
Abstract
De novo assembly is a commonly used application of next-generation sequencing experiments. The ultimate goal is to puzzle millions of reads into one complete genome, although draft assemblies usually result in a number of gapped scaffold sequences. In this paper we propose an automated strategy, called GapFiller, to reliably close gaps within scaffolds using paired reads. The method shows good results on both bacterial and eukaryotic datasets, allowing only few errors. As a consequence, the amount of additional wetlab work needed to close a genome is drastically reduced. The software is available at http://www.baseclear.com/bioinformatics-tools/.
Similar content being viewed by others

Background
De novo sequencing of unknown species and variants has become a major application of next-generation sequencing (NGS) technologies. As a consequence, there is a high demand for de novo assembly tools that can reconstruct the genomic sequence from millions of short reads. At present a vast number of tools is available to create a draft assembly that represents the genome in a number of contiguous sequences (contigs), such as Velvet [1], ABySS [2], and SOAPdenovo [3]. Nonetheless, the full closure of a complete genome remains a challenging and often expensive task. Generally, problems reside in the assembly of low-coverage areas and repetitive elements. Even though the distance information of paired-read sequences can help to bridge these difficult areas by linking the contigs into larger scaffolds [4, 5], scaffolding does not solve the inherent problem of low-coverage and repetitive elements: it does not add novel sequence information to the draft assembly. At present, two strategies have been proposed to automatically close draft assemblies: Li et al. [3] included a gap closure routine to their SOAPdenovo assembly software, whereas more recently Tsai et al. [6] introduced the IMAGE algorithm. Both methods use De Bruijn graphs to create a local assembly and seek to fill the gaps with the resulting contigs. A drawback of these strategies is that no prior knowledge of the estimated gap size is taken into account. Therefore, the local assembly might not reflect the true genomic situation. Also, the practical usability of these methods by a broad audience requires additional development. As a consequence, at present, most genome assemblies are still incomplete after scaffolding and would ideally require accurate gap closure. Given that manual closure of incomplete regions with Sanger sequencing is expensive, genome assemblies submitted to sequence databases often lack a significant number of nucleotides, especially in the case of larger eukaryotes. Importantly, the missing repetitive or low-coverage regions may contain essential functions for the organism studied.
In this study we aim to reduce the number of undefined nucleotides by automatically filling in the gapped regions within scaffolds. For this purpose, paired reads are (re)used. In brief, our GapFiller method seeks to find read pairs of which one member matches within a sequence region and the second member falls (partially) within the gap. The latter reads are then used to close the gap through sequence (k-mer) overlap. Gaps are entirely closed only if the size of the sequence insertion corresponds closely to the estimated gap size after scaffolding, which is based on the alignment of paired reads to the contigs. The process is iteratively repeated until no further gaps can be closed. We demonstrate that GapFiller gives promising results both on bacterial and eukaryotic datasets and accurately closes most of the gaps. From a comparative analysis with IMAGE and SOAPdenovo we show that our algorithm provides the most reliable outcomes. Finally, we investigate the nature of the closed regions in the human genome and show that these contain important coding and non-coding information. The software is designed to be accessible to a broad audience given its user-friendly design and limited memory usage. GapFiller can be downloaded from [7].
