interleukin-5, on human chromosome 5q and in the mouse on chromosome 11q, indicating a common evolutionary origin [23]. The cDNA that encodes murine interleukin-5 was cloned in 1986 from a T-cell line, followed by isolation of interleukin-5 cDNA from a human T-cell leukemia line [24,25] using a murine interleukin-5 cDNA as a probe. No overall significant amino acid sequence homology was found to exist with other cyokines, except for short stretches in the murine interleukin-3, murine GM-CSF, and murine interferon-γ proteins [25]. Furthermore, in the interleukin-5 promoter region there are short stretches of conserved sequence motifs, designated CLE 0, CLE 1 and CLE 2, which are also found in the 5'-flanking regions of the interleukin-3, interleukin-4, and GM-CSF genes [23,26].
Biologically active interleukin-5 is a disulfide-linked homodimer that is held together by the highly conserved cysteine residues that orient the monomers in an antiparallel arrangement [27,28]. The higher homology of mouse and human interleukin-5 found in the carboxyl-terminal compared with the amino-terminal half is consistent with the binding site for the interleukin-5 receptor that resides between helices C and D at an arginine-rich region that comprises residues 89 through 92 [29,30,31]. The broad range of apparent molecular weights (45-60 kDa) of recombinant murine interleukin-5 and human interleukin-5 results from differential glycosylation, but deglycosylated interleukin-5 retains full biologic activity [32]. A crystal structure shows that human interleukin-5 is a novel two-domain configuration with each domain requiring the participation of two chains, with a high degree of similarity to the cytokine fold found in GM-CSF, interleukin-3, and interleukin-4 [33].
Like interleukin-4, interleukin-5 is produced by T cells that belong to the Th2 but not the Th1 subset. By virtue of the pattern of cytokines that they synthesize, Th2 cells are thought to control the growth and effector function of those cell types that are involved in allergic inflammatory responses [34,35,36,37,38]. As with other cytokines, regulation of interleukin-5 production is thought to result from activation of gene transcription [37]. Interleukin-5 synthesis is also regulated at the level of mRNA stability [39]. Interleukin-5 gene expression requires de novo protein synthesis, and is effectively inhibited by glucocorticoids and cyclosporine [36,37,40]. Furthermore, in vivo depletion of T cells in a mouse model of pulmonary inflammation reduces pulmonary eosinophilia, and interleukin-5 and other cytokine mRNA levels [38]. Mast cells and eosinophils also synthesize interleukin-5, indicating that autocrine production of interleukin-5 may contribute to the chronicity of inflammation [41,42].
The interleukin-5 receptor is in the type I cytokine family, which includes receptors for interleukin-2 through interleukin-7, GM-CSF, granulocyte-colony stimulating factor, and erythropoietin [10,43]. These receptors are integral membrane glycoproteins with amino-termini directed extracellularly, a single membrane-spanning domain, and several conserved features [10,43]. The human interleukin-5 receptor has a Kd of 170–330 pmol/l, and is expressed on eosinophils and eosinophilic sublines of the HL60 cell [44,45]. The high-affinity interleukin-5 receptor is composed of two noncovalently associated subunits: α and β. The 60 kDa human interleukin-5 receptor α-chain binds mouse and human interleukin-5 with relatively high affinity (Kd = 1 nmol/l) [46], but does not induce signal transduction. Interaction of the α-subunit/interleukin-5 complex with the β-subunit, which is shared with the GM-CSF receptor and the interleukin-3 receptor, increases affinity to approximately 250 pmol/l and facilitates functional activity [9]. A soluble receptor form of the interleukin-5 receptor α has been identified, which antagonizes both binding and function of interleukin-5, and may protect against excessive eosinophil recruitment and activation [9].
Protein tyrosine kinases that physically associate with cytokine receptors and become activated after ligand binding have been identified [47]. Utilizing the β-subunit, interleukin-3, GM-CSF and interleukin-5 primarily activate Janus kinase (JAK)2 in response to ligand-receptor binding [47,48]. Activation of the JAK proteins is normally associated with autophosphorylation. Like interleukin-3 and GM-CSF, interleukin-5 induces rapid tyrosine phosphorylation of several proteins, further indicating that tyrosine kinases are involved in the cellular activation pathways [47,49]. JAK2 then induces tyrosine phosphorylation of STAT5, which activates its DNA-binding ability [47,49] and the ensuing cell activation [48].
Biology of interleukin-5
In the human, interleukin-5 is selective for eosinophils and basophils, whereas in the mouse it also acts on B lymphocytes [3,7,50]. Of course, eosinophils and basophils are two predominant effector cell types in allergic inflammation. By associating with its receptor, interleukin-5 effects eosinophil growth and differentiation [4,5,50,51], migration [8,50,52], activation and effector function [50,53,54], and survival [50,55]. As opposed to interleukin-3 or GM-CSF, only interleukin-5 promotes growth and differentiation to mature eosinophils in the bone marrow. Interleukin-3 and GM-CSF are also less selective than interleukin-5, stimulating the production of other granulocytes such as mast cells and neutrophils, respectively [50,56]. Because eosinophils are a dominant cell type in allergic reactions, this exquisite specificity makes interleukin-5 an excellent target for attenuating these responses. In fact, prolonged eosinophil survival and decreased eosinophil apoptosis caused by interleukin-5 are reversed by glucocorticoids [57,58], which accounts for at least some of the efficacy or these agents.
Activated eosinophils synthesize and release mediators, and secrete preformed granule constituents [59,60,61,62]. The eosinophil responds to a unique set of physiologic triggers, including secretory immunoglobulin A [59], which result largely from a Th2-type lymphocyte response. Eosinophils and neutrophils respond to many common stimulators, such as C5a, phorbol myristate acetate, zymosan, and formyl-methionyl-leucyl-phenylalanine [25, 60,61,62,63,64,65], with a respiratory burst, activation of phospholipases, production of eicosanoids, and secretion of preformed granule contents such as peroxidases and proteinases, including lysozyme and collagenases [63,64,65]. On the other hand, the ability to store and secrete the cationic low-molecular-weight proteins major basic protein, eosinophil cationic protein (ECP), and eosinophil-derived neurotoxin (EDN) is unique to the eosinophil [66]. Major basic protein and ECP can lyse cells and can cause tissue damage at low concentrations [67,68,69]. Although EDN also has a pI of 11, it is not as innately toxic to tissue, indicating that there is more to this cytotoxicity than just the positive charge [67].
