
Abstract
Background
Miniature inverted-repeat transposable elements (MITEs) are non-autonomous DNA-mediated transposable elements (TEs) derived from autonomous TEs. Unlike in many plants or animals, MITEs and other types of DNA-mediated TEs were previously thought to be either rare or absent in Drosophila. Most other TE families in Drosophila exist at low or intermediate copy number (around < 100 per genome).
Results
We present evidence here that the dispersed repeat Drosophila interspersed element 1 (DINE-1; also named INE-1 and DNAREP1) is a highly abundant DNA-mediated TE containing inverted repeats found in all 12 sequenced Drosophila genomes. All DINE-1s share a similar sequence structure, but are more homogeneous within species than they are among species. The inferred phylogenetic relationship of the DINE-1 consensus sequence from each species is generally consistent with the known species phylogeny, suggesting vertical transmission as the major mechanism for DINE-1 propagation. Exceptions observed in D. willistoni and D. ananassae could be due to either horizontal transfer or reactivation of ancestral copies. Our analysis of pairwise percentage identity of DINE-1 copies within species suggests that the transpositional activity of DINE-1 is extremely dynamic, with some lineages showing evidence for recent transpositional bursts and other lineages appearing to have silenced their DINE-1s for long periods of time. We also find that all species have many DINE-1 insertions in introns and adjacent to protein-coding genes. Finally, we discuss our results in light of a recent proposal that DINE-1s belong to the Helitron family of TEs.
Conclusion
We find that all 12 Drosophila species with whole-genome sequence contain the high copy element DINE-1. Although all DINE-1s share a similar structure, species-specific variation in the distribution of average pairwise divergence suggests that DINE-1 has gone through multiple independent cycles of activation and suppression. DINE-1 also has had a significant impact on gene structure evolution.
Similar content being viewed by others

Background
Interspersed repetitive sequences are ubiquitous to all eukaryotic organisms, and make up a significant portion of the genome [1–7]. These sequences are mostly transposable elements (TEs) or TE-derived sequences, and they play important roles in the evolution of chromosome organization and genome complexity [8].
Based on their mechanism of transposition, TEs can be divided into two classes: class I comprises retrotransposons, which transpose through RNA-mediated mechanisms, and class II comprises transposons, which mobilize through DNA-mediated mechanisms [9, 10]. Depending on their ability to direct their own transposition, each class of TEs can contain two types: autonomous and non-autonomous copies. Autonomous TEs code for the proteins that are required for their transposition, and are mobilized in cis. Non-autonomous TEs are mobilized in trans by enzymes produced from autonomous elements. Well-known examples include the vertebrate retroelements LINEs (long interspersed elements) and SINEs (short interspersed elements). The mobilization of non-autonomous SINEs requires retrotransposase from autonomous LINEs, and these elements co-evolve in a highly species-specific manner [11, 12]. Another example is the miniature inverted-repeat transposable elements (MITEs) found in many plant genomes. MITEs are non-autonomous DNA elements (class II) that originated from a subset of autonomous DNA transposons [13]. They are characterized by short sequences with no coding capacity, flanked by terminal (or occasionally subterminal) inverted repeats (TIRs) and very short direct repeats caused by target site duplication (TSD). MITEs have no internal homology to their parental autonomous transposons and often include non-homologous sequences in their internal regions. MITEs have also been found in several animal genomes, including Caenorhabditis elegans, mosquitoes, fish and humans (reviewed in [14]). Both SINEs and MITEs are highly abundant (usually > 1,000 copies per genome) in many host species across a broad taxonomic range. Because of their high abundance and active movement, and their frequent association with genes [15, 16], MITEs have had a significant impact on the evolution and complexity of eukaryotic genomes.
TE activity and evolution have been intensively studied in Drosophila and many families of TEs have been described [5, 6, 17–19]. Most TEs are at low or intermediate copy number in D. melanogaster. MITEs and SINEs have been previously reported as being either rare or absent in most species of this genus that have been examined [20]. D. melanogaster DINE-1 (Drosophila interspersed element 1; also named INE-1, DNAREP1) is an exception to these observations [21]. D. melanogaster contains thousands of copies of DINE-1 [19]. All copies appear to be non-autonomous, and analyses of their divergence patterns suggest that D. melanogaster DINE-1 has been inactive for over 4 million years [22]. Although DINE-1 was originally suggested to be a SINE-like retroelement, we have suggested that it is more likely to be a MITE, based on analysis of DINE-1 elements in D. yakuba that show evidence of recent transpositional activity [23]. We discuss below the structural features of DINE-1 supporting this designation, as well as the more recent proposal [24] that DINE-1s are members of the Helitron family of TEs.
Several earlier studies found high copy TEs that we here classify as DINE-1. Vivas et al. [25] discovered an element in D. subobscura called GEM that is composed of repetitive modules, one of which they also found in the D. melanogaster and D. virilis genomes. Miller et al. [26] characterized an abundant element called SGM in D. subobscura, D. guanche and D. madeirensis, noted its similarity to GEM, and also described that other species, including D. melanogaster and D. virilis, have similar sequences; GEM and SGM are the same as DINE-1. Wilder and Hollocher [27] subsequently discovered an element in a number of Drosophila species that they called mini-me and noted its similarity to D. melanogaster DINE-1. However, a comprehensive assessment of the abundance and transpositional dynamics of DINE-1 has not been reported. Here we expand our study of the evolutionary dynamics of DINE-1 using the recently available genome sequences of 12 Drosophila species [7]. We found that DINE-1-related sequences are not only highly abundant in all 12 species, but also share a similar sequence structure, suggesting that a common mechanism was used for their transposition. Different lineages, however, show different distributions of divergence, suggesting that DINE-1 has gone through multiple cycles of transposition and subsequent silencing.
Results
Identification and common sequence structure characteristics of DINE-1s in 12 Drosophilaspecies
Previously, we discovered that DINE-1 is highly abundant and appears to have experienced a recent transpositional burst in the lineage leading to D. yakuba [23]. Using the D. yakuba DINE-1 consensus sequence, we searched using BLAST for related sequences in 11 other sequenced genomes of Drosophila (Figure 1). This initial screen suggested that all 12 Drosophila species contain hundreds to thousands of copies of DINE-1-related sequences. To infer the structure of DINE-1 in each species, we manually aligned 50 sequences with the highest BLAST scores from each species. Then we aligned together these sequences from all the species. This analysis revealed that DINE-1-related sequences from the ten newly analyzed Drosophila species share a number of structural similarities with DINE-1 from D. yakuba and D. melanogaster (Figure 2; Table 1; also see Additional data file 1).