Results
Findings on the bacterial datasets
GapFiller was evaluated on four distinct bacterial datasets that are characterized by different genome sizes, levels of complexity and short-read sequence input. Also, the assembly strategy underlying the construction of the scaffolds varies and provides some good practical examples of draft assemblies (see Materials and methods for more information). Quality assessment was performed through comparison with the known (closed) reference genome and error types were classified into three categories - SNPs, indels and misjunctions - according to [8]. A first observation on these datasets is that automated gap closure can indeed be an important step in finishing (bacterial) genomes. In Table 1 we show that all methods can effectively close a significant amount of gaps and also that the total gap length is drastically reduced. However, from the Escherichia coli and Streptomyces coelicolor datasets it is apparent that the IMAGE strategy clearly underperforms the other two strategies on most quality measurements: the total gap length is reduced at most by 60% even though the number of errors increases significantly. This is partly due to the fact that IMAGE can not handle multiple libraries (nor mate pair libraries), and therefore the analysis was performed using only the library with the shortest insert size. In contrast, SOAPdenovo and GapFiller show more promising results: in E. coli the number of gaps (gap count) can be reduced by 97% and 98%, respectively; in S. coelicolor these numbers are 62% and 85%, respectively. Overall, the results indicate GapFiller closes a larger amount of gaps while making significantly less errors than the other methods. Arguably, the total gap length is shorter after gap closure with SOAPdenovo, but apparently at the cost of an increased number of misassemblies. The latter two tendencies are more pronounced in the Staphylococcus aureus and Rhodobacter sphaeroides datasets. A likely explanation resides in the fact that the raw input FASTQ reads are heavily filtered with Quake [9], leading to a relatively low genome coverage. For comparison purposes we reran our GapFiller algorithm with less stringent settings (GapFiller-LC: min coverage o = 1, ratio r = 0.5) to yield a more similar gap length to SOAPdenovo. We observe that applying less conservative settings leads (obviously) to more errors, but that these are still significantly less than those made with SOAPdenovo (regardless of the error type).
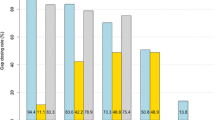
Another interesting observation concerns the computational resources needed to run the software. Figure 1 shows a comparative analysis between the runtimes and memory usage of SOAPdenovo and GapFiller based on one iterative cycle. Apparently SOAPdenovo can complete the process faster if the number of input reads is very low or high, but for intermediate data sizes (10 to 20 million reads) GapFiller needs somewhat less time. Differences are more pronounced when comparing the memory usage of both methods: SOAPdenovo requires an increasing amount of memory to analyze larger input datasets (up to 3.8 GB for S. coelicolor and 6.0 GB for human) whereas GapFiller shows stable memory usage of approximately 0.1 GB. This is because GapFiller seeks to store the intermediate output temporarily rather than maintaining it in the memory. As a consequence the method is well suited for computing systems with smaller amounts of memory.

Time and memory consumption of gap closure software. Comparative analysis of the runtime and memory usage per dataset based on a single iteration. SOAPdenovo needs a shorter time to complete the analysis if the amount of data is very small or large, whereas GapFiller is faster for intermediate data sizes (10 to 20 million reads). With regard to memory usage, GapFiller outperforms SOAPdenovo since intermediate output is temporarily stored (and not kept in the memory). For all datasets analyzed, GapFiller requires only 0.1 GB of memory, which is mostly consumed by the Burrows-Wheeler Aligner (BWA). Note that no results are displayed for IMAGE since the method can not handle multiple libraries and requires very large computation times to complete the process. M, million.
Summarizing, we provide strong evidence that bacterial draft assemblies can be accurately closed using an automated strategy. The few gaps that are left open by GapFiller mainly concern more difficult repeated areas. Here the extension method finds multiple alternatives with an equal ratio. These regions can eventually be closed through additional Sanger sequencing using only a limited number of reactions.
Gap closure in a small and large eukaryote: analysis of the yeast model organism Saccharomyces cerevisiae and the human chromosome 14
Saccharomyces cerevisiae is perhaps the most commonly used yeast and plays an important role in fermentation of beer and bread. We used the 288c reference genome to simulate two paired-end libraries, which were subsequently used for assembly (see Materials and methods for more specifications). In Table 2 we show that GapFiller can also be of value in closing genomes of smaller eukaryotes such as yeast: the number of gaps can be reduced with 84% (against 76% for SOAPdenovo) at a relatively low error rate. Nonetheless, as observed also for the bacterial datasets, SOAPdenovo more effectively reduces the total gap length (95% against 85% for GapFiller). The IMAGE method was not evaluated on eukaryotic datasets due to the very large computation time needed.