Animal models of interleukin-5 action
As a result of its efficacy and selectivity, interleukin-5 is an ideal drug development target for allergic eosinophil-mediated diseases. With the development of neutralizing monoclonal antibodies to interleukin-5, interleukin-5-deficient mice, in situ hybridization methodology, and immunocytochemical techniques, it has been possible to investigate the role of interleukin-5 in allergic inflammatory responses in animals and humans.
Because the activity of interleukin-5 is restricted to eosinophils, it should be an ideal target to block this response in the lungs of allergic animal models of asthma, and has been studied in several species. Sensitized guinea pigs respond to allergic challenge with bronchial hyperresponsiveness and infiltration of eosinophils into lung tissue and bronchoalveolar lavage (BAL) fluid [11,13,70]. Monoclonal antibodies to interleukin-5 inhibit these pulmonary responses [13]. In contrast, larger doses of an anti-interleukin-5 antibody are needed to block the hyperreactivity than are needed to block the eosinophilia. This suggests either that interleukin-5 has effects on bronchoconstrictor reactivity that are independent of its effects on eosinophils, or that eosinophil activation, degranulation and release of its cytotoxic products, which were not measured in these studies, are the relevant aspects of eosinophil function that correlate with the development of the hyperreactivity. Indeed, it has been shown [71] that delivery of recombinant human interleukin-5 to the lungs of naïve guinea pigs increases eosinophils and neutrophils in the lungs and bronchoalveolar fluid, but this condition is not associated with augmented bronchoconstrictor responsiveness. However, recent studies have shown that administration of recombinant interleukin-5 to isolated airway smooth muscle from both rabbits and humans results in increased reactivity to acetylcholine [72]. In these studies the interleukin-5-induced hyperreactivity was abated by blocking the activity of interleukin-1, and interleukin-1β mRNA and protein levels are increased by interleukin-5. Interleukin-5 may contribute to airway hyperreactivity by both indirect and direct mechanisms. In fact, it may work indirectly by releasing granule proteins from eosinophils that act as endogenous allosteric antagonists at inhibitory presynaptic muscarinic M2 receptors, augmenting bronchoconstrictor responses to vagal nerve stimulation [73]. It may also work directly by mediating synthesis of interleukin-1β in airway smooth muscle [72].
As with guinea pigs, antigen challenge to the lungs of sensitized mice causes an influx of eosinophils into the BAL fluid and lung tissue [74]. This effect is inhibited when monoclonal antibodies to interleukin-5 are given before the antigen challenge [75]. There is also increased expression of mRNA for Th2 cytokines such as interleukin-5 and interleukin-4 in the lungs of allergic mice [38]. To a lesser extent than are T lymphocytes, mast cells are involved in the development of pulmonary eosinophilia in allergic mice after single provocation by antigen [76], but are much less important in the pulmonary eosinophilia that occurs after a multiple antigen challenge paradigm [77]. Mice have been developed using standard technology that are deficient in interleukin-5 [20]. These mice produce constitutive levels of eosinophils driven by other cytokines, and have normal circulating levels of immunoglobulin E, but do not mount an eosinophilic response to allergic challenge.
After multiple exposure to inhaled antigen, sensitized mice exhibit airway hyperreactivity [19,20]. When challenged in this manner, both the lung and lavage eosinophilia and the airway hyperreactivity to cholinergic agents are blocked by anti-interleukin-5 antibodies [20]. In addition, in sensitized interleukin-5-deficient mice receiving multiple challenges, the hyperreactivity is eliminated along with the airway eosinophilia [19,20]. In a variety of knockout and transgenic mice that were further modified by the administration of cytokines, chemokines or antibodies, there appear to be significant interactions among these proteins with regard to establishing eosinophilia and airways hyperreactivity [78]. Whereas interleukin-4 and interleukin-13 are redundant with regard to these inflammatory parameters, interleukin-5 plays a distinct role in both. Furthermore, interleukin-5 and eotaxin synergistically enhance eosinophilia and airway hyperreactivity in allergic mice by a CD4+ T-cell-dependent mechanism [79]. To some degree, these observations are dependent on the background strain of mouse [20,78].
Interleukin-5 has also been identified as an important cytokine in regulating human eosinophil survival in asthmatic persons after antigen challenge [57]. Inhibition of interleukin-5 activity during an established pulmonary eosinophilia could, therefore, cause tissue damage due to destruction of eosinophils and release of their inflammatory mediators. However, in allergic mice, administration of an antibody to interleukin-5 after antigen challenge, when lung eosinophilia was already established, did not increase tissue damage in the lungs [75]. These results have important therapeutic implications for the potential use of interleukin-5 inhibitors in the treatment of inflammatory airway disorders.
Allergic cynomolgus monkeys are also subject to an inflammatory cell influx into the airways, an early and late-phase bronchoconstriction, and bronchial hyperresponsiveness [14,80]. Treatment with a monoclonal antibody to interleukin-5 inhibits these responses to antigen provocation [14]. TRFK5, a monoclonal anti-interleukin-5 antibody, at an intravenous dose of 0.3 mg/kg inhibits lavage eosinophilia to 70%, while completely blocking the hyperreactivity to histamine. Furthermore, inhibition of both the pulmonary eosinophilia and bronchial hyperresponsiveness lasted for at least 3 months after a single treatment because of the long circulating lifetime of the antibody. Hence, in several animal models of asthma, blockade of interleukin-5 activity suppressed both eosinophilia and changes in lung function, but the causal relationship between these two effects is somewhat tenuous.
Although there is often a correlation between lung eosinophilia, ECP in BAL fluid, and a decreased forced expiratory volume in 1 s (FEV1) in humans [81], this does not necessarily indicate that the eosinophils are responsible for the decreased lung function. In fact, in several animal models there is a lack of correlation between reduced levels of lung eosinophils and improved lung function, suggesting that a critical activation step is missing [13,14,15,16,17,18]. In reality, there are no animal models that precisely duplicate the chronic nature of asthma.
Interleukin-5 in human asthma
Atopic asthmatic persons have increased expression of Th2-type cytokine (interleukin-2, interleukin-3, interleukin-4, interleukin-5, and GM-CSF) mRNA in both BAL fluid and in bronchial biopsies as compared with healthy volunteers, but there is no difference between the two groups in the expression of Th1-type cytokine mRNA such as interferon-γ [82,83,84,85]. The predominant source of interleukin-4 and interleukin-5 mRNA in asthmatic persons is the T lymphocyte, and the CD4+ and CD8+ T-cell populations express elevated levels of activation markers including interleukin-2 receptor (CD25), human leukocyte antigen-DR, and the very late activation antigen-1 [84,86,87,88,89,90]. These results suggest that atopic asthma is associated with activation of the interleukin-3, interleukin-4, interleukin-5, and GM-CSF gene cluster, a pattern that is consistent with a Th2-like T-lymphocyte response [85]. Interleukin-5 mRNA is also found in activated eosinophils and mast cells in tissues from patients with atopic dermatitis [91,92,93], allergic rhinitis [94,95], and asthma [82,89], raising the possibility that interleukin-5 arises from multiple sources in atopic individuals.