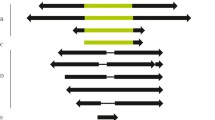
Flow chart of the strategy for identifying DINE-1 sequences in the 12 Drosophila genomes.

The generalized structure of DINE-1 sequences from 12 Drosophila genomes, and alignment of the DINE-1 consensus sequences from 12 species with each feature boxed. The element contains two conserved blocks, A and B. Within block A, the sequence can be further divided into two parts, A1 and A2, separated by a region of variable length containing the tandem repeats (CCGT)n(CTGT)n. Between blocks A and B is a region of central repeats, containing species-specific repeats. These central repeat sequences do not share homology among species; the length of the repeat unit can range from approximately 50 bp to approximately 500 bp, and the number of repeats is also variable within species. Locations of the subTIRs are shown as gray arrows; see Table 1 for precise designations of subTIR sequences. The 5' end also contains a second inverted repeat (IR) sequence that is partially complementary to the 5'-end terminal repeat and is shown as a gray arrowhead. An inverted repeat near the 3' end forms a potential stem-loop structure and is indicated by white arrowheads.
We previously defined D. yakuba DINE-1 as beginning at its 5' inverted repeat, based on the assumption that this sequence is a TIR [23]. We have here placed this repeat one nucleotide internal to D. yakuba DINE-1. This change in based on the recent designation of DINE-1 as a Helitron element [24, 28]. Our designation of the boundaries of DINE-1 differs by one nucleotide from that in [28], based on our analysis presented below of polymorphic insertions in D. yakuba (Figure 3). The precise boundaries of DINE-1 in each species are difficult to determine because of their preference for inserting in T-rich regions. We have annotated the sequences in Figure 2 in order to maximize similarity to D. yakuba DINE-1. Based on this alignment the 5' inverted repeat ranges from being terminal to 2 bp internal; in all species the corresponding 3' inverted repeat is clearly internal. We thus refer to these repeats as subterminal inverted repeats (subTIRs).

Analysis of ten sites that are polymorphic for DINE-1 insertions in natural populations of D. yakuba. For each site, the sequence from a strain containing a DINE-1 insertion is shown at the top, and the sequence from a strain lacking the insertion is shown at the bottom. Only the terminal sequences of DINE-1 and its flanking sequences are shown. The 5' subTIR is shown in bold italics. Insertions 1-3 were previously reported in [23]. The interpretation of these data depends on the designation of the DINE-1 termini and whether insertion causes a TSD. (a) Analysis using the annotation of DINE-1 structure presented in this paper. This annotation places the subTIR of D. yakuba 1 bp internal to the 5' end. It also assumes that no TSD is created, in accord with the proposed mechanism of Helitron-type replication [24]. Under these stipulations, all ten insertions occur between the dinucleotide TT in the consensus sequence WTT (where W = A or T), and eight of ten match a longer consensus sequence of insertion after the second nucleotide in the sequence of WTTTT. (b) Analysis assuming the DINE-1 termini of [28], and Helitron-type replication. Only sites 1 and 10 are shown. Under this annotation, DINE-1 would have an insertion preference for WT dinucleotides. (c) Analysis assuming that the DINE-1 5' end begins at its inverted repeat, inserts between the dinucleotide TT and causes a 2 bp TSD, as in MITE-like DNA transposons. The TSDs caused by DINE-1 are boxed.
The common features of DINE-1 from all species include: 13 bp subTIRs (the exact location of such repeats differs by 1-2 nucleotides among species); a partial inverted repeat next to the 5' subTIR; terminal regions that are relatively well-conserved within species, called blocks A and B; a GTCY-rich microsatellite repeat of variable length within block A; a variable central repeat region, which is responsible for most of the total length variation among elements; the lack of any significant open reading frames; and a propensity to insert between TT dinucleotides (discussed further below).
Our analysis revealed one novel feature not previously described for DINE-1, SGM or mini-me, namely a short hairpin stem-loop structure (with 7-11 nucleotide-long stems) located a few nucleotides downstream of the 3' subTIR (Figure 2). The sequence of the self-complementary stem differs among species, suggesting that compensatory mutations maintain its structure. This stem-loop may function as a terminator during rolling-circle replication (see Discussion).
Several of the features we characterized refine structural features inferred previously from SGM [26] and mini-me [27]. TIRs from mini-me were reported to vary from 10-20 bp in length from different species, while our analysis identified 13 bp TIRs in all species. These differences likely reflect the fact that we have analyzed many more sequences. The 13 bp subTIRs from D. melanogaster and D. virilis that we describe contain the 10 and 11 bp sequences reported by Wilder and Hollocher [27]. Likewise, the 17 bp TIR reported previously for D. subobscura mini-me contains the 13 bp subTIR reported here for D. pseudoobscura, with one internal base-pair difference. The partial inverted repeat flanking the 5' subTIR we identified is more variable than that reported based on two copies of mini-me, being a partial and/or interrupted repeat of the 5' subTIR in different species.
Wilder and Hollocher [27] also reported that mini-me elements from all species contain a highly conserved 33 bp core region. We find that this conserved core region actually extends over 90 bp, including the (TA)4 repeat and additional sequences 5' to this repeat. These sequences partially overlap with the LS module described for SGM elements [26]. The striking conservation of this core among the 12 species suggests that it is of functional significance for DINE-1 transposition.
We previously proposed that transposition of D. yakuba DINE-1 creates a dinucleotide (TT) TSD upon insertion [23]. However, based on analysis of the genome sequence from a single strain this conclusion was not definitive because some copies did not have a TT dinucleotide flanking both sides. This could be due either to accumulated mutations after insertion or because the TT site preference is not absolute. We performed a preliminary test of this hypothesis by comparing the sequences of three polymorphic insertion sites of DINE-1 in multiple strains of D. yakuba [23]. Here we extended this analysis with seven additional DINE-1 insertions that are polymorphic among different D. yakuba strains (see Materials and methods).