Similar tendencies are observed for human chromosome 14 (see Materials and methods for information about the datasets used), even though the amount of data and chromosome size differ largely in comparison to the yeast dataset. As expected, the number of scaffolds is higher for human, but notably the average scaffold length is relatively short (which is likely due to the use of ABySS for creation of the draft assembly). Consequently, the outcomes are less impressive, but still there seems to be an inverse relationship between the gap count (-29% after running SOAPdenovo, -40% with GapFiller) and the total gap length (-55% with SOAPdenovo, -26% with GapFiller). Given the observation that, after gap closure with SOAPdenovo, the total genome size is significantly reduced whereas the number of errors due to indels and misjoins has increased, it might be argued that a portion of these closures can be attributed to collapsing of (repeated) elements.
From a practical point of view it should be observed that both methods can run well on small scale systems: SOAPdenovo appears to be faster on larger input sets whereas GapFiller uses significantly less memory (Figure 1).
Functional analysis of the human chromosome 14
To further analyze the nature of the closed region, we extracted all annotations from the human GRCh37 chromosome 14 [GenBank:NC000014] and then aligned the corresponding sequences to the scaffolds (before and after closure with GapFiller). For each annotation type we counted the number of gapped nucleotides that were closed with GapFiller. Results are provided in Table 3 and show that novel nucleotides are incorporated in a wide range of functional regions, showing that gap closure adds mostly functional residues to the assembly. The largest relative increase of nucleotides is seen in coding regions (38.67% for genes, 30.53% for mRNA and 26.89% for coding sequences). Overall, the method is applicable on the relatively large genomic spectrum and does not just solve regions that are bound to specific functional characteristics.
Discussion
Dramatic advances in sequencing technologies have opened new possibilities for whole genome analysis. The increasing read length of next-generation sequencing platforms, as well as the promising perspectives of third generation sequencing platforms, will inevitably lead to better assemblies and represent genomes in large stretches of DNA. Also, third generation technologies (such as the PacBio and IonTorrent systems) will be capable of outputting sequencing reads with large undefined inserts, thus providing valuable paired read information for the assembly and scaffolding process. Concurrently, the development of genome closure software should also receive attention to overcome difficult genomic regions that cannot be covered by draft assemblies.
Our results with GapFiller indicate that gapped genomics regions can be reliably closed through an automatic protocol that uses only short sequencing reads. Costly Sanger sequencing can therefore be limited to a few difficult repeat areas. Also, we show that the method is suited for both bacterial and (large) eukaryotic datasets in terms of accuracy, time and memory usage. In the human dataset we underscore that gapped regions may contain diverse but crucial functional information, which is missed in the draft assembly.
From in-depth investigation of closed regions with GapFiller and SOAPdenovo, we argue that our GapFiller method has three main advantages. First, it takes into account the estimated gap size prior to closure, thus discarding erroneous closures that are shorter or larger than expected. Second, it does not try to fill a gap through local assembly of reads into contigs, but rather seeks to extend the contigs from each end through k-mer overlap. The latter point is especially important to overcome short tandem repeats. And third, it takes into account the contig edges, often a source of misassemblies, and re-evaluates these during gap closure. Importantly, GapFiller requires only limited computational resources and thus is also suited for (larger) eukaryotic genomes. Also, SOAPdenovo shows a good performance in terms of speed and memory usage and appears a valuable alternative to our strategy. From the results obtained on both bacterial and eukaryotic datasets, it appears SOAPdenovo is better able to close larger gaps. The explanation might reside in the fact that GapFiller implements a more conservative strategy than SOAPdenovo. Given that larger gaps usually represent more complex regions than smaller gaps, GapFiller may be simply more careful in avoiding dubious closures. This is also in line with the observation that GapFiller yields generally more accurate results than SOAPdenovo. Alternatively, a different explanation for GapFiller being less effective in closing larger gaps could be provided by the first advantage mentioned above. If the estimated gapsize in the scaffold does not meet the size of the closed fragment, such closure will be rejected. However, from a practical point of view it is still relatively difficult to estimate the exact gap size based on next-generation sequencing paired-read libraries. In particular, mate pair libraries can show a relatively large insert distribution, thus making it hard to define the correct distance between the pairs. Also paired-end background noise in mate pair data is commonly observed and leads to incorrect scaffolds. Consequently, there is a possibility that a correct closure is rejected because of the erroneous gap size estimation after scaffolding. Future research will hopefully lead to better insight into these issues.
