
Summary
Angiotensin-I-converting enzyme (ACE) is a monomeric, membrane-bound, zinc- and chloride-dependent peptidyl dipeptidase that catalyzes the conversion of the decapeptide angiotensin I to the octapeptide angiotensin II, by removing a carboxy-terminal dipeptide. ACE has long been known to be a key part of the renin angiotensin system that regulates blood pressure, and ACE inhibitors are important for the treatment of hypertension. There are two forms of the enzyme in humans, the ubiquitous somatic ACE and the sperm-specific germinal ACE, both encoded by the same gene through transcription from alternative promoters. Somatic ACE has two tandem active sites with distinct catalytic properties, whereas germinal ACE, the function of which is largely unknown, has just a single active site. Recently, an ACE homolog, ACE2, has been identified in humans that differs from ACE in being a carboxypeptidase that preferentially removes carboxy-terminal hydrophobic or basic amino acids; it appears to be important in cardiac function. ACE homologs (also known as members of the M2 gluzincin family) have been found in a wide variety of species, even in those that neither have a cardiovascular system nor synthesize angiotensin. X-ray structures of a truncated, deglycosylated form of germinal ACE and a related enzyme from Drosophila have been reported, and these show that the active site is deep within a central cavity. Structure-based drug design targeting the individual active sites of somatic ACE may lead to a new generation of ACE inhibitors, with fewer side-effects than currently available inhibitors.
Similar content being viewed by others
Gene organization and evolutionary history
Angiotensin-I-converting enzyme (ACE, also known as peptidyl-dipeptidase A or kininase II) was first isolated in 1956 and shown to be a chloride-dependent metalloenzyme that cleaves a dipeptide from the carboxyl terminus of the decapeptide angiotensin I to form the potent vasopressor (blood vessel constrictor) angiotensin II [1]. In addition, it inactivates the vasodilator bradykinin by sequential removal of two carboxy-terminal dipeptides. Indeed, it is a broad-specificity dipeptidyl carboxypeptidase and may also act on non-vasoactive peptides. There are two forms of ACE in humans, encoded by a single gene located on chromosome 17 at q23; it is 21 kb in length and contains 26 exons and 25 introns. The longer form, known as somatic ACE (sACE), is transcribed from exons 1-12 and 14-26, whereas the shorter form, known as germinal or testicular ACE (gACE), is transcribed from exons 13-26. The promoter for sACE is in the 5' flanking region of the first exon, whereas that for gACE is located within intron 12 [2].
Somatic ACE consists of an intracellular domain, a transmembrane domain and two similar extracellular domains, the amino or N domain and the carboxy or C domain (Figure 1). The structure of the ACE gene is the result of gene duplication; the N and C domains are similar in sequence, and the homologous exons encoding the N and C domains (exons 4-11 and 17-24, respectively) are very similar in size and have similar codon phases at exon-intron boundaries. Each of the domains contains a catalytically active site characterized by a consensus zinc-binding motif (HEXXH in the single-letter amino-acid code, where X is any amino acid) and a glutamine nearer the carboxyl terminus that also binds zinc; ACE and its homologs (see below) therefore make up the M2 gluzincin family [3].

Schematic representation of the primary structure of several members of the ACE protein family. The locations of the active-site zinc-binding motifs are indicated by HEXXH; transmembrane domains are in black. The sequence of gACE is identical to that of the C domain of sACE, except for its first 36 residues. Human gACE and sACE have the same carboxy-terminal transmembrane and cytosolic sequences, whereas ACE2 has a distinct transmembrane and cytosolic sequence. Neither of the Drosophila ACEs, AnCE and Acer, has a membrane-anchoring sequence. Dimensions are not to scale. N, amino terminus; C, carboxyl terminus. The single lines are regions of sequence with no similarity to other proteins. The carboxyl end of ACE2 is homologous to collectin, a non-enzymatic protein associated with renal injury [18].