Eosinophil infiltration into the airways after allergen challenge is a consistent feature of atopic asthmatic persons [96,97,98]. Interleukin-5 is predominantly an eosinophil-active cytokine in the late-phase response to antigen challenge [99,100], and is important for the recruitment and survival of eosinophils [57,99]. On the other hand, interleukin-5 is probably not important in the acute response to allergen challenge in asthmatic persons. Indeed, interleukin-5 is not detectable in the BAL fluid of mildly asthmatic persons shortly after allergen provocation [100]. Interleukin-5 may also be important for the recruitment of eosinophils from blood vessels into tissues, because topical administration of recombinant human interleukin-5 to the nasal airway of persons with allergic rhinitis induced eosinophil accumulation into the nasal mucosa [101,102]. Interleukin-5 may also induce activation of eosinophils that are resident to inflamed tissue, but this effect may be secondary to activation of secretory immunoglobulin A [103].
Several studies have demonstrated a correlation between the activation of T lymphocytes, increased concentration of interleukin-5 in serum and BAL fluid, and increased severity of the asthmatic response [87,104,105,106]. In a study of 30 asthmatic persons, Robinson et al [86] found a strong correlation between the number of BAL cells that expressed mRNA for interleukin-5, the magnitude of baseline airflow obstruction (FEV1), and bronchoconstrictor reactivity to methacholine. Furthermore, Zangrilli et al [106] found increased levels of interleukin-4 and interleukin-5 in the BAL fluid of asthmatic persons who had a late-phase response to antigen, but not in asthmatic persons who only demonstrated an early-phase response to antigen challenge. Motojima et al [104] compared serum levels of interleukin-5 in asthmatic patients during an exacerbation and in remission of asthma. Higher levels of serum interleukin-5 were found in each person during exacerbation, and patients with severe asthma had higher levels of serum interleukin-5 than did control individuals or patients with mild asthma. It is interesting to note that interleukin-5 levels were reduced in the serum of patients with moderate-to-severe asthma who were receiving oral glucocorticoids for control of their asthma [104,106]. These results are consistent with in vitro studies that show a potent inhibitory effect of corticosteroids on gene expression and on the release of pro-inflammatory cytokines, including interleukin-5, from inflammatory cells [107].
The link between interleukin-5, eosinophils, and asthma is currently under investigation using two humanized monoclonal antibodies specific for interleukin-5 that have been advanced into the clinic for evaluation as therapies for asthma. SCH55700 (reslizumab) is a humanized monoclonal antibody with activity against interleukin-5 from various species [108]. SB240563 (mepolizumab) is also a humanized antibody with specificity for human and primate interleukin-5 [109,110].
SCH55700 has an affinity for human interleukin-5 of 81 pmol/l and a 50% inhibitory concentration for inhibition of human interleukin-5-mediated TF-1 cell proliferation of 45 pmol/l. The efficacy of SCH55700 was further evaluated preclinically in a number of animal models. In a dose-dependent manner, SCH55700 inhibited total cell and eosinophil influx into BAL fluid, bronchi, and bronchioles of allergic mice for up to 8 weeks after a single 10 mg/kg dose and for 4 weeks after a single 2 mg/kg dose. Additional studies in allergic mice indicated that the combination of SCH55700 with an oral steroid (prednisolone) significantly increased the efficacy over that of either agent administered alone [108]. In allergic guinea pigs, SCH55700 caused a dose-dependent decrease in pulmonary eosinophilia and inhibited the development of allergen-induced airway hyperresponsiveness to substance P. It also inhibited the accumulation of total cells, eosinophils, and neutrophils in the lungs of guinea pigs exposed to human interleukin-5. SCH55700 had no effect on the numbers of inflammatory cells in unchallenged animals or in animals challenged with GM-CSF, and had no effect on the levels of circulating total leukocytes [108]. In cynomolgus monkeys naturally allergic to Ascaris suum, postchallenge pulmonary eosinophilia was significantly decreased for up to 6 months after a single 0.3 mg/kg intravenous dose of SCH55700 [108].
A rising single-dose phase I clinical trial was conducted with SCH55700 in patients with severe persistent asthma who remained symptomatic despite intervention with high-dose inhaled or oral steroids [22]. The two highest doses of SCH55700 significantly decreased peripheral blood eosinophils, with inhibition lasting up to 90 days after the 1 mg/kg dose. There was also a trend toward improvement in lung function at the higher doses 30 days after dosing, with mean FEV1 increasing by 11.2 and 8.6% in the 0.3 and 1.0 mg/kg groups, respectively, versus 4.0% in the placebo group [22].
Preclinical studies with SB240563 in cynomolgus monkeys indicated that peripheral blood eosinophils were decreased as a result of administration of the antibody [109,110]. Interestingly, maximal inhibition of peripheral blood eosinophils (80–90% of baseline) occurred 3–4 weeks after dosing (1 mg/kg subcutaneously), whereas maximal blood levels of the antibody were obtained 2–4 days after dosing, with a half-life of approximately 14 days.
SB240563 has also recently been tested in asthmatic persons in a clinical single-dose safety and activity study [21]. Patients with mild asthma were administered a single intravenous dose of SB240563 at either 2.5 or 10 mg/kg, or placebo. Patients were challenged with allergen 2 weeks before and 1 and 4 weeks after dosing. Peripheral blood and sputum eosinophil levels were measured, and early-phase and late-phase asthmatic responses were assessed by measuring the percentage fall in FEV1 induced by allergen challenge. Both doses of SB240563 caused a significant reduction in peripheral blood eosinophils. Eosinophil counts were reduced in the 10 mg/kg dose group by approximately 75% for up to 16 weeks, and in the 2.5 mg/kg dose group by approximately 65% for up to 8 weeks. Postchallenge sputum eosinophils were also reduced in the 10 mg/kg dose group. Neither dose of SB240563 attenuated the fall in induced by allergen challenge in these mildly FEV1 asthmatic persons.