The IMAGE method instead has several limitations in terms of quality, speed and ease of use (the method can handle only one library per analysis). Notably, the strategy produces relatively large genomes, and this is most likely a consequence of sequence extension of the left and right scaffold edges.
The findings in this paper have been derived using different short-read Illumina libraries. The length of the gapped regions is limited to the insert distance of paired reads. Nonetheless, the promising outcomes strongly indicate that long-range gaps can also be effectively closed with high quality 454 and/or single molecule paired sequence reads. Also, we have put emphasis on creating a user-friendly and fast tool so that it can be of wide use for the community. We feel GapFiller can make a significant contribution to (almost) closing genome assemblies in a reliable manner.
Materials and methods
GapFiller algorithm
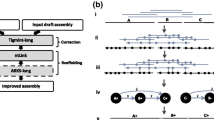
The GapFiller method works as follows (see Figure 2 for a schematic overview). The input consists of a set of scaffold sequences (in FASTA format) with gapped nucleotides represented as Ns and one or multiple sets of paired reads (in FASTQ or FASTA format). The data handling and analysis are setup in a multithreaded manner, thus allowing multiple datasets (libraries) to be processed simultaneously (by default one thread is used, but for this study we set this to eight). In a pre-filtering step the nucleotides at the edges of each gap are trimmed off (parameter t, default = 10 nucleotides) since these often contain misassemblies. Then all pairs are aligned to the trimmed input scaffolds with Bowtie [10] or the Burrows-Wheeler Aligner (BWA) [11]. The alignment method can be selected by the user, but we recommend using BWA in case of long reads. Bowtie instead is more suited for shorter reads, also because of the decreased runtime. For this study we used BWA on all datasets. Pairs are only kept if a) one read can be aligned and b) the second read (partially) falls within a gapped region. The latter is calculated based on the expected distance between the read pairs: the maximum allowed deviation used for this study was 25% of the pre-defined distance between the pairs. The final set of reads, which is expected to be (partially) in a gapped region, is then split into shorter k-mers that are used to gradually close the gap from each edge. The k-mer size is set by default to 30 nucleotides (1 + m, where m corresponds to a default sequence overlap of 29 nucleotides). Gaps are iteratively filled from the left and right edge by incorporating one overhang nucleotide at a time, provided the position is sufficiently covered (parameter o, default = 2). In case of ambiguous overhang nucleotides (that is, in a situation of allelic differences), a majority voting ratio is applied (parameter r, default = 0.7). If a nucleotide is incorporated, the k-mer(s) used for extension are removed and the algorithm checks if the gap can be closed. Gaps are only considered to be closed if a) an overlap can be found between the two extensions (parameter n, default = at least 10 nucleotides) and b) the sequence insertion corresponds to a pre-estimated gap length in the scaffold (parameter d, default = maximally 50 nucleotides difference). Otherwise, the next iteration is started. If no overhang nucleotides can be incorporated anymore, the whole process is repeated a second time by re-using all k-mers. Finally, the user can decide to repeat the complete analysis (alignment and gap closure) on the novel assembly. The number of such 'global' iterations can be specified using the (parameter i, default = 10).

