
Abstract
Introduction
Protein disulfide isomerase (PDI) is an important factor for the protein modification step in the post-translational event. PDI plays an essential role in cell survival under various stress conditions. It has been reported that PDI can serve as a negative regulator of nuclear factor-kappa-B (NF-κB) and that it can inhibit lipopolysaccharide (LPS)-induced proinflammatory cytokine production in macrophages. Thus, PDI may be an intracellular anti-inflammatory molecule. Although we have previously shown that Kupffer cell-derived proinflammatory cytokines cause liver injury in sepsis, the effect of sepsis on PDI expression as well as the effect of PDI inhibition on cytokine production have not been investigated. We therefore hypothesized that sepsis downregulates PDI expression and that the inhibition of PDI promotes proinflammatory cytokine production.
Method
Adult male rats were subjected to sepsis by cecal ligation and puncture (CLP) or endotoxemia (continuous infusion of 1 μg/kg body weight LPS by an osmotic pump) for 20 hours. Hepatic tissues were collected and PDI gene expression was determined. In additional experiments, cells from a macrophage-like cell line, RAW 264.7, were treated with 100 ng/mL LPS for 4 hours and protein expressions were measured. RAW 264.7 cells were also treated with bacitracin, a specific PDI inhibitor, for 24 hours, and tumor necrosis factor-alpha (TNF-α) gene and protein expression as well as its release in the cell supernatant were determined. To further confirm the beneficial effect of PDI in sepsis, RAW 264.7 cells were transfected with PDI short interfering RNA (siRNA) and PDI gene expression and TNF-α release were measured by quantitative polymerase chain reaction and enzyme-linked immunosorbent assay, respectively.
Results
PDI gene expression was significantly decreased by 28% and 69% at 20 hours after CLP or LPS infusion, respectively. LPS also decreased PDI protein expression by 33% in RAW 264.7 cells. Incubation of RAW 264.7 cells with bacitracin significantly increased TNF-α gene expression and TNF-α release as well as its cellular levels in a dose-dependent manner. Transfection of RAW 264.7 cells with PDI siRNA produced an average 36.8% inhibition of the PDI gene expression. This downregulation was correlated with a 3.19-fold increase in TNF-α release into the cell supernatant.
Conclusion
Taken together, these results suggest that downregulation of PDI by sepsis significantly increases proinflammatory cytokine production. Thus, prevention of PDI downregulation in sepsis may be a novel approach to attenuate hyperinflammation and to reduce tissue injury under such conditions.
Similar content being viewed by others


Introduction
Infection and sepsis continue to be the most common causes of death in noncardiac intensive care units [1–4]. Evidence indicates that, in the US alone, more than 750,000 patients develop sepsis and septic shock each year with an overall mortality of 28.6% [5]. Severe sepsis is a common, expensive, and frequently fatal condition with as many deaths annually as those from acute myocardial infarction. The sepsis model of cecal ligation and puncture (CLP) mimics many features of clinical sepsis-peritonitis [6–14]. By using the CLP model of sepsis in the rat, we have shown that organ dysfunction occurred early after sepsis [14–18] and that the liver residential macrophages, Kupffer cells, play an important role in producing proinflammatory cytokines (for example, tumor necrosis factor-alpha [TNF-α]) in sepsis [19, 20]. It is encouraging, however, that the complex pathophysiology of sepsis is becoming better understood as more studies are being reported. These studies are shedding light on the fundamental mechanisms of the pathogenesis of sepsis and are providing novel therapeutic approaches to modulate various pathological processes under such conditions.
Protein disulfide isomerase (PDI) catalyses the formation, breakage, and rearrangement of disulfide bonds within a molecule. This catalysis is an important post-translational event in the biosynthesis of many extracellular proteins that are usually coupled to the process of protein folding [21]. Disulfide formation involves the endogenous oxidized and reduced forms of glutathione and is catalysed by PDI in the endoplasmic reticulum[22]. The highly oxidative environment of the endoplasmic reticulum directs the catalytic action of the PDI-related proteins mainly toward the formation of disulfide bonds of proteins [23, 24]. Among various tissues, the liver contains the largest amount of PDI protein, followed by the kidneys and fat tissues, and it has been shown that fasting and refeeding affect the PDI protein and its enzyme activities [25]. PDI is one of the endoplasmic reticulum stress proteins and it plays an essential role in cell survival under stress conditions [26]. These proteins also have other properties, such as proteolytic activities and the capacity of binding calcium, ATP, or other small ligands [26].
Previous studies have demonstrated that proinflammatory cytokines play a critical role in the initiation and progression of sepsis syndrome and that TNF-α, interleukin (IL)-1β, and IL-6 are important mediators of hemodynamic, metabolic, and immunologic alterations in the host during sepsis [27–31]. In this regard, it has been reported that PDI is a negative regulator of nuclear factor-kappa-B (NF-κB) and can inhibit cytokine production in macrophages after lipopolysaccharide (LPS) stimulation, suggesting that PDI may serve as an intracellular anti-inflammatory molecule [32]. Although PDI has been implicated in tumor- or apoptosis-associated conditions [33, 34], its role in sepsis has not been investigated. In the present study, we determined PDI gene expression in the liver during sepsis and endotoxemia. Because previous studies have shown that Kupffer cell-derived proinflammatory cytokines play a major role in sepsis-induced liver injury [19, 20], we also investigated the expression of PDI in cells of the macrophage-like cell line, RAW 264.7, after incubation with LPS. In addition, the specific PDI inhibitor, bacitracin, was used to determine the effect of PDI inhibition on TNF-α gene expression and production in the RAW 264.7 cells.
