
Abstract
Cis-acting RNA elements control the accurate expression of human multi-exon protein coding genes. Single nucleotide variants altering the fidelity of this regulatory code and, consequently, pre-mRNA splicing are expected to contribute to the etiology of numerous human diseases.
Similar content being viewed by others
Introduction
Although genes span 33.4% of the human genome from start codon to stop codon, only 3.66% of their sequence comprises protein coding sequences [1]. Introns make up the rest of this gene space, separating adjacent protein coding exons from one another. To produce a mature mRNA that encodes a continuous string of codons, these exons must be put together following the precise excision of introns in a process referred to as precursor messenger RNA splicing (pre-mRNA splicing). Aberrant pre-mRNA splicing is now recognized as the underlying cause of many human diseases. Mutations in trans-acting factors or cis-acting regulatory elements compromise the expression of protein-coding genes by decreasing the specificity or fidelity of splice site selection, a fundamental step in expression of multi-exon genes.
At least three mechanisms can induce RNA-based disease. First, genetic variants such as point mutations can abolish cis-acting elements recognized by RNA binding proteins (RBPs), thereby inducing disease phenotypes in humans. Work on many disease genes, including the breast cancer gene BRCA1, the gene encoding the cystic fibrosis transmembrane conductance regulator (CFTR), the growth hormone gene GH1 and the ataxia telangiectasia mutated gene ATM, has demonstrated that all classes of point mutations, including nonsense mutations, can disrupt exonic splicing regulatory elements (ESRs) and induce aberrant pre-mRNA splicing [2–6]. Second, RBPs are implicated (either by mutation or aberrant expression) in numerous human diseases, including cancer, Alzheimer’s disease, frontotemporal dementia, spinal muscular atrophy (SMA) and retinitis pigmentosa (reviewed in [7]). These observations suggest that processing of specific transcripts or, more likely, families of related transcripts may be mis-regulated. Finally, RNAs transcribed from genes containing trinucleotide repeat expansions also induce disease. These toxic RNAs seem to function by sequestering RBPs and causing gross changes in post-transcriptional gene expression programs [8].
Given that other review articles have already done an exceptional job at summarizing the pleiotropic effects of RBPs and toxic RNA elements on pathogenesis [9–13], here we focus on aberrant protein-RNA interactions implicated in monogenic human diseases.
Splicing mechanism
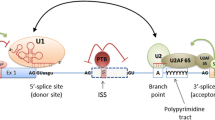
Pre-mRNA splicing is catalyzed by the spliceosome, a large ribonucleoprotein complex. The spliceosome assembles de novo on each and every transcribed intron and catalyzes two sequential trans-esterification reactions, which yield ligated exons and an excised intron-lariat [14]. The earliest stages of spliceosome assembly are critical in defining which exon sequences are to be joined during the splicing reaction [15]. Splicing of most human pre-mRNAs initiates in an exon-centric manner in which the upstream 3′ splice site and the downstream 5′ splice site are linked through interactions between U2 auxiliary factor (U2AF) and the U1 small ribonucleoprotein particle (U1 snRNP; Figure 1a) [16]. Later in the spliceosome assembly pathway these cross-exon interactions are replaced by intron-bridging interactions that connect the reactive 5′ and 3′ splice sites [16, 17].

Targets of single-nucleotide polymorphism-induced aberrant pre-mRNA splicing mutations. (a) Schematic diagram of the exon definition complex. 70 K, 70 kDa subunit of the U1 snRNP; ESE, exon splicing enhancer; RRM, RNA recognition motif protein domain; SR, serine-arginine rich protein domain; U1 snRNP, U1 small nuclear ribonucleoprotein; U2AF, U2 auxiliary factor; Y, pyrimidine nucleotide. Uppercase indicates donor and acceptor splice site dinucleotides, lowercase indicates adjacent consensus nucleotides. (b) Typical functionality of splicing regulatory elements in wild type (healthy) context. (c) Potential mechanisms for splicing-sensitive mutations. Green squares and arrows indicate splicing enhancers; red indicates silencers; solid boxes indicate constitutive or alternative exons; dashed boxes indicate pseudo-exons.