Schematic overview of the GapFiller algorithm. (a) The input data consist of a set of scaffold sequences containing gapped nucleotides and one or more sets of paired-end and/or mate-pair reads. (b) As a pre-processing step low quality nucleotides are removed from the sequence edges, thus enlarging the gap of ten nucleotides from each side. It should be stressed that the contig ends resulting from a draft assembly often contain misassemblies. (c) Paired-reads are aligned to the scaffolds and retained if one pair aligns to a scaffold sequence (dark grey) and one pair to a gapped region (black). (d) All pairs that are estimated to fall in the gapped regions are split into k-mers and used for gap filling. (e) The gap is closed from each edge by using k-mers that present a sequence overlap of size (k-mer - 1) and one nucleotide overhang. Gaps are closed if the right and left extensions can be merged and correspond to the estimated sequence gap.
Short-read datasets
To evaluate the results of our algorithm, we used short-read Illumina data from E. coli, S. coelicolor, S. aureus, R. sphaeroides, S. cerevisiae and Homo sapiens. Our aim was to create a divergent benchmark using a variety of organisms, dataset types and assembly strategies. For E. coli the dataset consists of a paired-end library taken from the NCBI Short Read Archive [SRA:SRR001665] (insert size of 200 bp, 36 cycles, 10.4 million reads). The dataset of S. coelicolor and S. cerevisiae consist of simulated paired-end libraries constructed from reference genomes deposited in GenBank. Random paired-end reads were created using wgsim (at default settings), which is part of the SAM tools package [12]. For S. coelicolor we used the A3(2) reference genome [GenBank:AL645882] to construct two paired-end libraries (insert sizes of 200 and 500 bp, 70 cycles, 10 million reads per library, whereas for S. cerevisiae we used all chromosomes of the S288c genome [GenBank:NC001133 to NC_001148] to construct two paired-end libraries (insert sizes of 200 and 500 bp, 70 cycles, 20 million reads per library). For S. aureus, R. sphaeroides and H. sapiens (chromosome 14) quakeCor-filtered [9] short-read datasets were taken from the Genome assembly Gold-standard Evaluations website [13]. For S. aureus this included a paired-end (insert size of 180 bp, 88 cycles, 0.8 million reads) and a mate-pair library (insert size of 3,500 bp, 37 cycles, 1.6 million reads). Similarly, for R. sphaeroides this also included a paired-end (insert size of 180 bp, 76 cycles, 1.5 millino reads) and a mate-pair library (insert size of 3,500 bp, 72 cycles, 1.5 million reads). For H. sapiens (chromosome 14) the dataset included one paired-end library (insert of 155 bp, 93 cycles, 32.6 million reads) and two mate-pair libraries (shortjump library: insert size of 2,283 to 2,803 bp, 82 cycles, 14.5 million reads; longjump library: insert size of 35,295 to 35,318 bp, 79 cycles, 2.0 million reads).
Assembly and scaffold construction
For three organisms a draft assembly was made using the paired-end libraries. For E. coli we used SOAPdenovo v1.3 [3] whereas for S. coelicolor and S. cerevisiae we used the CLCbio de novo assembler v4.9. Scaffolds were produced using the same libraries as follows. For E. coli the 1,111 contigs of the draft assembly were linked into 179 scaffolds using SSPACE Premium v1.0 [4]. For the S. coelicolor and S. cerevisiae genomes a similar strategy was adopted resulting in 115 scaffolds (430 contigs) and 334 scaffolds (733 contigs), respectively. For the remaining three species, S. aureus, R. sphaeroides and Homo sapiens, assemblies were retrieved from the GAGE website [13]. Based on the outcomes of the comparative analysis, we decided for the bacterial species to use the scaffolds produced with ALLPATHS-LG strategy [14] and for human the scaffolds (≥ 200 bp) produced with ABySS [2].
IMAGE and SOAPdenovo gap closure software
Comparative analysis with IMAGE [6] was performed using version 2.3, which was downloaded from [15]. The program was run at default settings using a k-mer size of 31 and with 10 iterations. The insert size of the paired-end libraries was specified accordingly. SOAPdenovo gap closer software version 1.10 was downloaded from [16]. The program was run at default settings using a k-mer size of 30 and in total 8 threads.