ACE homologs have also been found in other animal species, including chimpanzee, cow, rabbit, mouse, chicken, goldfish, electric eel, house fly, mosquito, horn fly, silk worm, Drosophila melanogaster and Caenorhabditis elegans, and in the bacteria Xanthomonas spp. and Shewanella oneidensis [3]. The cDNA of one form of D. melanogaster ACE (termed AnCE) encodes a protein of 615 amino acids that has a high degree of similarity to both domains of human sACE, indicating that the D. melanogaster protein is a single-domain enzyme [4]. It contains a signal peptide but no carboxy-terminal membrane-anchoring hydrophobic sequence. Kinetic studies suggest that it resembles the C domain of human sACE, as well as gACE. Other evidence suggests that it might resemble the ancestral form of the ACE gene before the gene duplication that is posited to have taken place in the deuterostome lineage and estimated to occur 330-350 million years ago (the time of the appearance and radiation of amphibians) [5].
A second ACE-related gene product, termed Acer, has also been identified in D. melanogaster. Selective inhibition by phosphinic peptides (containing -PO2-CH2- links instead of -CO-NH- links) indicates that Acer has active site features characteristic of the N domain of sACE. Four additional ACE-like genes have been found in the Adh region of Drosophila chromosome 2, and it has been suggested that this is a reflection of an ancestral gene structure present in both protostome and deuterostome lineages. It is further suggested that the duplication within the ACE gene in vertebrate genomes predates the divergence of these lineages [6].
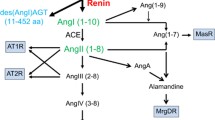
Recently, a further human homolog of ACE, referred to as ACE2, was identified and shown to be an essential regulator of cardiac function [7]. It differs from ACE in that it contains a single zinc-binding catalytic domain, is a carboxypeptidase with preference for carboxy-terminal hydrophobic or basic residues, and is not affected by ACE inhibitors. Angiotensin I and II, as well as numerous other biologically active peptides, are substrates for ACE2, but bradykinin is not. Genomic structure analysis indicates that ACE and ACE2 arose by duplication from a common ancestor.
Characteristic structural features
Human sACE is a type-I membrane-bound protein. It consists of a 28-residue carboxy-terminal cytosolic domain, a 22-residue hydrophobic transmembrane domain and a 1227-residue extracellular domain that is heavily glycosylated (30% by weight; see Figure 1). The extracellular domain is further divided into two homologous domains, a 612-residue N domain at the amino terminus linked by a 15-residue sequence to a 600-residue C domain. Each of the extracellular domains contains an HEXXH sequence in which the two histidine residues serve as zinc-binding ligands; together with a glutamine located 23-24 residues toward the carboxyl terminus and a water molecule, they provide the metal with tetrahedral coordination geometry. Detailed kinetic and mutational analyses have demonstrated that both zinc sites have catalytic activity [8, 9].
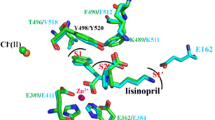
Human gACE corresponds to the C domain of sACE. It has the same 28-residue cytosolic and 22-residue transmembrane domains and, except for its first 36 residues, the same 615-residue extracellular domain. Recently, the three-dimensional structure of a deglycosylated, truncated version of gACE was determined by X-ray crystallography [10], as was the structure of intact Drosophila AnCE [11]. As might be expected, the two structures are quite similar: both have a preponderance of α-helices (over 70%) surrounding a deep central cavity that divides the molecule into two halves (Figure 2). The active site is deep within the cavity and access by substrates is limited by the cavity's dimensions. Although neither gACE nor AnCE has any sequence similarity to rat neurolysin or Pyrococcus furiosus carboxypeptidase, the three-dimensional structures of all these enzymes bear a striking resemblance to each other.

A schematic representation of the structure of truncated, deglycosylated human gACE in a complex with the inhibitor lisinopril [11]. The gACE molecule can be divided into two halves, subdomains I (light gray) and II (dark gray), that enclose the substrate-binding site. The active-site zinc atom is shown coordinated to lisinopril (in stick representation). Two bound chloride ions are designated Cl1 and Cl2. N, amino terminus; C, carboxyl terminus.