With both of these antibodies showing acceptable safety profiles, larger studies can be conducted to determine the impact of blocking interleukin-5 on the pathophysiology of asthma and other eosinophil-related diseases. Only when these clinical trials are conducted will we be able to determine whether interleukin-5-based therapy in humans will measure up to the promise that is projected from animal models.
Conclusion
There are circumstantial but compelling data that implicate interleukin-5 in diseases that involve eosinophils. Interleukin-5 is produced in lymphocytes, mast cells, eosinophils, and airway smooth muscle and epithelial cells, and is primarily responsible for the maturation and release of eosinophils in the bone marrow. In humans, it is a very selective cytokine because only eosinophils and basophils possess a type-1 cytokine receptor for interleukin-5 with a specific α-subunit and the βc-subunit that confers high-affinity binding and signal transduction. A specific inhibitor of interleukin-5 could, therefore, attenuate pulmonary inflammation and the consequent pathophysiology without the potential for immunosuppressive side effects that exist with steroids.
Interleukin-5 in the circulation has been reduced by potent, neutralizing anti-interleukin-5 monoclonal antibodies. As a result, eosinophils have been attenuated for long durations in various animal models of eosinophil accumulation. In some but not all of these animal models, inhibition of tissue or BAL eosinophilia correlates with decreased pathophysiology. In addition, interleukin-5-knockout mice do not respond to an allergic challenge with blood or tissue eosinophilia. Furthermore, these mice are not overly sensitive to parasitic infection and, as opposed to their normal counterparts, are not hyperreactive to cholinergic challenge to the lungs. By contrast, although eosinophil levels were reduced by an anti-interleukin-5 antibody in a human allergic challenge model, there was no reduction in hyperreactivity. In a phase I clinical trial with another humanized anti-interleukin-5 antibody, eosinophils were reduced for 90 days in severe steroid-dependent asthmatic persons. Nevertheless, ongoing phase II studies are required to determine the utility of this approach in treating asthma and other eosinophilic diseases.
Abbreviations
- BAL:
-
bronchoalveolar lavage
- ECP:
-
eosinophil cationic protein
- FEV1:
-
forced expiratory volume in 1 s
- GM-CSF:
-
granulocyte macrophage-colony stimulating factor
- JAK:
-
Janus kinase
- Th:
-
T-helper (cell).
References
Kay AB: Asthma and inflammation. J Allergy Clin Immunol. 1991, 87: 893-910.
Gleich GJ, Kita H, Adolphson CR: Eosinophils. In: Samters Immunologic Diseases, edn 5. Edited by Frank MN, Austen KF, Cloman HN, Inanue ER. Boston: Little Brown Co.;. 1995, 205-245.
Clutterbuck E, Shields JG, Gorden J, Smith SH, Boyd A, Callard RE, Campbell HD, Young IG, Sanderson CJ: Recombinant human interleukin-5 is an eosinophil differentiation factor but has no activity in standard human B cell growth factor assays. Eur J Immunol. 1987, 17: 1743-1750.
Clutterbuck EJ, Hirst EMA, Sanderson CJ: Human interleukin-5 (IL-5) regulates the production of eosinophils in human bone marrow cultures: comparison and interaction with IL-1, IL-3, IL-6 and GM-CSF. Blood. 1989, 73: 1504-1512.
Clutterbuck EJ, Sanderson CJ: Regulation of human eosinophil precursor production by cytokines: a comparison of recombinant human interleukin-1 (rhIL-1), rhIL-3, rhIL-5, rhIL-6 and rh granulocyte-macrophage colony stimulating factor. Blood. 1990, 75: 1774-1779.
Mckenzie ANJ, Ely B, Sanderson CJ: Mutated interleukin-5 monomers are biologically inactive. Mol lmmunol. 1991, 28: 155-158. 10.1016/0161-5890(91)90099-6.
Hirai K, Yamaguchi M, Misaki Y, Takaishi T, Ohfa K, Morita Y, Ito K, Miyamoto T: Enhancement of human basophil histamine release by interleukin 5. J Exp Med. 1990, 172: 1525-1528.
Resnick MB, Weller PF: Mechanisms of eosinophil recruitment. Am J Respir Cell Mol Biol. 1993, 8: 349-355.
Tavernier J, Devos R, Cornelis S, Tuypens T, van der Heyden J, Fiers W, Plaetinck G: A human high affinity interleukin-5 receptor (IL5R) is composed of an IL5 specific α chain and a β chain shared with the receptor for GM-CSF. Cell. 1991, 66: 1175-1184.
Miyajima A, Kitamura T, Harada N, Yokota T, Arai KI: Cytokine receptors and signal transduction. Annu Rev lmmunol. 1992, 10: 295-331. 10.1146/annurev.iy.10.040192.001455.
Gulbenkian AR, Egan RW, Fernandez X, Jones H, Kreutner W, Kung TT, Payvandi F, Sullivan L, Zurcher JA, Watnick AS: lnterleukin-5 modulates eosinophil accumulation in allergic guinea pig lung. Am Rev Respir Dis. 1992, 146: 263-265.
Kung TT, Stelts D, Zurcher JA, Watnick AS, Jones H, Mauser PJ, Fernandez X, Umland S, Kreutner W, Chapman RW, Egan RW: Mechanisms of allergic pulmonary eosinophilia in the mouse. J Allergy Clin lmmunol. 1994, 94: 1217-1224.
Mauser PJ, Pitman A, Witt A, Fernandez X, Zurcher J, Kung TT, Jones H, Watnick AS, Egan RW, Kreutner W, Adams GK: Inhibitory effect of the TRFK-5 anti-IL-5 antibody in a guinea pig model of asthma. Am Rev Respir Dis. 1993, 148: 1623-1627.
Mauser PJ, Pitman AM, Fernandez X, Foran SK, Adams GK, Kreutner W, Egan RW, Chapman RW: Effects of an antibody to IL-5 in a monkey model of asthma. Am J Respir Crit Care Med. 1995, 152: 467-472.
Demonchy JG, Kauffman HF, Venge P, Koeter GH, Jansen HM, Sluiter HJ, Devries K: Bronchoalveolar eosinophilia during antigen-induced late asthmatic reactions. Am Rev Respir Dis. 1985, 131: 373-376.
Gibson PG, Manning PJ, O'Byrne PM, Girgis-Gabardo A, Dolovich J, Denburg JA, Hargreave FE: Allergen-induced asthmatic responses. Relationship between increases in airway responsiveness and increases in circulating eosinophils, basophils and their progenitors. Am Rev Respir Dis. 1991, 143: 331-335.