The decision of whether to splice or not to splice is typically modeled as a stochastic rather than deterministic process, such that even the most defined splicing signals can sometimes splice incorrectly [18]. However, under normal conditions, pre-mRNA splicing proceeds at surprisingly high fidelity [19]. This is attributed in part to the activity of adjacent cis-acting auxiliary exonic and intronic splicing regulatory elements (ESRs or ISRs) [20–24]. Typically, these functional elements are classified as either exonic or intronic splicing enhancers (ESEs or ISEs) or silencers (ESSs or ISSs) based on their ability to stimulate or inhibit splicing, respectively. Although there is now evidence that some auxiliary cis-acting elements may act by influencing the kinetics of spliceosome assembly, such as the arrangement of the complex between U1 snRNP and the 5′ splice site, it seems very likely that many elements function in concert with trans-acting RBPs [25]. For example, the serine- and arginine-rich family of RBPs (SR proteins) are a conserved family of proteins [26] that have a key role in defining exons [27]. SR proteins promote exon recognition by recruiting components of the pre-spliceosome to adjacent splice sites or by antagonizing the effects of ESSs in the vicinity [28–30]. The repressive effects of ESSs can be mediated by members of the heterogeneous nuclear ribonucleoprotein (hnRNP) family and can alter recruitment of core splicing factors to adjacent splice sites [31]. In addition to their roles in splicing regulation, silencer elements are suggested to have a role in repression of pseudo-exons, sets of decoy intronic splice sites with the typical spacing of an exon but without a functional open reading frame [32]. ESEs and ESSs, in cooperation with their cognate trans-acting RBPs, represent important components in a set of splicing controls that specify how, where and when mRNAs are assembled from their precursors [30, 33, 34].
Alternative splicing
The sequences marking the exon-intron boundaries are degenerate signals of varying strengths that occur at high frequency within human genes [35]. In multi-exon genes, different pairs of splice sites can be linked together in many different combinations, creating a diverse array of transcripts from a single gene [36, 37]. This is commonly referred to as alternative pre-mRNA splicing, and is classified into several discrete event types that have been observed both in vitro and in vivo[38].
Recent studies suggest that 86 to 94% of human multi-exon genes undergo alternative splicing [39, 40] and a considerable portion of human functional variation within the population is likely to cause changes at the transcript level [41]. The sheer abundance of this phenomenon is remarkable, raising the question of how many of the isoforms produced by a single gene encode functional messages. Although most mRNA isoforms produced by alternative splicing will be exported from the nucleus and translated into functional polypeptides, different mRNA isoforms from a single gene can vary greatly in their translation efficiency [42]. Those mRNA isoforms with premature termination codons at least 50 bp upstream of an exon junction complex are likely to be targeted for degradation by the nonsense-mediated mRNA decay (NMD) pathway [43]. Although this type of unproductive splicing is typically thought to be a byproduct of splicing as a stochastic process, the SR genes are clear examples of how this can be exploited as an essential regulatory mechanism [44–46]. SR proteins have been shown to regulate the splicing of their own genes, each of which contain an ultraconserved sequence [47] such as a poison exon containing a premature termination codon; when spliced into the mature RNA, these exons can trigger transcript degradation by NMD [48, 49]. The first example of this form of splicing factor autoregulation coupled to mRNA surveillance was characterized in the SRSF2/SC35 gene (a member of the SR family): high levels of the SRSF2/SC35 protein promote a 3′ untranslated region splicing event that destabilizes the SRSF2/SC35 mRNA [46].
Mis-splicing and monogenic diseases
Given that exon-intron boundaries can occur at any of the three positions of a codon, it is clear that only a subset of alternative splicing events can maintain the canonical open reading frame. For example, only exons that are evenly divisible by 3 can be skipped or included in the mRNA without any alteration of reading frame. Splicing events that do not have compatible phases will induce a frame-shift. Unless reversed by downstream events, frame-shifts will almost certainly lead to one or more premature termination codons, probably resulting in subsequent degradation by NMD. The most common frame-preserving alternative event type is compatibly phased exon skipping; however, 20% of all frame-preserving alternative splicing events involve the alternative use of adjacent 3′ NAGNAG splice sites [50, 51]. Several studies have investigated the evolution of multi-exon gene architectures and found significant correlation of the edges of exons with protein domain boundaries [52, 53]. Furthermore, exons whose edges correlated with protein domain boundaries were significantly enriched for compatible splice site phase. These observations have been used as evidence for the evolutionary hypothesis of exon shuffling, a mechanism for diversification of modular protein functions [54, 55]. Moreover, the data clearly support the postulate that evolutionary history of a gene will affect its susceptibility to alternative splicing-induced frame-shifting.
Following a model of neutral genetic drift, some genes are under greater selective constraints than others. Genes encoding proteins that have vital and non-redundant roles may impart a major loss of fitness to an organism if disrupted by germline and somatic mutation. Depending on protein structure and function and exon-intron architecture, these genes may be more or less susceptible to aberrant function by different means. For example, different mutations causing loss of function in CFTR can cause varying levels of severity of cystic fibrosis (CF) [56]. Although 70% of CF cases are at least heterozygous for a deletion of phenylalanine 508 (ΔF508) that impairs protein folding and subsequent function [57], only four other mutations (G542X, N1303K, G551D and W1282X) have allele frequencies above 1% [58]. This leaves a percentage of atypical CF-associated mutations that are rare or unique to individuals or families, resulting in roughly 15% of all CF cases having mutations with unknown functions [59]. Moreover, about 13 to 20% of all the CF-associated mutations are thought to cause pre-mRNA splicing defects by aberrant inclusion or exclusion of several of the 27 exons as a primary mechanism of disease causation [10]. At least one of these, exon 9, has been studied in great detail, illuminating a complex set of regulatory elements that regulate its alternative splicing [60–62].