Quality assessment of assemblies
In order to assess the quality of the assemblies, we followed the criteria set by [8]. Errors were classified into three categories: SNPs, indels (including indels ≤ 5 bp and indels > 5 bp) and misjoins (including inversions, relocations and translocations).
Compute server specifications
All analyses were performed on a 32 GB Linux machine (Intel Xeon X7350, 2.93 GHz).
Availability of software
The software can be downloaded from [7]and is free of charge for academic users.
Abbreviations
- bp:
-
base pair
- BWA:
-
Burrows-Wheeler Aligner
- LC:
-
low coverage
- SNP:
-
single nucleotide polymorphism.
References
Zerbino DR, Birney E: Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2011, 18: 821-829.
Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJ, Birol I: ABySS: A parallel assembler for short read sequence data. Genome Res. 2009, 19: 1117-1123. 10.1101/gr.089532.108.
Li R, Fan W, Tian G, Zhu H, He L, Cai J, Huang Q, Cai Q, Li B, Bai Y, Zhang Z, Zhang Y, Wang W, Li J, Wei F, Li H, Jian M, Li J, Zhang Z, Nielsen R, Li D, Gu W, Yang Z, Xuan Z, Ryder OA, Leung FC, Zhou Y, Cao J, Sun X, Fu Y, et al: The sequence and de novo assembly of the giant panda genome. Nature. 2010, 463: 311-317. 10.1038/nature08696.
Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W: Scaffolding pre-assembled contigs using SSPACE. Bioinformatics. 2011, 27: 578-579. 10.1093/bioinformatics/btq683.
Dayarian A, Michael TP, Sengupta AM: SOPRA: Scaffolding algorithm for paired reads via statistical optimization. BMC Bioinformatics. 2010, 11: 345-10.1186/1471-2105-11-345.
Tsai IJ, Otto TD, Berriman M: Improving draft assemblies by iterative mapping and assembly of short reads to eliminate gaps. Genome Biol. 2010, 11: R41-10.1186/gb-2010-11-4-r41.
GapFiller. [http://www.baseclear.com/bioinformatics-tools/]
Langmead B, Trapnell C, Pop M, Salzberg SL: Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10: R25-10.1186/gb-2009-10-3-r25.
Li H, Durbin R: Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009, 25: 1754-1760. 10.1093/bioinformatics/btp324.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup: The Sequence alignment/map (SAM) format and SAMtools. Bioinformatics. 2009, 25: 2078-2079. 10.1093/bioinformatics/btp352.
Kelley DR, Schatz MC, Salzberg SL: Quake: quality-aware detection and correction of sequencing errors. Genome Biol. 2010, 11: R116-10.1186/gb-2010-11-11-r116.
Salzberg SL, Phillippy AM, Zimin A, Puiu1 D, Magoc T, Koren S, Treangen TJ, Schatz MC, Delcher AL, Roberts M, Marçais G, Pop M, Yorke JA: GAGE: a critical evaluation of genome assemblies and assembly algorithms. Genome Res. 2012, 22: 557-567. 10.1101/gr.131383.111.
MacCallum I, Przybylski D, Gnerre S, Burton J, Shlyakhter I, Gnirke A, Malek J, McKernan K, Ranade S, Shea TP, Williams L, Young S, Nusbaum C, Jaffe DB: ALLPATHS 2: small genomes assembled accurately and with high continuity from short paired reads. Genome Biol. 2009, 10: R103-10.1186/gb-2009-10-10-r103.
SOAPdenovo. [http://soap.genomics.org.cn/soapdenovo.html]
Acknowledgements
This research was partially funded by the WBSO (BaseClear grants SO201101 and SO201209).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
MB conceived of the study and wrote the program, WP participated in its design and coordination and wrote the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Boetzer, M., Pirovano, W. Toward almost closed genomes with GapFiller. Genome Biol 13, R56 (2012). https://doi.org/10.1186/gb-2012-13-6-r56
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/gb-2012-13-6-r56