Hutson PA, Church MK, Clay TP, Miller P, Holgate ST: Early and late-phase bronchoconstriction after antigen challenge of nonanesthetized guinea pigs. 1. The association of disordered airway physiology to leukocyte infiltration. Am Rev Respir Dis. 1988, 137: 548-557.
Ishida K, Thompson RJ, Beattie LL, Wiggs B, Schellenberg RR: Inhibition of antigen-induced airway hyperresponsiveness but not acute hypoxia nor airway eosinophilia, by an antagonist of platelet-activating factor. J Immunol. 1990, 144: 3907-3911.
Foster PS, Hogan SP, Ramsay AJ, Matthaei KI, Young IG: Interleukin 5 deficiency abolishes eosinophilia, airways hyperreactivity, and lung damage in a mouse asthma model. J Exp Med. 1996, 183: 195-201.
Kopf M, Brombacher F, Hodgkin PD, Ramsay AJ, Milbourne EA, Dai WJ, Ovington KS, Behm CA, Kohler G, Young IG, Matthaei KI: IL-5-deficient mice have a developmental defect in CD5+ B-1 cells and lack eosinophilia but have normal antibody and cytotoxic T cell responses. Immunity. 1996, 4: 15-24.
Leckie MJ, ten Brinke A, Khan J, Diamant Z, O'Connor BJ, Walls CM, Mathur AK, Cowley H, Chung KF, Djukanovic R, Hansel TT, Holgate ST, Sterk PJ, Barnes PJ: Effects of an interleukin-5 blocking monoclonal antibody on eosinophils, airway hyperresponsiveness, and the late asthmatic response. Lancet. 2000, 356: 2144-2148. 10.1016/S0140-6736(00)03496-6.
Kips JC, O'Connor BJ, Langley SJ, Woodcock A, Kerstjens HAM, Postma DS, Danzig M, Cuss F, Pauwels RA: Results of a phase I trial with SCH5 a humanized anti-IL-5 antibody, in severe persistent asthma [abstract]. Am J Respir Crit Care Med. 2000, 161: A505-
van Leeuwen BH, Martinson ME, Webb GC, Young IG: Molecular organization of the cytokine gene cluster, involving, the human IL-3, IL-4, IL-5, and GM-CSF genes, on human chromosome 5. Blood. 1989, 73: 1142-1148.
Azuma C, Tanabe T, Konishi M, Kinashi T, Noma T, Matsuda F, Yaoita Y, Takatsu K, Hammarstrom L, Edvard-Smith Cl, Severinson E, Honio T: Cloning of cDNA for human T-cell replacing factor (interleukin-5) and comparison with the murine homologue. Nucl Acids Res. 1986, 14: 9149-9158.
Kinashi T, Harada N, Severinson E, Tanabe T, Sideras P, Konishi M, Azuma C, Tomiaga A, Bergstedt-Lindqvist S, Takahashi M, Matsuda F, Yaoita Y, Takatsu K, Honjo T: Cloning of complementary DNA encoding T-cell replacing factor and identity with B-cell growth factor II. Nature. 1986, 324: 70-73.
Miyatake S, Shlomai J, Ken-Ichi A, Ari N: Characterization of the mouse granulocyte macrophage colony-stimulating factor (GM-CSF) gene promoter: nuclear factors that interact with an element shared by three lymphokine genes - those for GM-CSF, interleukin-4 (IL-4), and IL-5. Mol Cell Biol. 1991, 12: 5894-5901.
Minamitake Y, Kodama S, Katayama T, Adachi H, Tanaka H, Tsujimoto M: Structure of recombinant human interleukin 5 produced by chinese hamster ovary cell. J Biochem. 1990, 107: 292-297.
Takahashi T, Yamaguchi N, Mita S, Yamaguchi Y, Suda T, Tominaga A, Kikuchi Y, Miura Y, Takatsu K: Structural comparison of murine T-cell (Bl 51 Kl 2) derived T-cell-replacing factor (IL-5) with rIL-5: dimer formation is essential for the expression of biological activity. Mol lmmunol. 1990, 27: 911-920. 10.1016/0161-5890(90)90158-V.
Dickason RR, Huston MM, Huston DP: Delineation of IL-5 domains predicted to engage the IL-5 receptor complex. J Immunol. 1996, 156: 1030-1037.
Zhang J, Kuvelkar R, Murgolo NJ, Taremi SS, Chou CC, Wang P, Billah MM, Egan RW: Mapping and characterization of the epitope(s) of Sch 5 a humanized mAb, that inhibits human IL-5. Int Immunol. 1999, 11: 1935-1944. 10.1093/intimm/11.12.1935.
Kodama S, Tsuruoka N, Tsujimoto M: Role of the C-terminus in the biological activity of human interleukin 5. Biochem Biophys Res Commun. 1991, 178: 514-519.
Tominaga A, Takahashi T, Kikuchi Y, Mita S, Naomi S, Harada N, Yamaguchi N, Takatsu K: Role of carbohydrate moiety of IL-5. J Immunol. 1990, 144: 1345-1352.
Milburn MV, Hassell AM, Lambert MH, Jordan SR, Proudfoot AEI, Graber P, Wells TNC: A novel dimer configuration revealed by the crystal structure at 2.4 Å resolution of human interleukin-5. Nature. 1993, 363: 172-176. 10.1038/363172a0.
Mosmann TR, Coffman RL: TH1 and TH2 cells: Different patterns of lymphokine secretion lead to different functional properties. Ann Rev lmmunol. 1989, 7: 145-173. 10.1146/annurev.iy.07.040189.001045.
Altman A, Coggeshall KM, Mustelin T: Molecular events mediating T cell activation. Adv Immunol. 1990, 48: 227-360.
Naora H, Altin JG, Young IG: TCR-dependent and independent signaling mechanisms differentially regulate lymphokine gene expression in the murine T helper clone DlO.G4.1. J lmmunol. 1994, 152: 5691-5702.
van Straaten JFM, Dokter WHA, Stulp BK, Vellenga BK: The regulation of interleukin-5 and interleukin-3 gene expression in human T cells. Cytokine. 1994, 6: 229-234.
Garlisi CG, Falcone A, Kung TT, Stelts D, Pennline KG, Beavis AJ, Smith SR, Egan RW, Umland SP: T cells are necessary for TH2 cytokine production and eosinophil accumulation in airways of antigen-challenged allergic mice. Clin lmmunol lmmunopathol. 1995, 75: 75-83. 10.1006/clin.1995.1055.
Umland SP, Razac S, Shah H, Nehrebne DK, Egan RW, Billah MM: Interleukin-5 mRNA stability in human T cells is regulated differently than interleukin-2, interleukin-3, interleukin-4, granulocyte/macrophage colony-stimulating factor, and interferon-γ. Am J Respir Cell Mol Biol. 1998, 18: 631-642.