High-throughput DNA sequencing is now revealing the extent of human genetic variation on a comprehensive scale. However, because of the complexity of these data, it is often unclear which variants are functional and which biochemical mechanisms they affect [63]. For genes that are highly susceptible to aberrant splicing by a number of different mechanisms (such as CFTR; Figure 1), determining the penetrance associated with de novo atypical mutations is a crucial gap towards comprehensive molecular diagnosis for their associated diseases. To tackle this problem for CFTR and other genes with pre-mRNA splicing defects, it is necessary to consider the possible mechanistic impacts of a point mutation on the splicing machinery. Figure 1b,c illustrates some of the architectural features of a generic wild-type (healthy) gene, such as: the presence of one or more exonic splicing enhancers; splicing silencers that work to repress intronic pseudo-exons; and cryptic splice sites. Mutation of 5′ and 3′ splice site dinucleotides and adjacent bases can render them inactive; this is the most easily recognized mechanism of splicing disruption, accounting for 10% of all human inherited disease mutations [64]. For this reason, disruption of the GU and AG splice site dinucleotides are recognized as deleterious by most of the recent single-nucleotide polymorphism functional classification tools, such as those based on SIFT [65, 66]. However, the need for methods or tools to evaluate the impact of genetic variants towards the loss or gain of both ISRs and ESRs remains critical.
Work from our group and another suggest that 22 to 25% of exonic human inherited disease mutations are likely to be splicing sensitive [67, 68]. We find that this percentage is unevenly distributed across different diseases (Tables 1 and 2), suggesting that there is a spectrum of susceptibility towards aberrant gene regulation through loss or gain of ESRs. In the case of the Duchenne muscular dystrophy (DMD) gene, for example, rather than the approximately 22 to 25% presented previously [67, 68], we find that as many as 120 mutations, representing nearly half of all the missense and nonsense disease-causing mutations targeting this gene, cause the loss or gain of disease enriched ESRs. This suggests an expanded role for splicing mutations relating to Duchenne or the less severe Becker muscular dystrophy. Future studies that include mutations affecting intronic cis-acting elements may shed light on an additional class of splicing-sensitive variants.
We find it intriguing that the architecture of different genes renders some more sensitive to mutation-induced aberrant splicing than others. Within this subset of splicing-sensitive genes we noted that the proportion of cancer-related genes [68] increases with the percentage of putative splicing-sensitive mutations (Figure 2). Of the 492 genes from the Human Gene Mutation Database considered by our analysis, only 11% are cancer related [69] while this proportion is three-fold higher within the top 50 splicing-sensitive genes (χ2 goodness-of-fit P-value of 1.2 × 10-4). Summarizing the data in Tables 1 and 2, it becomes apparent that the majority of these are known tumor suppressor genes, providing support for the motion to recognize aberrant splicing as a hallmark of cancer [70]. Aberrant splicing has already been directly implicated as a causative mechanism for disruption of many of these genes previously [71]. Missense and nonsense mutations in the mismatch repair genes MLH1 and MSH2 have both been shown to cause aberrant splicing in multiple contexts [72–75]. Likewise, mutations in the APC tumor suppressor gene have aberrant isoforms that are thought to be degraded by NMD [76]. These data suggest the intriguing hypothesis that cancer-related genes may have a greater susceptibility towards aberrant splicing than other genes.

Splicing susceptibility of cancer related genes. The positive increase in cancer-associated genes is dependent on the overall percentage of putative splicing mutations within the gene (black line). Independent permutations followed by re-plotting without any putative splicing sensitivity measure results in expected numbers of cancer-associated genes (dotted gray line). Cancer-associated genes are defined as the 510 genes represented in the Cancer Census [69].
Future therapeutic potential
The susceptibility of many disease genes (such as DMD, ATM, NPC1 (Niemann-Pick disease), F9 (hemophilia A), F8 (hemophilia B)) to aberrant pre-mRNA splicing has spawned creative therapeutic approaches that have been the focus of a great deal of time and effort [77–84]. Of these, one of the most successful cases has been the reversal of aberrant exon 7 skipping in the SMA-related gene SMN2 by antisense oligonucleotides [79, 83, 84]. SMA is an autosomal recessive disorder that is characterized by varying severity due to the loss of function of SMN1, of which humans have one copy on each chromosome 5. A nearly identical paralog, SMN2, has only five single nucleotide differences, all of which are non-coding except one C > T synonymous mutation six bases from the 3′ splice site within exon 7. The mechanistic impact of this C > T transition has been studied extensively, and has been shown to be associated with both the loss of an ESE that binds SRFS1 to stimulate exon definition [85, 86] and the antagonistic gain of an ESS that binds hnRNP A1 to repress exon definition [5]. In vivo selection studies and antisense oligonucleotide tiling experiments have additionally discovered a number of other regulatory elements within and adjacent to this exon [80, 87, 88].