We found that all of these ten sites have a similar sequence structure (Figure 3). The interpretation of these data, however, depends on where precisely DINE-1 starts. Our previous interpretation of DINE-1 beginning at the 5' subTIR is consistent with insertions causing a TT TSD (Figure 3c). With the re-designation of the 5' end of DINE-1, these two nucleotides are instead part of the element, and DINE-1 would not create a TSD. The lack of a TSD is consistent with the proposal that DINE-1s are Helitrons (see below). Based on our designation of the DINE-1 boundaries, all D. yakuba insertions occur between the dinucleotide TT (Figure 3a). Using the DINE-1 boundaries from [28], the insertion site preference is more variable (Figure 3b). We then examined the sequences flanking the putative DINE-1s identified in the other Drosophila species. The majority (> 80%) of DINE-1s are flanked by TT dinucleotides. The conservation of this site preference in all 12 species, combined with the numerous other similarities described above, strongly suggests that each of these elements is in fact a species-specific DINE-1 and that they likely share a common mechanism of transposition.
Relationships of DINE-1s within and among species
Two pieces of evidence demonstrate that DINE-1 is highly homogeneous within 11 of the 12 species, with D. willistoni discussed below as being exceptional. First, we performed BLAST searches using the 90 bp sequence of the core region, which is conserved among all types of DINE-1s. By comparing the TIRs and block A sequences we found that each species contains only one type of DINE-1. Second, we searched for DINE-1s in one genome using the DINE-1 consensus sequences from other genomes as queries, and found only the same sets of sequences.
Among the 11 species (again excluding D. willistoni), there are 5 different subTIRs (Table 1). All five melanogaster subgroup species have the same subTIR sequence. D. ananassae has a unique subTIR, while the closely related species D. pseudoobscura and D. persimilis share the same subTIR, as do D. virilis and D. mojavensis. The DINE-1 subTIR from D. grimshawii shares 12/13 bp with D. virilis and D. mojavensis. The central repeat is the most diverse region of DINE-1 among species. Even species sharing the same subTIRs, such as D. virilis and D. mojavensis, have unrelated central repeat regions.
Analysis of D. willistoni gave uniquely different results. D. willistoni contains three different subtypes of DINE-1s, each with different subTIRs and different central repeat sequences (Figure 2). Phylogenetic evidence presented below suggests that they have at least two independent evolutionary origins.
Abundance and divergence of DINE-1s within species
DINE-1 is highly abundant in all 12 Drosophila species. It is difficult to determine an exact number because each species contains small and fragmented copies that cannot always be unambiguously identified as DINE-1s. We therefore used stringent criteria to identify DINE-1s in order to obtain a reliable comparison among species (Table 1). For example, this search identified 355 copies in D. melanogaster compared to previous analyses that suggested that D. melanogaster has approximately 1,000 copies [22]. Using identical search criteria, we found vast differences in the copy number of DINE-1s among species, ranging from 334 in D. grimshawii to 6,297 in D. willistoni.
We identified similar numbers of DINE-1s in the D. melanogaster sister species D. simulans and D. sechellia compared to D. melanogaster. In contrast, more than ten-fold more copies were identified in D. yakuba. This high copy number is due to the large number of closely related copies in D. yakuba, and is consistent with previous work that suggested that DINE-1 has been inactive in D. melanogaster but underwent a recent transpositional burst in D. yakuba [23].
We therefore sought to determine whether other species with high copy number also show evidence of recent transpositional bursts. We used BLAST percent identity scores as an approximate method to estimate divergence among individual copies within species (Table 1; Figure 4). This method accurately recapitulates previous analyses for D. yakuba and D. melanogaster that were based on estimates of per-site divergence [23]: DINE-1s from D. melanogaster have a broad peak of identities centered approximately around 90%, while D. yakuba shows a peak from approximately 96-100%, with a long tail of more diverged copies. These differences are highly significant (Mann-Whitney U test, two-tailed, p < 0.001).

The frequency distribution of sequence identity of DINE-1 in different species. The percentage identity was based on BLAST search, using consensus sequences of part A1 of block A from each species as the query. To exclude short and fragmented sequences from our analysis, only hits > 100 bp were used. Note that the y-axis scale differs among species.
D. simulans and D. sechellia show distributions similar to D. melanogaster (Figure 4; p > 0.05) and have average percent identities around 90-91% (Table 1). D. erecta has an average percent identity more similar to D. melanogaster than to D. yakuba; however, its distribution is significantly different from both species (p < 0.001). These data suggest that D. yakuba is the only melanogaster subgroup species that experienced a recent transpositional burst. D. grimshawii also has a distribution with very few copies of high similarity, and a similar copy number to D. melanogaster, suggesting that DINE-1 has not been recently active in this species. In contrast, DINE-1s from D. pseudoobscura, D. persimilis, and D. ananassae have average percent identities > 95% with distributions highly skewed toward young copies, suggesting recent transpositional bursts in these species. The distributions in D. pseudoobscura and D. persimilis are significantly different (p < 0.001; see Discussion). D. ananassae in particular stands out for having many copies identical to the consensus sequence (in the block A region). D. virilis and D. mojavensis also have substantial numbers of young copies but more broad distributions, suggesting the possibility that multiple rounds of transposition, silencing and reactivation may have occurred in these species.
D. willistoni has more than 1,000 copies of each of its three subtypes (Table 1), with subtype 1 and 3 having about twice as many copies as subtype 2. Each subtype has a peak near 100% identity, suggesting recent transpositional activity; however, their distributions are significantly different from each other (p < 0.001). Interestingly, these subtypes also have different phylogenetic patterns (see below).
Phylogenetic relationship of DINE-1s
These very different estimates of DINE-1 divergence within different species, and in particular our evidence for recent transpositional bursts, raises the question of whether DINE-1 may have undergone horizontal transmission into some Drosophila species. To understand the evolutionary dynamics of DINE-1s and their association with their host species, we analyzed the phylogenetic relationship of DINE-1 consensus sequences from the 12 Drosophila species (Additional data file 1) and compared it with the known phylogeny of Drosophila [29]. Because of the rapid evolution in the central repeat region, reliable alignment for phylogenetic reconstruction could be obtained only for blocks A and B.
With the exceptions of D. willistoni and D. ananassae, the phylogenetic relationships of the DINE-1 sequences are, in general, consistent with the host species phylogeny (Figure 5). The grouping of DINE-1 in a separate clade containing D. ananassae and two of the three types from D. willistoni is surprising. The fact that this clade is an outgroup suggests that this result is not due to horizontal transfer from other Drosophila into these species. One possibility is that DINE-1 was horizontally transferred from non-Drosophila into these species. Alternatively, DINE-1 sequences resembling ancestral copies may have become reactivated in D. ananassae and D. willistoni. These sequences would be related to ancestral copies that were vertically inherited in the common ancestor of Drosophila.