Umland SP, Shah H, Jakway JP, Shortall J, Razac S, Garlisi CF, Falcone A, Kung TT, Stelts D, Hegde V, Patel M, Billah MM, Egan RW: Effects of cyclosporin A and dinactin on T-cell proliferation, interleukin-5 production, and murine pulmonary inflammation. Am J Respir Cell Mol Biol. 1999, 20: 481-492.
Bradding P, Roberts JA, Britten KM, Montefort S, Dujukanovic R, Mueller R, Heusser CH, Howarth PH, Holgate ST: Interleukin-4, -5, and -6 and tumor necrosis factor-α in normal and asthmatic airways: evidence for the human mast cell as a source of these cytokines. Am J Respir Cell Mol Biol. 1994, 10: 471-480.
Dubucquoi S, Desreumaux P, Janin A, Klein O, Goldman M, Tavernier J, Capron A, Capron M: Interleukin 5 synthesis by eosinophils: association with granules and immunoglobulin-dependent secretion. J Exp Med. 1994, 179: 703-708.
Bazan JF: Structural design and molecular evaluation of a cytokine receptor superfamily. Proc Natl Acad Sci USA. 1990, 87: 6934-6938.
Migita M, Yamaguchi N, Mita S, Higuchi S, Hitoshi Y, Yosha Y, Tomonaga M, Matsuda I, Tominaga A, Takatsu K: Characterization of the human IL-5 receptors on eosinophils. Cell lmmunol. 1991, 133: 484-497.
Plaetinck G, van der Heyden J, Tavernier J, Fache I, Tuypens T, Fischkoff S, Fiers W, Devos R: Characterization of interleukin 5 receptors on eosinophilic sublines from human promyelocytic leukemia (HL-60) cells. J Exp Med. 1990, 172: 683-691.
Murata Y, Takaki S, Migita M, Kikuchi Y, Tominaga A, Takatsu K: Molecular cloning and expression of the human interleukin 5 receptor. J Exp Med. 1992, 175: 341-351.
Ihle JN, Witthuhn BA, Quelle FW, Yamamoto K, Thierfelder WE, Kreider B, Silvennoinen O: Signaling by the cytokine receptor superfamily: JAKs and STATs. Trends Biochem Sci. 1994, 19: 222-227. 10.1016/0968-0004(94)90026-4.
Darnell JE, Kerr AM, Stark GR: Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994, 262: 1415-1420.
Mul Al-F, Wakao H, O'Farrell A-M, Harada N, Miyajima A: lnterleukin-3, granulocyte-macrophage colony stimulating factor and interleukin-5 transduce signals through two STAT5 homologues. EMBO J. 1995, 14: 1166-1175.
Lopez AF, Shannon MF, Chia M-M, Park L, Vadas MA: Regulation of human eosinophil production and function by interleukin-5. lmmunol Ser. 1992, 57: 549-571.
Yamaguchi Y, Suda T, Suda J, Eguchi M, Miura Y, Harad N, Taminaga A, Takatsu K: Purified interleukin 5 supports the terminal differentiation and proliferation of murine eosinophilic precursors. J Exp Med. 1988, 167: 43-56.
Warringa RAJ, Schweizer RC, Maikoe T, Kujiper PHM, Bruijnzeel PLB, Koenderman L: Modulation of eosinophil chemotaxis by interleukin-5. Am J Respir Cell Mol Biol. 1992, 7: 631-636.
Carlson M, Peterson C, Venge P: The infuence of IL-3, IL-5 and GM-CSF on normal human eosinophil and neutrophil C3b-induced degranulation. Allergy. 1993, 48: 437-442.
Kita H, Weiler D, Abu-Ghazaleh R, Sanderson CJ, Gleich GJ: Release of granule proteins from eosinophils cultured with IL-5. J lmmunol. 1992, 149: 629-635.
Yamaguchi Y, Suda T, Oha S, Toinaga K, Miura Y, Kasahara T: Analysis of the survival of mature human eosinophils: interleukin-5 prevents apoptosis in mature human eosinophils. Blood. 1991, 78: 2542-2547.
Woolley MJ, Denburg JA, Ellis R, Dahlback M, O'Byrne P: Allergen-induced changes in bone marrow progenitors and airway responsiveness in dogs and the effect of inhaled budesonide on these parameters. Am J Respir Cell Mol Biol. 1994, 11: 600-606.
Ohnishi T, Sur S, Collins DS, Fish J, Gleich GJ, Peters SP: Eosinophil survival activity identified as interleukin-5 is associated with eosinophil recruitment and degranulation and lung injury twenty-four hours after segmental antigen lung challenge. J Allergy Clin Immunol. 1993, 92: 607-615.
Wallen N, Kita H, Weiler D, Gleich GJ: Glucocorticoids inhibit cytokine-mediated eosinophil survival. J lmmunol. 1991, 147: 3490-3495.
Abu-Ghazaleh RI, Fujisawa T, Mestecky J, Kyle RA, Gleich GJ: IgA-induced eosinophil degranulation. J Immunol. 1989, 142: 2393-2400.
Minnicozzi M, Anthes JC, Siegel Ml, Billah MM, Egan RW: Activation of phospholipase D in normodense human eosinophils. Biochem Biophys Res Commun. 1990, 170: 540-547.
Petreccia DC, Nauseef WM, Clark RA: Respiratory burst of normal human eosinophils. J Leuk Biol. 1987, 41: 283-288.
Yazdanbakhsh M, Eckmann CM, Koenderman L, Verhoeven AJ, Roos D: Eosinophils do respond to FMLP. Blood. 1987, 70: 379-383.
Bruynzeel PLB, Kok PTM, Hamelink ML, Kijne AM, Verhagen J: Exclusive leukotrine C4 synthesis by purified human eosinophils induced by opsonized zymosan. FEBS Lett. 1985, 189: 350-354. 10.1016/0014-5793(85)81054-1.
Cockcroft S: G-protein-regulated phospholipase C, D, and A2-mediated signalling in neutrophils. Biochim Biophys Acta. 1992, 1113: 135-160.
Lehrer RI, Ganz T, Selsted ME, Babior BM, Curnutte JT: Neutrophils and host defense. Ann Intern Med. 1988, 109: 127-142.
Gleich GJ, Adolphson CR: The eosinophilic leukocyte: structure and function. Adv lmmunol. 1986, 39: 177-253.