Because individuals with SMA typically have loss of SMN1 but normal copies of SMN2, research into a general treatment for SMA has been targeted towards methods to increase splicing of endogenous SMN2 exon 7 as a means to increase functional SMN protein. Recent studies have robustly ameliorated symptoms of severe SMA mouse models through delivery of antisense oligonucleotides masking the ESS-N1 element [79, 83, 84], demonstrating that antisense approaches may represent an effective treatment for SMA. Although most inherited disease-related genes do not have a backup copy similar to SMN2 to serve as a template for RNA targeted therapies, this scenario does illuminate the potential feasibility of rational nucleic acid-based therapeutics in the coming years.
Conclusion
Functional characterization of both germline and somatic variants remains a considerable challenge. This is due in part to the limited understanding of the gene architectural contexts that give rise to varying degrees of susceptibility to aberrant processing. How different degrees of susceptibility contribute to the etiology of inherited and somatic diseases remains a crucial question in the field. This question is becoming increasingly important for several inherited and somatic diseases, including cancer. The root of this question lies at the heart of unraveling the networks of protein-RNA interaction that are active in various cellular contexts. Beyond this task lies the promise of potentially groundbreaking therapeutic approaches based on correcting aberrant protein-RNA interactions within a cell.
Abbreviations
- CF:
-
Cystic fibrosis
- CFTR:
-
Cystic fibrosis transmembrane conductance regulator
- ESE:
-
Exonic splicing enhancer
- ESR:
-
Exonic splicing regulator
- ESS:
-
Exonic splicing silencer
- ISE:
-
Intronic splicing enhancer
- ISS:
-
Intronic splicing silencer
- NMD:
-
Nonsense-mediated decay
- RBP:
-
RNA binding protein
- SMA:
-
Spinal muscular atrophy
- snRNP:
-
small ribonucleoprotein particle
- SR:
-
Protein serine- and arginine-rich protein.
References
Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S, Barnes I, Bignell A, Boychenko V, Hunt T, Kay M, Mukherjee G, Rajan J, Despacio-Reyes G, Saunders G, Steward C, Harte R, Lin M, Howald C, Tanzer A, Derrien T, Chrast J, Walters N, Balasubramanian S, Pei B, Tress M, et al: GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 2012, 22: 1760-1774. 10.1101/gr.135350.111.
Teraoka SN, Telatar M, Becker-Catania S, Liang T, Onengut S, Tolun A, Chessa L, Sanal O, Bernatowska E, Gatti RA, Concannon P: Splicing defects in the ataxia-telangiectasia gene, ATM: underlying mutations and consequences. Am J Hum Genet. 1999, 64: 1617-1631. 10.1086/302418.
Liu HX, Cartegni L, Zhang MQ, Krainer AR: A mechanism for exon skipping caused by nonsense or missense mutations in BRCA1 and other genes. Nat Genet. 2001, 27: 55-58.
Moseley CT, Mullis PE, Prince MA, Phillips JA: An exon splice enhancer mutation causes autosomal dominant GH deficiency. J Clin Endrocrinol Metab. 2002, 87: 847-852.
Kashima T, Manley JL: A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy. Nat Genet. 2003, 34: 460-463. 10.1038/ng1207.
Pagani F, Buratti E, Stuani C, Baralle FE: Missense, nonsense, and neutral mutations define juxtaposed regulatory elements of splicing in cystic fibrosis transmembrane regulator exon 9. J Biol Chem. 2003, 278: 26580-26588. 10.1074/jbc.M212813200.
Wang GS, Cooper TA: Splicing in disease: disruption of the splicing code and the decoding machinery. Nat Rev Genet. 2007, 8: 749-761. 10.1038/nrg2164.
Du H, Cline MS, Osborne RJ, Tuttle DL, Clark TA, Donohue JP, Hall MP, Shiue L, Swanson MS, Thornton CA, Ares M: Aberrant alternative splicing and extracellular matrix gene expression in mouse models of myotonic dystrophy. Nat Struct Mol Biol. 2010, 17: 187-193. 10.1038/nsmb.1720.
Faustino NA, Cooper TA: Pre-mRNA splicing and human disease. Genes Dev. 2003, 17: 419-437. 10.1101/gad.1048803.
Garcia-Blanco MA, Baraniak AP, Lasda EL: Alternative splicing in disease and therapy. Nat Biotechnol. 2004, 22: 535-546. 10.1038/nbt964.