Phylogenetic relationship of DINE-1 consensus sequences compared to their host species. (a) Phylogenetic tree based on pooled sequences of block A and B (Additional data file 1) and constructed using the neighbor-joining method with the Jukes-Cantor one parameter substitution model [55]. Bootstrap resampling percentages based on 500 replications are indicated. Scale bar represents the estimated number of substitutions. (b) The host species phylogeny is adapted from [29]. Myrs, million years.
DINE-1insertions in or near to genes
In situ hybridizations to polytene chromosomes from several species using species-specific DINE-1 probes revealed strong signals in the heterochromatic chromocenter region, with additional hybridization observed along chromosome arms (data not shown). These non-chromocenter sites of hybridization led us ask whether DINE-1 insertions (whole or partial) are found in or near to protein-coding genes. SGM-related sequences were previously found in introns or adjacent to a number of D. melanogaster genes [26]. We found hundreds of DINE-1 insertions in predicted introns in all species (Table 2; Additional data file 1). We also found many DINE-1 copies within 1 kb of genes, which could potentially be in either untranslated regions or in regulatory regions. These results suggest that DINE-1 has had a significant impact on gene structure evolution throughout the Drosophilidae. Few DINE-1 insertions were found in predicted coding sequences (CDSs). Strikingly, the largest number (15) was found in D. ananassae, a species that has a very high copy number of highly similar (young) DINE-1 copies (Table 1; Figure 4). This result suggests that recent transpositional activity of DINE-1 in D. ananassae has resulted in mildly deleterious insertions into coding regions that have not yet been removed by selection.
Analysis of Helitron sequences in D. yakuba and D. virilis
Kapitonov and Jurka [24, 28] recently proposed that DINE-1 is related to Helitron, a family of DNA-mediated TEs. They reported consensus sequences of autonomous and non-autonomous copies of Helitron in D. yakuba and D. virilis. The non-autonomous consensus sequences are closely similar to our consensus sequences reported here. The consensus autonomous copies have an open reading frame (ORF) encoding the RepHel protein found in many other Helitrons, and sequences at each end similar to what we report here for DINE-1. These include block A at the 5' end and block B at the 3' end. We searched these two species using the RepHel portion of the autonomous consensus sequence as a query to determine whether these species contain potentially active copies. Among the top ten hits in D. yakuba, none have a fully intact RepHel ORF. Three copies have DINE-1 sequences flanking both sides of the RepHel sequences; two of these have over 500 bp of DINE-1 sequence at each end while the third has only approximately 50-60 bp of DINE-1 sequence at each end. Six of the remaining copies have DINE-1 sequences flanking one side of the RepHel sequence, and the last hit has no flanking DINE-1 sequences. Among the top ten hits in D. virilis we again found no copies with a fully intact RepHel ORF. One copy has DINE-1 sequences flanking both sides and five copies have DINE-1 sequences flanking one side. Among the remaining copies, one is in a highly repetitive region and could not be further analyzed, and the remaining four copies have no flanking DINE-1 sequences. We conclude that D. yakuba and D. virilis are unlikely to contain currently active autonomous Helitrons.
Discussion
DINE-1 is the most abundant repetitive sequence in the Drosophila genome. DINE-1 was first identified on the fourth chromosome of D. melanogaster [21], and was suggested to be a non-autonomous retroelement, analogous to vertebrate SINEs. This argument was based on its high abundance, composing > 1% of the total genome, its small size and its lack of significant ORFs [19, 21, 22, 27, 30]. However, unlike known SINEs, D. melanogaster DINE-1 did not appear to have polymerase III promoter consensus sequences or similarity to tRNAs or other small RNAs.
Subsequently, DINE-1-related sequences were found in other Dipteran species and were classified as novel TE families. Miller et al. [26], following earlier observations by Vivas et al. [25], identified SGM from several obscura group species as well as related sequences in GenBank from at least eight other Drosophila species, and noted its possible similarity to MITEs [26]. They further suggested that SGM elements composed approximately 10% of the D. guanche genome. Wilder and Hollocher [27] identified 'mini-me' and characterized its sequence structure based on approximately 80 clones isolated from 2 species of the cardini group, D. dunni and D. nigrodunni, and 28 sequences from 14 different species obtained from GenBank. mini-me was classified as a non-autonomous retroelement, although no direct relationship to previously known retroelements was observed.
Previously, Yang et al. [23] identified a recent transpositional burst of DINE-1 in the genome of D. yakuba. The analysis of highly similar, newly inserted DINE-1s in this species allowed for a more detailed characterization of DINE-1 sequence structure. We concluded that DINE-1 is more likely to be a non-autonomous DNA transposon, similar to MITEs first described in maize [13], rather than a SINE-like retroelement, based on the existence of perfect terminal and subterminal inverted repeats and a TSD (TT), which are typical characteristics of DNA transposons. Moreover, the lack of polymerase III binding sites or tRNA-related structures in these recently inserted copies argued against DINE-1 being similar to SINEs [23]. Bergman et al. [6] also characterized DINE-1 as being a TIR transposon.
In order to understand the origin and distribution of DINE-1 in the Drosophilidae, we expanded our search to ten additional partial or complete Drosophila genome databases using the consensus sequence of D. yakuba DINE-1. Strikingly, we found that sequences related to D. yakuba DINE-1 are very abundant in all these genomes (Table 1). BLAST searches did not find any related sequences in the mosquito, silk worm or other eukaryotic genomes, suggesting that DINE-1 is unique to Diptera. DINE-1-related sequences from all the Drosophila species share the same sequence structure that was defined from D. yakuba DINE-1 or from mini-me, with each containing: highly conserved blocks A and B at both ends, including a core region of approximately 90 bp in block A; a central repeat region of variable length; inverted repeats 13 nucleotides long at or near the 5' end and close to the 3' end; and insertion preference for T-rich regions (Figure 2). The sequences of the central repeat region from different species are very different, suggesting non-homologous origins of this region among species. In contrast, the within species divergence of this region is much smaller.