Hamann KJ, Barker RI, Ten RM, Gleich GJ: The molecular biology of eosinophil granule proteins. Int Arch Allergy Appl lmmunol. 1991, 94: 202-209.
Minnicozzi M, Duran WN, Gleich GJ, Egan RW: Eosinophil granule proteins increase microvascular macromolecular transport in the hamster cheek pouch. J Immunol. 1994, 153: 2664-2670.
Frigas E, Loegering DA, Gleich GJ: Cytotoxic effects of the guinea pig eosinophil major basic protein on tracheal epithelium. J Lab Invest. 1980, 42: 35-43.
Coeffier E, Joseph D, Vargaftig BB: Role of interleukin-5 in enhanced migration of eosinophils from airways of immunized guinea pigs. Br J Pharmacol. 1994, 113: 749-756.
Lilly CM, Chapman RW, Sehring SJ, Mauser PJ, Showell HJ, Egan RW, Drazen JM: Effects of interleukin 5-induced pulmonary eosinophilia on airway reactivity in the guinea pig. Am J Physiol. 1996, 270: L368-L375.
Hakonarson H, Maskeri N, Carter C, Chuang S, Grunstein MM: Autocrine interaction between IL-5 and IL-1β mediates altered responsiveness of atopic asthmatic sensitized airway smooth muscle. J Clin Invest. 1999, 104: 657-667.
Elbon CL, Jacoby DB, Fryer AD: Pretreatment with an antibody to interleukin 5 prevents loss of pulmonary M2 muscarinic receptor function in antigen challenged guinea pigs. Am J Respir Cell Mol Biol. 1995, 12: 320-328.
Kung TT, Jones H, Adams GK, Umland SP, Kreutner W, Egan RW, Chapman RW, Watnick AS: Characterization of a murine model of allergic pulmonary inflammation. Int Arch Allergy Immunol. 1994, 105: 83-90.
Kung TT, Stelts DM, Zurcher JA, Adams GK, Egan RW, Kreutner W, Watnick AS, Jones H, Chapman RW: Involvement of IL-5 in a murine model of allergic pulmonary inflammation: prophylactic and therapeutic effect of an anti-IL-5 antibody. Am J Respir Cell Mol Biol. 1995, 13: 360-365.
Kung TT, Stelts D, Zurcher JA, Jones H, Umland SP, Kreutner W, Egan RW, Chapman RW: Mast cells modulate allergic pulmonary eosinophilia in mice. Am J Respir Cell Biol. 1995, 12: 404-409.
Brusselle GG, Kips JC, Tavernier JH, van der Heyden JG, Cavelier CA, Pauwels RA, Bluethmann H: Attenuation of allergic airway inflammation in IL-4 deficient mice. Clin Exp Allergy. 1994, 24: 73-80.
Webb DC, McKenzie ANJ, Koskinen AML, Yang M, Mattes J, Foster PS: Integrated signals between IL-13, IL-4, and IL-5 regulate airways hyperreactivity. J Immunol. 2000, 165: 108-113.
Mould AW, Ramsay AJ, Matthaei KI, Young IG, Rothenberg ME, Foster PS: The effect of IL-5 and eotaxin expression in the lung on eosinophil trafficking and degranulation and the induction of bronchial hyperreactivity. J Immunol. 2000, 164: 2142-2150.
Gundel RH, Wegner CD, Letts LG: Antigen-induced acute and late-phase responses in primates. Am Rev Respir Dis. 1992, 146: 369-373.
Adelroth E, Rosenhall L, Johansson S, Linden M, Venge P: Inflammatory cells and eosinophilic activity in asthmatics investigated by bronchoalveolar lavage. Am Rev Respir Dis. 1990, 142: 91-99.
Broide DH, Pain MM, Firestein GS: Eosinophils express interleukin 5 and granulocyte macrophage-colony-stimulating factor mRNA at sites of allergic inflammation in asthmatics. J Clin Invest. 1992, 90: 1414-1424.
Fukuda T, Nakajima H, Fukushima Y, Akutsu I, Namao T, Majima K, Motojima S, Sato Y, Takatsu K, Makino S: Detection of interleukin-5 messenger RNA and interleukin-5 protein in bronchial biopsies from asthma by nonradioactive in situ hybridization and immunohistochemistry. J Allergy Clin lmmunol. 1994, 94: 584-593.
Hamid Q, Azzawi M, Ying S, Moqbel R, Wardlaw AJ, Corrigan CJ, Bradley B, Durham SR, Collins JV, Jeffery PK, Quint DJ, Kay AB: Interleukin-5 mRNA in mucosal bronchial biopsies from asthmatic subjects. Int Arch Allergy Appl lmmunol. 1991, 94: 169-170.
Krishnaswamy G, Liu MC, Su S-N, Kumai M, Ziao H-Q, Marsh DG, Huang SK: Analysis of cytokine transcripts in the bronchoalveolar lavage cells of patients with asthma. Am J Respir Cell Mol Biol. 1993, 9: 279-286.
Robinson DS, Ying S, Bentley AM, Meng Q, North J, Durham SR, Kay AB, Hamid Q: Relationships among numbers of bronchoalveolar lavage cells expressing messenger ribonucleic acid for cytokines, asthma symptoms, and airway methacholine responsiveness in asthma. J Allergy Clin Immunol. 1993, 92: 397-403.
Robinson DS, Hamid Q, Ting S, Tsicopoulos A, Barkans J, Bentley AM, Corrigan C, Durham SR, Kay AB: Predominant TH2-like bronchoalveolar T-lymphocyte population in atopic asthma. N Engl J Med. 1992, 326: 298-304.
Bentley AM, Qiu Meng DS, Robinson DS, Hamid Q, Kay AB, Durham SR: Increases in activated T lymphocytes, eosinophils and cytokine mRNA expression for interleukin-5 and granulocyte/macrophage colony-stimulating factor in bronchial biopsies after allergen inhalation challenge in atopic asthmatics. Am J Respir Cell Mol Biol. 1993, 8: 35-42.
Walker C, Bauer W, Braun RK, Menz G, Braun P, Schwarz F, Hansel T-F, Villiger B: Activated T cells and cytokines in bronchoalveolar lavages from patients with various lung diseases associated with eosinophilia. Am J Respir Crit Care Med. 1994, 150: 1030-1048.
Ying S, Durham SR, Corrigan CJ, Hamid Q, Kay AB: Phenotype of cells expressing mRNA for TH2-type (interleukin 4 and interleukin 5) and TH1-type (interleukin 2 and interferon γ cytokines in bronchoalveolar lavage and bronchial biopsies from atopic asthmatic and normal control subjects. Am J Respir Cell Mol Biol. 1995, 12: 477-487.