Tazi J, Bakkour N, Stamm S: Alternative splicing and disease. Biochim Biophys Acta. 2009, 1792: 14-26. 10.1016/j.bbadis.2008.09.017.
Singh RK, Cooper TA: Pre-mRNA splicing in disease and therapeutics. Trends Mol Med. 2012, 18: 472-482. 10.1016/j.molmed.2012.06.006.
Cooper TA, Wan L, Dreyfuss G: RNA and disease. Cell. 2009, 136: 777-793. 10.1016/j.cell.2009.02.011.
Fica SM, Tuttle N, Novak T, Li N-S, Lu J, Koodathingal P, Dai Q, Staley JP, Piccirilli JA: RNA catalyses nuclear pre-mRNA splicing. Nature. 2013, doi:10.1038/nature12734
Berget SM: Exon recognition in vertebrate splicing. J Biol Chem. 1995, 270: 2411-2414.
Schneider M, Will CL, Anokhina M, Tazi J, Urlaub H, Luhrmann R: Exon definition complexes contain the tri-snRNP and can be directly converted into B-like precatalytic splicing complexes. Mol Cell. 2010, 38: 223-235. 10.1016/j.molcel.2010.02.027.
Lim SR, Hertel KJ: Commitment to splice site pairing coincides with A complex formation. Mol Cell. 2004, 15: 477-483. 10.1016/j.molcel.2004.06.025.
Melamud E, Moult J: Stochastic noise in splicing machinery. Nucleic Acids Res. 2009, 37: 4873-4886. 10.1093/nar/gkp471.
Fox-Walsh KL, Hertel KJ: Splice-site pairing is an intrinsically high fidelity process. Proc Natl Acad Sci U S A. 2009, 106: 1766-1771. 10.1073/pnas.0813128106.
Goren A, Ram O, Amit M, Keren H, Lev-Maor G, Vig I, Pupko T, Ast G: Comparative analysis identifies exonic splicing regulatory sequences–the complex definition of enhancers and silencers. Mol Cell. 2006, 22: 769-781. 10.1016/j.molcel.2006.05.008.
Zhang XH, Kangsamaksin T, Chao MS, Banerjee JK, Chasin LA: Exon inclusion is dependent on predictable exonic splicing enhancers. Mol Cell Biol. 2005, 25: 7323-7332. 10.1128/MCB.25.16.7323-7332.2005.
Yeo G, Hoon S, Venkatesh B, Burge CB: Variation in sequence and organization of splicing regulatory elements in vertebrate genes. Proc Natl Acad Sci U S A. 2004, 101: 15700-15705. 10.1073/pnas.0404901101.
Fairbrother WG, Yeh RF, Sharp PA, Burge CB: Predictive identification of exonic splicing enhancers in human genes. Science. 2002, 297: 1007-1013. 10.1126/science.1073774.
Wang Z, Rolish ME, Yeo G, Tung V, Mawson M, Burge CB: Systematic identification and analysis of exonic splicing silencers. Cell. 2004, 119: 831-845. 10.1016/j.cell.2004.11.010.
Blencowe BJ: Exonic splicing enhancers: mechanism of action, diversity and role in human genetic diseases. Trends Biochem Sci. 2000, 25: 106-110. 10.1016/S0968-0004(00)01549-8.
Zahler AM, Lane WS, Stolk JA, Roth MB: SR proteins: a conserved family of pre-mRNA splicing factors. Genes Dev. 1992, 6: 837-847. 10.1101/gad.6.5.837.
Liu HX, Zhang M, Krainer AR: Identification of functional exonic splicing enhancer motifs recognized by individual SR proteins. Genes Dev. 1998, 12: 1998-2012. 10.1101/gad.12.13.1998.
Kohtz JD, Jamison SF, Will CL, Zuo P, Luhrmann R, Garcia-Blanco MA, Manley JL: Protein-protein interactions and 5′-splice-site recognition in mammalian mRNA precursors. Nature. 1994, 368: 119-124. 10.1038/368119a0.
Graveley BR, Hertel KJ, Maniatis T: The role of U2AF35 and U2AF65 in enhancer-dependent splicing. RNA. 2001, 7: 806-818. 10.1017/S1355838201010317.
Zhu J, Mayeda A, Krainer AR: Exon identity established through differential antagonism between exonic splicing silencer-bound hnRNP A1 and enhancer-bound SR proteins. Mol Cell. 2001, 8: 1351-1361. 10.1016/S1097-2765(01)00409-9.
Yu Y, Maroney PA, Denker JA, Zhang XH, Dybkov O, Luhrmann R, Jankowsky E, Chasin LA, Nilsen TW: Dynamic regulation of alternative splicing by silencers that modulate 5′ splice site competition. Cell. 2008, 135: 1224-1236. 10.1016/j.cell.2008.10.046.