Localization and function
Human sACE, like all ACE or ACE-like gene products, has a signal peptide that directs it to an extracellular localization. Each vertebrate ACE also has a carboxy-terminal hydrophobic sequence that anchors it to the cell membrane whereas non-vertebrate ACEs lack such a sequence and are extracellular soluble enzymes. Human sACE is expressed strongly in many types of endothelial cells, especially in the capillaries of the lung, as well as in epithelial cells in the kidney, small intestine and epididymis. Microarray analysis of gene expression indicates that sACE mRNA is expressed in virtually all tissues [12].
The extracellular localization of sACE on endothelial cells positions it optimally for interaction with its substrate angiotensin I. It has been shown, in fact, that a single passage of blood through the pulmonary vasculature is sufficient to convert all circulating angiotensin I to angiotensin II [13]. Despite this high efficiency, angiotensin I is not the sole substrate for ACE. In addition to bradykinin, other bioactive peptides can be acted upon by ACE. N-acetyl-Ser-Asp-Lys-Pro, a negative regulator of the recruitment of pluripotent hematopoetic stem cells into the cell cycle, is specifically degraded by the N-domain active site of sACE [14]. The localization of ACE on the epithelial cells of the small intestine and the renal proximal tubule also suggests that peptides affecting these tissues may well serve as substrates. Moreover, the fact that ACE-like enzymes are found in species that do not produce angiotensin I indicates that they must be involved in processing other types of peptides. The effects of disrupting the mouse ACE gene are consistent with this hypothesis: ACE-/- mice have low blood pressure but also exhibit severe abnormalities in renal structure and function. Mice expressing only the N domain of sACE, and in soluble form, have much the same phenotype as ACE-/- mice [15].
The gACE isoform is expressed only in developing sperm cells and mature sperm. Sperm lacking gACE are deficient in transport and attachment to the zonae pellucidae of oocytes, and male ACE-/- mice have markedly diminished fertility [16], even though they have normal testis structure, sperm count, sperm morphology and sperm motility [17]. The specific substrate of gACE, if it has one, is unknown, and the precise role of the enzyme in reproduction remains to be established.
ACE2 is a type-I membrane-bound glycoprotein present on endothelial and epithelial cells. It has been found mostly in heart, kidney and testis with lesser amounts in colon, small intestine and ovary [18–20]. It has been implicated in cardiac function [7] but details of its specific role are just beginning to emerge.
Mechanism
Details of the catalytic mechanism of ACE have yet to be elucidated, but other zinc metalloenzymes with the same HEXXH metal-binding motif are thought to initiate substrate hydrolysis by an attack at the scissile carbonyl group of the substrate by the water molecule coordinated to the metal and assisted by the glutamate (E) in the HEXXH sequence. The X-ray structure of gACE shows two histidine residues within hydrogen-bonding distance of the amide carbonyl group of the inhibitor lisinopril bound at the active site, and these may function much like corresponding residues in the active site of the extensively studied metalloproteinase thermolysin [21]. The C-domain active site of sACE, as well as that of ACE2 and to a lesser extent also gACE, are activated by high concentrations of chloride. The effect is primarily an enhancement of substrate binding but its physiological significance is not well understood.
Using the assumed mechanistic analogy to other zinc metallopeptidases, plus the knowledge that several snake-venom peptides potentiate the action of bradykinin by inhibiting ACE, efforts were undertaken to develop orally-active ACE inhibitors based on metal-binding versions of venom peptide analogs for potential use in the treatment of hypertension. The first such compound, captopril, was approved for use in 1981, and since that time many other similar compounds have been introduced and studied extensively [22]. ACE inhibitors are now first-line therapy for hypertension and also for congestive heart failure, left ventricular systolic dysfunction and myocardial infarction (heart attack), and they are recommended to slow the progression of diabetic and non-diabetic nephropathy [23, 24]. They have also been shown to have a direct anti-atherogenic effect, thereby slowing the progression of atherosclerotic vascular disease [25].