Hamid Q, Boguniewicz M, Leung DYM: Differential in situ cytokine gene expession in acute versus chronic atopic dermatitis. J Clin Invest. 1994, 94: 870-876.
Kay AB, Ying S, Varney J, Gaga M, Durham SR, Moqbel R, Wardlaw AJ, Hamid Q: Messenger RNA expression of the cytokine gene cluster, interleukin 3 (IL-3), IL-4, IL-5, and granulocyte/macrophage colony-stimulating factor, in allergen-induced late-phase cutaneous reactions in atopic subjects. J Exp Med. 1991, 173: 775-778.
Tanaka Y, Delaporte E, Dubucquoi S, Gounni AS, Porchet E, Capron A, Capron M: lnterleukin-5 messenger RNA and immunoreactive protein expression by activated eosinophils in lesional atopic dermatitis skin. J Invest Dermatol. 1994, 103: 589-592.
Bradding P, Feather IH, Wilson S, Bardin PG, Heusser CH, Holgate ST, Howarth PH: Immunolocalization of cytokines in the nasal mucosa of normal and perennial rhinitic subjects. J Immunol. 1993, 151: 3853-3865.
Ying S, Durham SR, Barkans J, Masuyama K, Jacobson M, Rak S, Lowhagen O, Moqbel R, Kay AB, Hamid QA: T cells are the principal source of interleukin 5 mRNA in allergen-induced rhinitis. Am J Respir Cell Mol Biol. 1993, 9: 356-360.
Durham SR, Kay AB: Eosinophils, bronchial hyperreactivity and late phase asthmatic reactions. Clin Allergy. 1985, 13: 411-418.
Wardlaw AJ, Dunnett S, Gleich GJ, Collins JV, Kay AB: Eosinophils and mast cells in bronchoalveolar lavage in subjects with mild asthma: relationship to bronchial hyperreactivity. Am Rev Respir Dis. 1988, 137: 62-69.
Ohnishi T, Kita H, Weiler D, Sur S, Sedgwick JB, Calhoun WJ, Busse WW, Abrams JS, Gleich GJ: IL-5 is the predominant eosinophil-active cytokine in the antigen induced pulmonary late-phase reaction. Am Rev Respir Dis. 1993, 147: 901-907.
Robinson D, Hamid Q, Bentley A, Ying S, Kay AB, Durham SR: Activation of CD4+ T cells, increased Th2-type cytokine mRNA expression and eosinophil recruitment in bronchoalveolar lavage after allergen inhalation challenge in patients with atopic asthma. J Allergy Clin Imunol. 1993, 92: 313-324.
Sedgwick JB, Calhoun WC, Gleich GJ, Kita H, Abrams JS, Schwartz LB, Volovitz B, Ben-Yaakov M, Busse WW: Immediate and late airway response of allergic rhinitis patients to segmental antigen challenge. Characterization of eosinophil and mast cell mediators. Am Rev Respir Dis. 1991, 144: 1274-1281.
Terada N, Konno A, Natori T, Tada H, Togawa K: lnterleukin-5 preferentially recruits eosinophils from vessels in nasal mucosa. Acta Otolaryngol Suppl. 1993, 506: 57-60.
Terada N, Konno A, Tada H, Shirotori K, Ishikawa K, Togawa K: The effect of recombinant human interleukin-5 on eosinophil accumulation and degranulation in human nasal mucosa. J Allergy Clin Immunol. 1992, 90: 160-168.
Corrigan CJ, Haczku A, Gemou-Engesaeth V, Dol S, Kukuchi Y, Takatsu K, Durham SR, Kay AB: CD4 T-Lymphocyte activation in asthma is accompanied by increased serum concentrations of interleukin-5. Am Rev Respir Dis. 1993, 147: 540-547.
Motojima S, Akutsu I, Fukuda T, Makino S, Takatsu K: Clinical significance of measuring levels of sputum and serum ECP and serum IL-5 in bronchial asthma. Allergy. 1993, 48: 98-106.
Walker C, Bode E, Boer L, Hansel TT, Blaser K, Virchow J-C: Allergic and nonallergic asthmatics have distinct patterns of T-cell activation and cytokine production in peripheral blood and bronchoalveolar lavage. Am Rev Respir Dis. 1992, 146: 109-115.
Zangrilli JG, Shaver JR, Cirelli RA, Cho SK, Garlisi CG, Falcone A, Cuss FM, Fish JE, Peters SP: sVCAM-1 levels after segmental antigen challenge correlate with eosinophil influx, IL-4 and IL-5 production, and the late phase response. Am J Respir Crit Care Med. 1995, 151: 1346-1353.
Rolfe FG, Hughes JM, Armour CL, Sewell WA: Inhibition of interleukin-5 gene expression by dexamethasone. lmmunol. 1992, 77: 494-499.
Egan RW, Athwal E, Bodmer MW, Carter JM, Chapman RW, Chou C-C, Cox MA, Emtage JS, Fernandez X, Genatt N, Indelicato SR, Jenh C-H, Kreutner W, Kung TT, Mauser PJ, Minnicozzi M, Murgolo NJ, Narula SK, Petro ME, Schilling A, Sehring S, Stelts D, Stephens S, Taremi SS, Weiner SH, Zavodny PJ, Zurcher J: Effect of SCH 5 a humanized monoclonal antibody to human interleukin-5, on eosinophilic responses and bronchial hyperreactivity. Arzneimittel-Forschung. 1999, 49: 779-790.
Hart TK, Cook RM, Herzyk DJ, Zia-Amirhosseini P, Williams DM, Bugelski PJ: Inhibition of eosinophilia in monkeys with SB-24 a humanized anti-human IL-5 monoclonal antibody [abstract]. Am J Respir Crit Care Med. 1998, 157: A744-
Zia-Amirhosseini P, Minthorn E, Benincosa LJ, Hart TK, Hottenstein CS, Tobia LAP, Davis CB: Pharmacokinetics and pharmacodynamics of SB-24 a humanized monoclonal antibody directed to human interleukin-5, in monkeys. J Pharmacol Exp Ther. 1999, 291: 1060-1067.
Acknowledgement
The authors thank Mrs Maureen Frydlewicz for preparing the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Greenfeder, S., Umland, S.P., Cuss, F.M. et al. Th2 cytokines and asthma The role of interleukin-5 in allergic eosinophilic disease. Respir Res 2, 71 (2001). https://doi.org/10.1186/rr41
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/rr41