Sironi M, Menozzi G, Riva L, Cagliani R, Comi GP, Bresolin N, Giorda R, Pozzoli U: Silencer elements as possible inhibitors of pseudoexon splicing. Nucleic Acids Res. 2004, 32: 1783-1791. 10.1093/nar/gkh341.
Wang Z, Burge CB: Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA. 2008, 14: 802-813. 10.1261/rna.876308.
Barash Y, Calarco JA, Gao W, Pan Q, Wang X, Shai O, Blencowe BJ, Frey BJ: Deciphering the splicing code. Nature. 2010, 465: 53-59. 10.1038/nature09000.
Keren H, Lev-Maor G, Ast G: Alternative splicing and evolution: diversification, exon definition and function. Nat Rev Genet. 2010, 11: 345-355. 10.1038/nrg2776.
Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, Xue C, Marinov GK, Khatun J, Williams BA, Zaleski C, Rozowsky J, Röder M, Kokocinski F, Abdelhamid RF, Alioto T, Antoshechkin I, Baer MT, Bar NS, Batut P, Bell K, Bell I, Chakrabortty S, Chen X, Chrast J, Curado J, et al: Landscape of transcription in human cells. Nature. 2012, 489: 101-108. 10.1038/nature11233.
Matlin AJ, Clark F, Smith CW: Understanding alternative splicing: towards a cellular code. Nat Rev Mol Cell Biol. 2005, 6: 386-398. 10.1038/nrm1645.
Black DL: Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem. 2003, 72: 291-336. 10.1146/annurev.biochem.72.121801.161720.
Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ: Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet. 2008, 40: 1413-1415. 10.1038/ng.259.
Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB: Alternative isoform regulation in human tissue transcriptomes. Nature. 2008, 456: 470-476. 10.1038/nature07509.
Lappalainen T, Sammeth M, Friedlander MR, Hoen PA T, Monlong J, Rivas MA, Gonzalez-Porta M, Kurbatova N, Griebel T, Ferreira PG, Barann M, Wieland T, Greger L, van Iterson M, Almlöf J, Ribeca P, Pulyakhina I, Esser D, Giger T, Tikhonov A, Sultan M, Bertier G, MacArthur DG, Lek M, Lizano E, Buermans HP, Padioleau I, Schwarzmayr T, Karlberg O, Ongen H, et al: Transcriptome and genome sequencing uncovers functional variation in humans. Nature. 2013, 501: 506-511. 10.1038/nature12531.
Sterne-Weiler T, Martinez-Nunez RT, Howard JM, Cvitovik I, Katzman S, Tariq MA, Pourmand N, Sanford JR: Frac-seq reveals isoform-specific recruitment to polyribosomes. Genome Res. 2013, 23: 1615-1623. 10.1101/gr.148585.112.
Nagy E, Maquat LE: A rule for termination-codon position within intron-containing genes: when nonsense affects RNA abundance. Trends Biochem Sci. 1998, 23: 198-199. 10.1016/S0968-0004(98)01208-0.
Jumaa H, Nielsen PJ: The splicing factor SRp20 modifies splicing of its own mRNA and ASF/SF2 antagonizes this regulation. EMBO J. 1997, 16: 5077-5085. 10.1093/emboj/16.16.5077.
Lejeune F, Cavaloc Y, Stevenin J: Alternative splicing of intron 3 of the serine/arginine-rich protein 9G8 gene. Identification of flanking exonic splicing enhancers and involvement of 9G8 as a trans-acting factor. J Biol Chem. 2001, 276: 7850-7858. 10.1074/jbc.M009510200.
Sureau A, Gattoni R, Dooghe Y, Stevenin J, Soret J: SC35 autoregulates its expression by promoting splicing events that destabilize its mRNAs. EMBO J. 2001, 20: 1785-1796. 10.1093/emboj/20.7.1785.
Bejerano G, Pheasant M, Makunin I, Stephen S, Kent WJ, Mattick JS, Haussler D: Ultraconserved elements in the human genome. Science. 2004, 304: 1321-1325. 10.1126/science.1098119.
Ni JZ, Grate L, Donohue JP, Preston C, Nobida N, O’Brien G, Shiue L, Clark TA, Blume JE, Ares M: Ultraconserved elements are associated with homeostatic control of splicing regulators by alternative splicing and nonsense-mediated decay. Genes Dev. 2007, 21: 708-718. 10.1101/gad.1525507.
Lareau LF, Inada M, Green RE, Wengrod JC, Brenner SE: Unproductive splicing of SR genes associated with highly conserved and ultraconserved DNA elements. Nature. 2007, 446: 926-929. 10.1038/nature05676.
Akerman M, Mandel-Gutfreund Y: Alternative splicing regulation at tandem 3′ splice sites. Nucleic Acids Res. 2006, 34: 23-31. 10.1093/nar/gkj408.