Frontiers
The recent crystal-structure determination of gACE opens the way to the design of a new generation of ACE inhibitors [11]. Although ACE inhibitors have been in clinical use for over 20 years, are very effective, and clearly have had a major impact on antihypertensive therapy, they are not totally without side-effects. The most frequent one is a dry cough that can occur in from 5-20% of patients and that can be so debilitating in certain instances as to result in the cessation of treatment [23]. A more serious problem is angioedema (swellings caused by leakage from blood vessels), which has an incidence of only 0.1-0.5% but can be life-threatening [26]. The basis for these side effects is still a matter of discussion, but it is generally thought that decreased ACE activity leads to altered concentrations of bradykinin, other bioactive peptides and even other angiotensin-related peptides, and that these are responsible for the side-effects.
All clinical ACE inhibitors developed to date have been based on the original assumption of an active site related to that of pancreatic carboxypeptidase A but organized to cleave a dipeptide rather than a single amino acid from the carboxyl terminus of its substrate. It is now known that sACE has two active sites, neither of which resembles that of carboxypeptidase A, and that these sites are not identical. Indeed, the N-domain active site preferentially cleaves bradykinin and other peptides. Clinical ACE inhibitors show little discrimination between these two active sites, however, and it is probable that an inhibitor specific for one or the other could have special benefits. The side effects of conventional ACE inhibitors might not occur with a C-domain-specific inhibitor, for example.
At present, only the C-domain structure is available, and although the structure of the N domain can be deduced by analogy, it would be ideal to have crystal structures for both the N domain and intact sACE. The complicating effects of glycosylation and the hydrophobic membrane anchor on crystallization, which delayed X-ray analysis for many years, may not be overcome easily, but deglycosylation and truncation may again prove successful. Structure-based design of active site-specific inhibitors may well be possible in the near future.
The discovery of ACE2 and its importance in cardiac function will certainly spur new efforts toward understanding the complex and delicately balanced relationships between the many bioactive peptides on which it may act. Greater insight into the possible metabolic pathways involving both angiotensin-related and bradykinin-related peptides, as well as their receptors and downstream signaling events, could lead to more finely-tuned therapies appropriate for individual patients.
It is somewhat ironic that the first ACE crystal structure was obtained with a version of gACE, which is so much less well understood in terms of biological function than sACE. The C domain of sACE is primarily responsible, albeit indirectly, for maintaining the angiotensin II blood levels that regulate blood pressure and fluid balance, yet it is also the C domain, in the guise of gACE, that is found in developing and mature sperm. There is no indication that conventional ACE inhibitors have any effect on male fertility or that gACE normally acts on angiotensin I. It remains to be seen whether inhibitors can be specifically targeted to gACE, but it does seem likely that the availability of a crystal structure will focus attention on this very interesting member of the ACE protein family.
References
Skeggs LT, Kahn JR, Shumway NP: Preparation and function of the hypertensin converting enzyme. J Exp Med. 1956, 103: 295-299. The first report on the isolation and partial characterization of ACE, in this case from horse plasma.
Hubert C, Houot AM, Corvol P, Soubrier F: Structure of the angiotensin I-converting enzyme gene. Two alternate promoters correspond to evolutionary steps of a duplicated gene. J Biol Chem. 1991, 266: 15377-15383. The complete description of the gene structure of sACE and evidence for alternative promoters for the expression of the mRNAs for somatic and germinal ACE.
Corvol P, Williams TA: Peptidyl-dipeptidase A/angiotensin 1-converting enzyme. In Handbook of Proteolytic Enzymes. Edited by: Barrett AJ, Rawlings ND, Woessner JF. 1998, San Diego: Academic Press, 1066-1076. An excellent summary of ACE, including sequence data bank codes and a thorough analysis of the chemical and biological properties of ACE.