Our comparison of DINE-1 from 12 species revealed a previously unobserved 3' inverted repeat structure that could potentially form a stem-loop (Figure 2). It is important to note that in the absence of any internal ORFs, the designation of 5' and 3' for DINE-1 is arbitrary. The presence of potential stem-loops near both ends of DINE-1 raises the possibility that these structures are recognized by a reverse transcriptase, which would imply that DINE-1 is in fact a non-autonomous retroelement. However, considering all the evidence outlined above, we suggest that DINE-1 transposition is DNA mediated.
It was thought previously that MITE-like DNA transposons are rare in Drosophila, with only a few having been identified. One example of a Drosophila MITE is derived from pogo-like transposons in D. melanogaster [31]. Other examples are Vege and Mar, derived from the autonomous TE hobo of the hAT superfamily in D. willistoni [32]. However, unlike most MITEs, which are usually highly abundant in the host genome, only a few copies (< 10) of Vege and Mar were found in the genomes of their Drosophila hosts [32].
Non-autonomous DNA transposons require an external source of transposase for transposition. For many TEs transposase initiates transposition by recognizing and binding to the TIR sequence, and this interaction is highly specific [33, 34] Recently, Feschotte et al. [35] have shown that autonomous mariner-like transposase can not only interact with its own TIR, but can also interact with the TIR of Stowaway MITEs in rice. This provides strong evidence that Stowaway MITEs may use mariner-like TEs as their source for transposase.
Casola et al. [36] recently identified several Drosophila PIF-like transposons (DPLTs), which are found among Drosophila in both apparently autonomous and non-autonomous forms. Neither the TIR nor TSD sequences of these transposons match that of DINE-1, which suggests that they are not the autonomous parental copies of DINE-1. Intriguingly, however, DPLT1 has apparently active copies in D. yakuba, D. pseudoobscura, D. persimilis and D. willistoni and only inactive MITE-like copies in D. melanogaster, D. simulans, D. sechellia, D. erecta and D. mojavensis. This pattern closely resembles the division seen here for species that either do or do not show evidence for recent transpositional bursts of DINE-1 (Figure 4). These shared patterns suggest that species such as D. yakuba have experienced recent and ongoing movement of several DNA transposon families.
DINE-1: MITE or Helitron?
DINE-1 has many features characteristic of MITEs - small size, lack of coding potential, high copy number, and frequent association with genes. On the other hand, most MITEs have TIRs, which are presumably sites of transposase binding. A few MITE-like elements have been discovered that have subTIRs rather than TIRs but their corresponding autonomous elements have not been identified [37–39]. DINE-1 has inverted repeats and their conservation in structure despite ongoing changes in primary sequence argues strongly that they are of functional importance. We have placed the 5' inverted repeat 0-2 nucleotides internal to the end of DINE-1 in different species. Under this annotation, D. yakuba DINE-1 insertions would not cause a TSD (Figure 3). If the true 5' end of DINE-1 instead corresponds to the 5' inverted repeat, then D. yakuba DINE-1 insertion would cause a 2 bp TSD, as seen in other MITEs. The 3' inverted repeat, however, is clearly subterminal, which would be unusual for a MITE element.
Kapitonov and Jurka [24, 28] have recently proposed that DINE-1 is instead a non-autonomous Helitron element. They noted that DINE-1 has a number of features unusual for Helitrons. One was the absence of a short hairpin or palindrome at the 3' end, which is thought to function as a replication terminator. We have identified here a 3' hairpin structure in all 12 species that may fulfill this function. A number of unusual features remain. Foremost are the termini. Helitrons do not contain TIRs but instead have highly conserved 5' TC or 3' CTRR sequences. In contrast, DINE-1 lacks these short termini sequences but instead contains conserved subTIRs. The presence of relatively long blocks of conserved sequence between non-autonomous DINE-1 and the proposed autonomous copies also contrasts with other species. For example, bats contain several families of very high copy number non-autonomous Helitrons, which differ almost entirely from their autonomous master copies other than at their di- and tetra-nucleotide termini [40].
The most decisive evidence favoring the Helitron hypothesis is the association of DINE-1 elements with non-functional but recognizable partial ORFs of the RepHel protein in D. yakuba and D. virilis, making these copies the candidate autonomous elements responsible for the recent transpositional bursts of non-autonomous DINE-1s in these species. Considering some of the unusual features mentioned above, it will be of great interest to investigate experimentally the mechanism of DINE-1 transposition.
DINE-1 in the melanogastersubgroup
From our previous study [23], we found that D. melanogaster and D. yakuba contain structurally similar types of DINE-1s. The species differed significantly, however, in their distributions of sequence divergence and the chromosomal location of their DINE-1 copies. D. yakuba contains many similar copies, and these apparently younger copies have a higher relative frequency in euchromatic regions compared to older, more diverged copies. We hypothesized that DINE-1s in D. melanogaster and D. yakuba derive from a common ancestor that existed before the divergence of the melanogaster subgroup species. This hypothesis was tested here by our identification of DINE-1 from three other species of the melanogaster subgroup, D. simulans, D. sechellia and D. erecta. All five subgroup species share the same TIRs, core, central repeat unit, and 3' end stem-loop sequences. DINE-1s from the three newly characterized species have similar copy numbers and distributions of sequence divergence, an observation consistent with the hypothesis that DINE-1 was active and then silenced in the common ancestor of the melanogaster subgroup. D. yakuba is the only species showing evidence of a second, recent transpositional burst. We did find that DINE-1s from D. erecta have a different sequence in the region joining the central repeat to block B, suggesting that this is the most rapidly evolving region of DINE-1.
Dynamic nature and genomic impact of DINE-1
Our analysis reveals that several species outside the melanogaster subgroup have distributions of DINE-1 identity similar to that described above for D. yakuba, suggesting that DINE-1 has undergone multiple, independent transpositional bursts. D. ananassae and D. willistoni show the strongest evidence, with distributions skewed toward 100% (Figure 4). D. virilis has a somewhat broader distribution, with many similar copies suggestive of recent transpositional activity. D. mojavensis shows a broad distribution that is suggestive of multiple rounds of transposition and silencing at different times.
D. pseudoobscura and D. persimilis have distributions with peaks around 98-99% identity. These species diverged less than one million years ago [41], which might suggest that the similarly high identity in both species reflects activity of DINE-1 before or during their speciation. However, the distributions are significantly different, with D. pseudoobscura retaining proportionally more copies of high divergence. One possible explanation is that DINE-1 remained active more recently in D. persimilis. Alternatively, the strength of selection against older copies may differ between the species.