Bradley RK, Merkin J, Lambert NJ, Burge CB: Alternative splicing of RNA triplets is often regulated and accelerates proteome evolution. PLoS Biol. 2012, 10: e1001229-10.1371/journal.pbio.1001229.
Kaessmann H, Zollner S, Nekrutenko A, Li WH: Signatures of domain shuffling in the human genome. Genome Res. 2002, 12: 1642-1650. 10.1101/gr.520702.
Liu M, Grigoriev A: Protein domains correlate strongly with exons in multiple eukaryotic genomes–evidence of exon shuffling?. Trends Genet. 2004, 20: 399-403. 10.1016/j.tig.2004.06.013.
Kolkman JA, Stemmer WP: Directed evolution of proteins by exon shuffling. Nat Biotechnol. 2001, 19: 423-428. 10.1038/88084.
Long M, Betran E, Thornton K, Wang W: The origin of new genes: glimpses from the young and old. Nat Rev Genet. 2003, 4: 865-875.
Noone PG, Knowles MR: ‘CFTR-opathies’: disease phenotypes associated with cystic fibrosis transmembrane regulator gene mutations. Respir Res. 2001, 2: 328-332. 10.1186/rr82.
Lukacs GL, Verkman AS: CFTR: folding, misfolding and correcting the DeltaF508 conformational defect. Trends Mol Med. 2012, 18: 81-91. 10.1016/j.molmed.2011.10.003.
Rowntree RK, Harris A: The phenotypic consequences of CFTR mutations. Ann Hum Genet. 2003, 67: 471-485. 10.1046/j.1469-1809.2003.00028.x.
Farrell PM, Rosenstein BJ, White TB, Accurso FJ, Castellani C, Cutting GR, Durie PR, Legrys VA, Massie J, Parad RB, Rock MJ, Campbell PW, Cystic Fibrosis Foundation: Guidelines for diagnosis of cystic fibrosis in newborns through older adults: cystic Fibrosis foundation consensus report. J Pediatr. 2008, 153: S4-S14. 10.1016/j.jpeds.2008.05.005.
Chu CS, Trapnell BC, Curristin S, Cutting GR, Crystal RG: Genetic basis of variable exon 9 skipping in cystic fibrosis transmembrane conductance regulator mRNA. Nat Genet. 1993, 3: 151-156. 10.1038/ng0293-151.
Buratti E, Dork T, Zuccato E, Pagani F, Romano M, Baralle FE: Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. EMBO J. 2001, 20: 1774-1784. 10.1093/emboj/20.7.1774.
Niksic M, Romano M, Buratti E, Pagani F, Baralle FE: Functional analysis of cis-acting elements regulating the alternative splicing of human CFTR exon 9. Hum Mol Genet. 1999, 8: 2339-2349. 10.1093/hmg/8.13.2339.
Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA: An integrated map of genetic variation from 1,092 human genomes. Nature. 2012, 491: 56-65. 10.1038/nature11632.
Krawczak M, Thomas NS, Hundrieser B, Mort M, Wittig M, Hampe J, Cooper DN: Single base-pair substitutions in exon-intron junctions of human genes: nature, distribution, and consequences for mRNA splicing. Hum Mutat. 2007, 28: 150-158. 10.1002/humu.20400.
Cingolani P, Platts A, Wang Le L, Coon M, Nguyen T, Wang L, Land SJ, Lu X: Ruden DM: A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 2012, 6: 80-92. 10.4161/fly.19695.
Medina I, De Maria A, Bleda M, Salavert F, Alonso R, Gonzalez CY, Dopazo J: VARIANT: command line, web service and web interface for fast and accurate functional characterization of variants found by next-generation sequencing. Nucleic Acids Res. 2012, 40: W54-W58. 10.1093/nar/gks572.
Lim KH, Ferraris L, Filloux ME, Raphael BJ, Fairbrother WG: Using positional distribution to identify splicing elements and predict pre-mRNA processing defects in human genes. Proc Natl Acad Sci U S A. 2011, 108: 11093-11098. 10.1073/pnas.1101135108.
Sterne-Weiler T, Howard J, Mort M, Cooper DN, Sanford JR: Loss of exon identity is a common mechanism of human inherited disease. Genome Res. 2011, 21: 1563-1571. 10.1101/gr.118638.110.
Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, Rahman N, Stratton MR: A census of human cancer genes. Nat Rev Cancer. 2004, 4: 177-183. 10.1038/nrc1299.
Ladomery M: Aberrant alternative splicing is another hallmark of cancer. Int J Cell Biol. 2013, 2013: 463786-
Miura K, Fujibuchi W, Sasaki I: Alternative pre-mRNA splicing in digestive tract malignancy. Cancer Sci. 2011, 102: 309-316. 10.1111/j.1349-7006.2010.01797.x.