Williams TA, Michaud A, Houard X, Chauvet M-T, Soubrier F, Corvol P: Drosophila melanogaster angiotensin I-converting enzyme expressed in Pichia pastoris resembles the C domain of the mammalian homologue and does not require glycosylation for secretion and enzymatic activity. Biochem J. 1996, 318: 125-131. This paper shows that AnCE has catalytic properties that resemble those of human C-domain ACE.
Cornell MJ, Williams TA, Lamango NS, Coates D, Corvol P, Soubrier F, Hoheisel J, Lehrach H, Isaac RE: Cloning and expression of an evolutionary conserved single-domain angiotensin converting enzyme from Drosophila melanogaster. J Biol Chem. 1995, 270: 13613-13619. 10.1074/jbc.270.29.17627. This paper presents an interesting analysis of the evolution of the ACE gene and its duplication.
Coates D, Isaac RE, Cotton J, Siviter R, Williams TA, Shirras A, Corvol P, Dive V: Functional conservation of the active sites of human and Drosophila angiotensin I-converting enzyme. Biochemistry. 2000, 39: 8963-8969. 10.1021/bi000593q. Further discussion of the ancestral ACE gene structure and evidence that the two active sites of human ACE have different functions.
Crackower MA, Sarao R, Oudit GY, Yagil C, Kozieradzki I, Scanga SE, Oliveira-dos-Santos AJ, da Costa J, Zhang L, Pei Y, et al: Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature. 2002, 417: 822-828. 10.1038/nature00786. The gene for ACE2 maps to a quantitative trait locus on the X chromosome in rat models of hypertension, and disruption of the gene results in severe cardiac dysfunction. Concomitant disruption of the ACE gene rescues cardiac function.
Wei L, Alhenc-Gelas F, Corvol P, Clauser E: The two homologous domains of human angiotensin I-converting enzyme are both catalytically active. J Biol Chem. 1991, 266: 9002-9008. This paper and [9] provide direct evidence that both catalytic domains of sACE are enzymatically active.
Jaspard E, Wei L, Alhenc-Gelas F: Differences in the properties and enzymatic specificities of the two active sites of angiotensin I-converting enzyme (kininase II). Studies with bradykinin and other natural peptides. J Biol Chem. 1993, 268: 9496-9503. Further evidence that both the N and C domains of sACE are catalytically active, but with different specificities.
Natesh R, Schwager SLU, Sturrock ED, Acharya KR: Crystal structure of the human angiotensin-converting enzyme-lisinopril complex. Nature. 2003, 421: 551-554. 10.1038/nature01370. The first crystal structure of a truncated version of human sACE.
Kim HM, Shin DR, Yoo OJ, Lee H, Lee J-O: Crystal structure of Drosophila angiotensin I-converting enzyme bound to captopril and lisinopril. FEBS Lett. 2003, 538: 65-70. 10.1016/S0014-5793(03)00128-5. After years of effort to obtain crystals of ACE, the crystal structure of Drosophila AnCE was published within weeks of that for human sACE.
Coates D: The angiotensin converting enzyme (ACE). Int J Biochem Cell Biol. 2003, 35: 769-773. 10.1016/S1357-2725(02)00309-6. A short review that examines the recently emerging factors underlying the therapeutic basis for ACE inhibition.
Ng KK, Vane JR: Fate of angiotensin I in the circulation. Nature. 1968, 218: 144-150. A classic analysis of angiotensin pharmacokinetics.
Rousseau A, Michaud A, Chauvet M-T, Lenfant M, Corvol P: The hemoregulatory peptide N-acetyl-Ser-Asp-Lys-Pro is a natural and specific substrate of the N-terminal active site of human angiotensin-converting enzyme. J Biol Chem. 1995, 270: 3656-3661. 10.1074/jbc.270.8.3656. The first report of a natural substrate for the N domain catalytic site of sACE.