The discovery of multiple and relatively distant species each showing evidence for recently active DINE-1 copies raises the question of whether this element has been transmitted vertically or horizontally. The phylogenetic relationship among different DINE-1s (Figure 5) suggests vertical inheritance, with transpositional bursts resulting from existing copies escaping from host suppression. DINE-1 from D. ananassae and two subtypes from D. willistoni give a pattern discordant from the accepted species phylogeny but this pattern is also not consistent with a simple model of horizontal transfer among Drosophila species. Instead, we suggest that the phylogenetic pattern is likely to reflect reactivation of a related ancestral element in both D. ananassae and D. willistoni. Our analysis is necessarily limited by the relatively short sequences available for analysis. We suggest that further phylogenetic analysis of the autonomous elements from each species will help to further understand the evolution of DINE-1. Nevertheless, the combination of our phylogenetic analysis and the divergence data indicates that the activity of DINE-1 is extremely dynamic. The activation and suppression of the element seems to have evolved rapidly and repeatedly in multiple lineages leading to the 12 species.
Similar dynamics of transposition and suppression are found in LINEs and SINEs of mammalian genomes [42–44] and MITEs in plant genomes (see review in [14]). Some insertions of MITEs in plants have been shown to affect gene regulation [45, 46]. We have found that DINE-1 insertions are frequently found in the flanking regions and introns of genes, suggesting that some copies may also influence gene regulation (Table 2).
Highly abundant interspersed repetitive sequences can also serve as targets for ectopic recombination. Such recombination may be deleterious by promoting genome instability [47, 48], but may also catalyze structural evolution of existing genes and contribute to new gene formation [49]. DINE-1 is a candidate for causing analogous phenomenon in Drosophila. The testis-expressed gene hydra is one well-characterized example [50]. hydra exists only in the melanogaster subgroup, and its exon 1 has undergone multiple independent duplications. Many of these duplicated exon 1s are flanked by DINE-1 insertions, which suggests that DINE-1 may have facilitated some of these duplications by providing homologous target sequences for unequal crossing over. Given its high abundance and evidence for multiple rounds of transpositional activity, DINE-1 has clearly had a significant impact on Drosophila genome evolution, and we suggest that other examples of gene structural evolution associated with DINE-1 will be found among these species.
DINE-1 can also be a valuable system for studying rates and patterns of mutations. One can study de novo mutations in species that have had recent transpositional bursts by comparing the sequence variation among young, recently inserted DINE-1 copies. One can also use DINE-1 to examine substitution patterns between species. Previous comparative analysis of the chromosome distribution of DINE-1 in D. yakuba and D. melanogaster suggests that most new insertions are eliminated from the genome by negative selection [23]. Old copies that remain are thus likely to be evolving neutrally. One could therefore identify orthologous insertions between D. melanogaster and D. simulans, whose insertions must predate the divergence of these species, in order to infer the substitution pattern along lineages leading to both species. The ability to perform similar studies in multiple Drosophila species will allow unprecedented power for determining whether and how patterns of mutations vary in different lineages.
Materials and methods
Identifying DINE-1-related sequences from 12 Drosophilagenomes
Using the D. yakuba DINE-1 consensus sequence as a query, we searched for DINE-1-related sequences in all 12 Drosophila genome databases (from Comparative Assembly Freeze 1 (CAF1) [51]) using BLAST with the default setting of the parameters (Figure 1). Note that D. persimilis and D. sechellia were sequenced at only approximately three- to four-fold coverage and, thus, are incomplete. We retrieved the 50 copies of DINE-1 with the lowest E-value in each species, aligned them, and derived a consensus sequence for each species (Additional data file 1).
Sequence divergence and copy number among DINE-1s
We then BLAST-searched each genome using part of the consensus sequences of DINE-1 (5' end to end of core sequence) from its own species, using the default settings of the program. All BLAST hits greater than 100 bp were retained. The frequencies of percent identity between the query sequence and all hits were plotted for each species.
Sequence alignment and phylogenetic analysis
Sequences were aligned using ClustalW [52] with the default parameter settings. Alignments were further improved by manual adjustment. Inferred phylogenetic trees of the species consensus of DINE-1 were constructed using the neighbor-joining method with bootstrap resampling (500 replicates) using MEGA 3.0 [53].
Searching for DINE-1within or near genes
The UCSC Genome Browser Gateway [54] was used to obtain locations of DINE-1 in the annotated genomes, with the exception of D. willistoni. DNA sequences were retrieved using the Genes and Gene Prediction tracks (track setting: Other RefSeq) and grouped into the following categories: category 1, 1,000 bp upstream of CDS; category 2, CDS; category 3, introns; and category 4, 1,000 bp downstream of CDS. Categories 1 and 4 were then merged into a single class of flanking sequences. We then performed BLAST search to each of these three classes of sequences, using the block A regions of the DINE-1 consensus of each species. Only hits longer than 40 bp with an E-value lower than 10-5 were included.
Characterizing target site duplication of DINE-1
From the D. yakuba genome database, seven sites of DINE-1 insertion with sequence similarity > 97% to the D. yakuba DINE-1 consensus were chosen for analysis. PCR primers complementary to the 100 bp flanking sequence of each site were designed (Additional data file 2). A total of ten lines of D. yakuba, including nine Cy lines from a natural population (gift from Dr Peter Andolfatto at UCSD) and the strain Tai18E2, which was used for whole-genome sequencing, were checked for the presence of DINE-1 insertions at each site. Genomic DNA was phenol-chloroform extracted from 20-30 flies per line followed by ethanol precipitation. The program for PCR reaction was: 94°C for 5 minutes, followed by 30 cycles of 94°C (30s), 60°C (30s), and 70°C (1 minute), and extension at 70°C for 7 minutes. For lines not containing the DINE-1 insertion, PCR products were directly sequenced using ABI BigDye (Applied Biosystems, Foster City, CA, USA) technologies.
Additional data files
The following additional data are available with the online version of this paper. Additional data file 1 is the alignment of DINE-1 consensus sequences from the 12 Drosophila species. Additional data file 2 is a table listing the genome locations and sequences of primers used for the presence/absence screen of DINE-1s in D. yakuba. Additional data file 3 is a table listing DINE-1 insertions in or near to genes.