Stella A, Wagner A, Shito K, Lipkin SM, Watson P, Guanti G, Lynch HT, Fodde R, Liu B: A nonsense mutation in MLH1 causes exon skipping in three unrelated HNPCC families. Cancer Res. 2001, 61: 7020-7024.
McVety S, Li L, Gordon PH, Chong G, Foulkes WD: Disruption of an exon splicing enhancer in exon 3 of MLH1 is the cause of HNPCC in a Quebec family. J Med Genet. 2006, 43: 153-156.
Pagenstecher C, Wehner M, Friedl W, Rahner N, Aretz S, Friedrichs N, Sengteller M, Henn W, Buettner R, Propping P, Mangold E: Aberrant splicing in MLH1 and MSH2 due to exonic and intronic variants. Hum Genet. 2006, 119: 9-22. 10.1007/s00439-005-0107-8.
Lastella P, Surdo NC, Resta N, Guanti G, Stella A: In silico and in vivo splicing analysis of MLH1 and MSH2 missense mutations shows exon- and tissue-specific effects. BMC Genomics. 2006, 7: 243-10.1186/1471-2164-7-243.
De Rosa M, Morelli G, Cesaro E, Duraturo F, Turano M, Rossi GB, Delrio P, Izzo P: Alternative splicing and nonsense-mediated mRNA decay in the regulation of a new adenomatous polyposis coli transcript. Gene. 2007, 395: 8-14. 10.1016/j.gene.2006.10.027.
Meyer K, Marquis J, Trub J, Nlend Nlend R, Verp S, Ruepp MD, Imboden H, Barde I, Trono D, Schumperli D: Rescue of a severe mouse model for spinal muscular atrophy by U7 snRNA-mediated splicing modulation. Hum Mol Genet. 2009, 18: 546-555.
Hua Y, Vickers TA, Okunola HL, Bennett CF, Krainer AR: Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am J Hum Genet. 2008, 82: 834-848. 10.1016/j.ajhg.2008.01.014.
Hua Y, Sahashi K, Hung G, Rigo F, Passini MA, Bennett CF, Krainer AR: Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev. 2010, 24: 1634-1644. 10.1101/gad.1941310.
Hua Y, Vickers TA, Baker BF, Bennett CF, Krainer AR: Enhancement of SMN2 exon 7 inclusion by antisense oligonucleotides targeting the exon. PLoS Biol. 2007, 5: e73-10.1371/journal.pbio.0050073.
Coady TH, Lorson CL: Trans-splicing-mediated improvement in a severe mouse model of spinal muscular atrophy. J Neurosci. 2010, 30: 126-130. 10.1523/JNEUROSCI.4489-09.2010.
Baughan TD, Dickson A, Osman EY, Lorson CL: Delivery of bifunctional RNAs that target an intronic repressor and increase SMN levels in an animal model of spinal muscular atrophy. Hum Mol Genet. 2009, 18: 1600-1611. 10.1093/hmg/ddp076.
Passini MA, Bu J, Richards AM, Kinnecom C, Sardi SP, Stanek LM, Hua Y, Rigo F, Matson J, Hung G, Kaye EM, Shihabuddin LS, Krainer AR, Bennett CF, Cheng SH: Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci Transl Med. 2011, 3: 72ra18-
Hua Y, Sahashi K, Rigo F, Hung G, Horev G, Bennett CF, Krainer AR: Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature. 2011, 478: 123-126. 10.1038/nature10485.
Cartegni L, Krainer AR: Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat Genet. 2002, 30: 377-384. 10.1038/ng854.
Cartegni L, Hastings ML, Calarco JA, de Stanchina E, Krainer AR: Determinants of exon 7 splicing in the spinal muscular atrophy genes, SMN1 and SMN2. Am J Hum Genet. 2006, 78: 63-77. 10.1086/498853.
Kashima T, Rao N, Manley JL: An intronic element contributes to splicing repression in spinal muscular atrophy. Proc Natl Acad Sci U S A. 2007, 104: 3426-3431. 10.1073/pnas.0700343104.
Singh NN, Androphy EJ, Singh RN: An extended inhibitory context causes skipping of exon 7 of SMN2 in spinal muscular atrophy. Biochem Biophys Res Comm. 2004, 315: 381-388. 10.1016/j.bbrc.2004.01.067.
Acknowledgments
We wish to thank our funding agencies, including the Ellison Foundation for Medical Research (JRS), NIH NIA (JRS) and The Santa Cruz Cancer Benefit Group (JRS). We also thank Jon Howard for his thoughtful comments on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Sterne-Weiler, T., Sanford, J.R. Exon identity crisis: disease-causing mutations that disrupt the splicing code. Genome Biol 15, 201 (2014). https://doi.org/10.1186/gb4150
Published:
DOI: https://doi.org/10.1186/gb4150