Esther CR, Marino EM, Howard TE, Machaud A, Corvol P, Capecchi M, Bernstein KE: The critical role of tissue angiotensin-converting enzyme as revealed by gene targeting in mice. J Clin Invest. 1997, 99: 2375-2385. This paper describes the changes in renal structure that result from disrupting the C-domain of sACE.
Hagaman JR, Moyer JS, Bachman ES, Sibony M, Magyar PL, Welch JE, Smithies O, Krege JH, O'Brien DA: Angiotensin-converting enzyme and male fertility. Proc Natl Acad Sci USA. 1998, 95: 2552-2557. 10.1073/pnas.95.5.2552. Demonstration that sACE is essential for male fertility but that angiotensin I is not a necessary substrate.
Ramaraj P, Kessler SP, Colmenares C, Sen GC: Selective restoration of male fertility in mice lacking angiotensin-converting enzymes by sperm-specific expression of the testicular enzyme. J Clin Invest. 1998, 102: 371-378. The fertility defect in male ACE knockout mice is restored by selective expression of ACE only in sperm.
Turner AJ, Hooper NM: The angiotensin-converting enzyme gene family: genomics and pharmacology. Trends Pharmacol Sci. 2002, 23: 177-183. 10.1016/S0165-6147(00)01994-5. A review of the ACE gene family and an integrated analysis of the reninangiotensin system and its relationship to ACE2.
Tipnis SR, Hooper MN, Hyde R, Karran E, Christie G, Turner AJ: A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J Biol Chem. 2000, 275: 33238-33243. 10.1074/jbc.M002615200. The first report of a homolog of human ACE that turns out to be a carboxypeptidase and tissue-specific. The authors refer to it as ACEH.
Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N, Donovan M, Woolf B, Robison K, Jeyaseelan R, et al: A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ Res. 2000, 87: e1-e9. A report, almost concomitant with [19], on the identification of ACE2 and delineation of its enzymatic characteristics.
Matthews BW: Structural basis of the action of thermolysin and related zinc peptidases. Acc Chem Res. 1988, 21: 333-340. A classic review of the mechanism of action of the prototype zinc proteinase, thermolysin.
Zaman MA, Oparil S, Calhoun DA: Drugs targeting the renin-angiotensin-aldosterone system. Nat Rev Drug Discov. 2002, 1: 621-636. 10.1038/nrd873. An excellent summary of numerous clinical studies of drugs targeting one or more components of reninangiotensin-aldosterone system.
Bicket DP: Using ACE inhibitors appropriately. Am Fam Physician. 2002, 66: 461-468. A helpful review of the side-effects encountered with the use of ACE inhibitors.
Sleight P: The reninangiotensin system: a review of trials with angiotensin-converting enzyme inhibitors and angiotensin receptor blocking agents. Eur Heart J. 2002, 4 (Suppl A): A53-A57. A summary of clinical trials with ACE inhibitors and ACE receptor blockers.
Lonn E, Gerstein HC, Smieja M, Mann JFE, Yusuf S: Mechanisms of cardiovascular risk reductions with ramipril: insights from HOPE and HOPE substudies. Eur Heart J. 2003, 5 (Suppl A): A43-A48. How one ACE inhibitor, ramipril, can affect the risk of cardiovascular disease.
Adam A, Cagno M, Molinaro G, Perez M, Lepage Y, Agostoni A: Aminopeptidase P in individuals with a history of angio-oedema on ACE inhibitors. Lancet. 2002, 359: 2088-2089. 10.1016/S0140-6736(02)08914-6. A short summary of the incidence of angioedema as a side-effect of taking ACE inhibitors and the role of aminopeptidase P in the etiology of the disease.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Riordan, J.F. Angiotensin-I-converting enzyme and its relatives. Genome Biol 4, 225 (2003). https://doi.org/10.1186/gb-2003-4-8-225
Published:
DOI: https://doi.org/10.1186/gb-2003-4-8-225