Abbreviations
- CDS:
-
coding sequence
- DINE-1 :
-
Drosophila interspersed element 1
- LINE:
-
long interspersed elements
- MITE:
-
miniature inverted-repeat transposable element
- ORF:
-
open reading frame
- SINE:
-
short interspersed elements
- TE:
-
transposable element
- TIR:
-
terminal inverted repeat
- TSD:
-
target site duplication.
References
Deininger P, Roy-Engel A: Mobile elements in animal and plant genomes. Mobile DNA II. Edited by: Craig NL. 2002, Washington, DC: ASM Press, 1074-1092.
Bartolome C, Maside X, Charlesworth B: On the abundance and distribution of transposable elements in the genome of Drosophila melanogaster. Mol Biol Evol. 2002, 19: 926-937.
Kidwell MG, Lisch D: Transposable elements as sources of variation in animals and plants. Proc Natl Acad Sci USA. 1997, 94: 7704-7711. 10.1073/pnas.94.15.7704.
Finnegan DJ: Eukaryotic transposable elements and genome evolution. Trends Genet. 1989, 5: 103-107. 10.1016/0168-9525(89)90039-5.
Kaminker J, Bergman C, Kronmiller B, Carlson J, Svirskas R, Patel S, Frise E, Wheeler D, Lewis S, Rubin G: The transposable elements of the Drosophila melanogaster euchromatin: a genomics perspective. Genome Biol. 2002, 3: R0084-10.1186/gb-2002-3-12-research0084.
Bergman C, Quesneville H, Anxolabehere D, Ashburner M: Recurrent insertion and duplication generate networks of transposable element sequences in the Drosophila melanogaster genome. Genome Biol. 2006, 7: R112-10.1186/gb-2006-7-11-r112.
Drosophila 12 Genomes Consortium: Evolution of genes and genomes on the Drosophila phylogeny. Nature. 2007, 450: 203-218. 10.1038/nature06341.
Kazazian HH: Mobile elements: drivers of genome evolution. Science. 2004, 303: 1626-1632. 10.1126/science.1089670.
Berg D, Howe M: Mobile DNA. 1989, Washington, DC: American Society of Microbiology
McDonald JF: Transposable Elements and Evolution. 1993, Dodrect, The Netherlands: Kluver
Kajikawa M, Okada N: LINEs mobilize SINEs in the eel through a shared 3' sequence. Cell. 2002, 111: 433-444. 10.1016/S0092-8674(02)01041-3.
Okada N, Hamada M, Ogiwara I, Ohshima K: SINEs and LINEs share common 3' sequences: a review. Gene. 1997, 205: 229-243. 10.1016/S0378-1119(97)00409-5.
Bureau TE, Wessler SR: Tourist: a large family of small inverted repeat elements frequently associated with maize genes. Plant Cell. 1992, 4: 1283-1294. 10.1105/tpc.4.10.1283.
Feschotte C, Jiang N, Wessler SR: Plant transposable elements: where genetics meets genomics. Nat Rev Genet. 2002, 3: 329-341. 10.1038/nrg793.
Bureau T, Wessler S: Mobile inverted-repeat elements of the tourist family are associated with the genes of many cereal grasses. Proc Natl Acad Sci USA. 1994, 91: 1411-1415. 10.1073/pnas.91.4.1411.
Feschotte C, Jiang N, Wessler SR: Plant transposable elements: where genetics meets genomics. Nat Rev Genet. 2002, 3: 329-341. 10.1038/nrg793.
Quesneville H, Bergman CM, Andrieu O, Autard D, Nouaud D, Ashburner M, Anxolabehere D: Combined evidence annotation of transposable elements in genome sequences. PLoS Comput Biol. 2005, 1: 166-175. 10.1371/journal.pcbi.0010022.
Capy P: Classification and nomenclature of retrotransposable elements. Cytogenet Genome Res. 2005, 110: 457-461. 10.1159/000084978.
Kapitonov VV, Jurka J: Molecular paleontology of transposable elements in the Drosophila melanogaster genome. Proc Natl Acad Sci USA. 2003, 100: 6569-6574. 10.1073/pnas.0732024100.
Biemont C, Vieira C: What transposable elements tell us about genome organization and evolution: the case of Drosophila. Cytogenet Genome Res. 2005, 110: 25-34. 10.1159/000084935.
Locke J, Howard LT, Aippersbach N, Podemski L, Hodgetts RB: The characterization of DINE-1, a short, interspersed repetitive element present on chromosome and in the centric heterochromatin of Drosophila melanogaster. Chromosoma. 1999, 108: 356-366. 10.1007/s004120050387.
Singh ND, Petrov DA: Rapid sequence turnover at an intergenic locus in Drosophila. Mol Biol Evol. 2004, 21: 670-680. 10.1093/molbev/msh060.
Yang H-P, Hung T-L, You T-L, Yang T-H: Genome-wide comparative analysis of the highly abundant transposable element DINE-1 suggests a recent transpositional burst in Drosophila yakuba. Genetics. 2006, 173: 189-196. 10.1534/genetics.105.051714.
Kapitonov VV, Jurka J: Helitrons on a roll: eukaryotic rolling-circle transposons. Trends Genet. 2007, 23: 521-529. 10.1016/j.tig.2007.08.004.
Vivas MV, Garcia-Planells J, Ruiz C, Marfany G, Paricio N, Gonzalez-Duarte R, de Frutos R: GEM, a cluster of repetitive sequences in the Drosophila subobscura genome. Gene. 1999, 229: 47-57. 10.1016/S0378-1119(99)00031-1.
Miller WJ, Nagel A, Bachmann J, Bachmann L: Evolutionary dynamics of the SGM transposon family in the Drosophila obscura species group. Mol Biol Evol. 2000, 17: 1597-1609.
Wilder J, Hollocher H: Mobile elements and the genesis of microsatellites in Dipterans. Mol Biol Evol. 2001, 18: 384-392.
Kapitonov VV, Jurka J: Helitrons in fruit flies. Repbase Reports. 2007, 7: 127-132.
Powell J, DeSalle R: Drosophila molecular phylogenies and their uses. Evol Biol. 1995, 28: 87-138.
Singh ND, Arndt PF, Petrov DA: Genomic heterogeneity of background substitutional patterns in Drosophila melanogaster. Genetics. 2005, 169: 709-722. 10.1534/genetics.104.032250.
Tudor M, Lobocka M, Goodell M, Pettitt J, O'Hare K: The pogo transposable element family of Drosophila melanogaster. Mol Gen Genet. 1992, 232: 126-134. 10.1007/BF00299145.
Holyoake AJ, Kidwell MG: Vege and Mar: two novel hAT MITE families from Drosophila willistoni. Mol Biol Evol. 2003, 20: 163-167. 10.1093/molbev/msg023.
Zhang L, Dawson A, Finnegan DJ: DNA-binding activity and subunit interaction of the mariner transposase. Nucleic Acids Res. 2001, 29: 3566-3575. 10.1093/nar/29.17.3566.
Auge-Gouillou C, Hamelin M-H, Demattei M-V, Periquet G, Bigot Y: The ITR binding domain of the Mariner Mos-1 transposase. Mol Genet Genomics. 2001, 265: 58-65. 10.1007/s004380000386.
Feschotte C, Osterlund MT, Peeler R, Wessler SR: DNA-binding specificity of rice mariner-like transposases and interactions with Stowaway MITEs. Nucleic Acids Res. 2005, 33: 2153-2165. 10.1093/nar/gki509.
Casola C, Lawing AM, Betran E, Feschotte C: PIF-like transposons are common in Drosophila and have been repeatedly domesticated to generate new host genes. Mol Biol Evol. 2007, 24: 1872-1888. 10.1093/molbev/msm116.
Song WY, Pi LY, Bureau TE, Ronald PC: Identification and characterization of 14 transposon-like elements in the noncoding regions of members of the Xa21 family of disease resistance genes in rice. Mol Gen Genet. 1998, 258: 449-456. 10.1007/s004380050755.
Akagi H, Yokozeki Y, Inagaki A, Mori K, Fujimura T: Micron, a microsatellite-targeting transposable element in the rice genome. Mol Genet Genomics. 2001, 266: 471-480. 10.1007/s004380100563.
Tu Z, Orphanidis SP: Microuli, a family of miniature subterminal inverted-repeat transposable elements (MSITEs): transposition without terminal inverted repeats. Mol Biol Evol. 2001, 18: 893-895.
Pritham EJ, Feschotte C: Massive amplification of rolling-circle transposons in the lineage of the bat Myotis lucifugus. Proc Natl Acad Sci USA. 2007, 104: 1895-1900. 10.1073/pnas.0609601104.
Tamura K, Subramanian S, Kumar S: Temporal patterns of fruit fly (Drosophila) evolution revealed by mutation clocks. Mol Biol Evol. 2004, 21: 36-44. 10.1093/molbev/msg236.
Singer MF: SINEs and LINEs: highly repeated short and long interspersed sequences in mammalian genomes. Cell. 1982, 28: 433-434. 10.1016/0092-8674(82)90194-5.
Gentles AJ, Wakefield MJ, Kohany O, Gu W, Batzer MA, Pollock DD, Jurka J: Evolutionary dynamics of transposable elements in the short-tailed opossum Monodelphis domestica. Genome Res. 2007, 17: 992-1004. 10.1101/gr.6070707.
Jurka J, Kohany O, Pavlicek A, Kapitonov VV, Jurka MV: Duplication, coclustering, and selection of human Alu retrotransposons. Proc Natl Acad Sci USA. 2004, 101: 1268-1272. 10.1073/pnas.0308084100.
Wessler SR, Bureau TE, White SE: LTR-retrotransposons and MITEs: important players in the evolution of plant genomes. Curr Opin Genet Dev. 1995, 5: 814-821. 10.1016/0959-437X(95)80016-X.
Tikhonov AP, Bennetzen JL, Avramova ZV: Structural domains and matrix attachment regions along colinear chromosomal segments of maize and sorghum. Plant Cell. 2000, 12: 249-264. 10.1105/tpc.12.2.249.
Sen SK, Han K, Wang J, Lee J, Wang H, Callinan PA, Dyer M, Cordaux R, Liang P, Batzer MA: Human genomic deletions mediated by recombination between Alu elements. Am J Hum Genet. 2006, 79: 41-53. 10.1086/504600.
Bailey JA, Eichler EE: Primate segmental duplications: crucibles of evolution, diversity and disease. Nat Rev Genet. 2006, 7: 552-564. 10.1038/nrg1895.
Ciccarelli FD, von Mering C, Suyama M, Harrington ED, Izaurralde E, Bork P: Complex genomic rearrangements lead to novel primate gene function. Genome Res. 2005, 15: 343-351. 10.1101/gr.3266405.
Chen S-T, Cheng H-C, Barbash DA, Yang H-P: Evolution of hydra, a recently evolved testis-expressed gene with nine alternative first exons in Drosophila melanogaster. PLoS Genet. 2007, 3: e107-10.1371/journal.pgen.0030107.
Assembly/Alignment/Annotation of 12 related Drosophila species. [http://rana.lbl.gov/drosophila/]
Thompson JD, Higgins DG, Gibson TJ: CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, positions-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22: 4673-4680. 10.1093/nar/22.22.4673.
Kumar S, Tamura K, Nei M: MEGA 3: integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief Bioinform. 2004, 5: 150-163. 10.1093/bib/5.2.150.
The UCSC Genome Browser. [http://genome.ucsc.edu/]
Jukes TH, Cantor CR: Evolution of protein molecules. Mammalian Protein Metabolism. Edited by: Munro HN. 1969, New York: Academic Press, 21-132.
Acknowledgements
We gratefully acknowledge Dr Pierre Capy, who pointed out the possibility of species-specific DINE-1s in Drosophila, and Dr Michael Ashburner and the anonymous reviewers for helpful comments on the manuscript. We thank Hsin-Chien Cheng and Jin-Yen Huang for helping with sequence alignments, Pei-San Li for technical support, and Dr David Waxman for statistical consultation. This work was supported by a grant to H-PY from National Science Council, Taiwan (NSC 93-2311-B-010-007-).
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
H-PY designed the research, performed the research and analyzed the data. H-PY and DAB wrote the paper.
Electronic supplementary material
13059_2007_1901_MOESM2_ESM.doc
Additional data file 2: Genome locations and sequences of primers used for the presence/absence screen of DINE-1s in D. yakuba. (DOC 32 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Yang, HP., Barbash, D.A. Abundant and species-specific DINE-1 transposable elements in 12 Drosophila genomes. Genome Biol 9, R39 (2008). https://doi.org/10.1186/gb-2008-9-2-r39
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/gb-2008-9-2-r39