Materials and methods
Experimental model of sepsis
Polymicrobial sepsis was induced in adult male rats by CLP as we have previously described [35–37]. Briefly, male Sprague-Dawley rats (275 to 325 g; Charles River Laboratories, Wilmington, MA, USA) were housed in a temperature-controlled room on a 12-hour light/dark cycle and fed on a standard Purina rat chow diet (Nestlé Purina PetCare Company, St. Louis, MO, USA). Prior to the experiment, rats were fasted overnight but were allowed water ad libitum. The animals were anesthetized by isoflurane inhalation and a 2-cm ventral midline abdominal incision was made. The cecum was then exposed, ligated just distal to the ileocecal valve to avoid intestinal obstruction, punctured twice with an 18-gauge needle, and returned to the abdominal cavity. The incision was closed in layers and the animals were resuscitated by 3 mL/100 g body weight (BW) normal saline subcutaneously immediately after CLP to provide fluid resuscitation. Sham-operated animals underwent the same surgical procedure with the exception that the cecum was neither ligated nor punctured. Hepatic tissues were then harvested at 5 hours (early sepsis) and 20 hours (late sepsis) after CLP or sham operation for further analysis. This project was approved by the Animal Care and Use Committee of the Feinstein Institute for Medical Research (Manhasset, NY, USA).
Administration of lipopolysaccharides
Male rats were fasted overnight but were allowed water ad libitum. The animals were anesthetized with isoflurane inhalation and a 1-cm ventral midline abdominal incision was made. A 200-μL mini-osmotic pump (Model 2ML1; Durect Corporation, Cupertino, CA, USA) was prefilled with LPS (Escherichia coli O55:B5; Sigma-Aldrich, St. Louis, MO, USA) solution (2 μg/mL in saline) and connected to a silastic catheter. The prefilled pump was primed in sterile normal saline for 2 hours at 37°C. The primed osmotic pump was then implanted subcutaneously in the rat and the silastic catheter was inserted into the abdominal cavity for the continuous infusion of LPS at a rate of 8 μL/hour for 20 hours (total dose: 1 μg/kg BW). Following the closure of the incision, the animals received 3 mL/100 g BW normal saline subcutaneously. Control animals underwent the same surgical procedure except that normal saline was infused. Hepatic tissues were collected at 20 hours after the infusion for further analysis.
Cell culture and tumor necrosis factor-alpha measurement
Cells of the murine macrophage-like cell line, RAW 264.7, were obtained from the American Type Culture Collection (Manassas, VA, USA) and cultured in Dulbecco's modified Eagle's medium containing 10% heat-inactivated fetal bovine serum, supplemented with 15 mM HEPES (pH 7.4), 2 mM L-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin, and placed in an incubator at 37°C in 5% CO2/95% air. Cells were incubated for 4 hours with LPS (100 ng/mL) and PDI gene expression was determined by reverse transcription-polymerase chain reaction (RT-PCR), as described below. In addition, RAW 264.7 cells were incubated for 24 hours with bacitracin, a specific PDI inhibitor (Sigma-Aldrich, 0.25, 1.25, and 3.75 mM) and the supernatant and cell lysate were collected for the measurement of TNF-α. The levels of TNF-α were determined by using commercially obtained enzyme-linked immunosorbent assay (ELISA) kits specific for rat TNF-α (BioSource International, Camarillo, CA, USA). The assay was carried out according to the instructions provided by the manufacturer.
Assessment of protein disulfide isomerase and tumor necrosis factor-alpha gene expression
Hepatic tissues harvested from animal experiments or cells from the in vitro studies were fixed in RNAlate solution (Ambion, Inc., Austin, TX, USA). Total RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) and 4 μg RNA from hepatic tissues was reverse-transcribed to cDNA. The resulting cDNAs were amplified by PCR using specific primers for rat PDI (forward CTA CGA TGG CAA ATT GAG CA and reverse CTT CCA CCT CAT TGG CTG TT) and rat glyceraldehyde 3-phosphate dehydrogenase (G3PDH) (forward TTG TAA CCA ACT GGG ACG ATA TGG and reverse GAT CTT GAT CTT CAT GGT GCT AGG). For TNF-α gene expression, 1.8 μg RNA from RAW 264.7 cells was reverse-transcribed to cDNA and amplified by PCR using specific primers for mouse TNF-α (forward TTC TGT CCC TTT CAC TCA CTG G and reverse TTG GTG GTT TGC TAC GAC GTG G) and mouse β-actin (forward GTG GGC CGC TCT AGG CAC CAA and reverse CTC TTT GAT GTC ACG CAC GAT TTC). For both PDI and the TNF-α gene expression, the PCR was conducted at 30 cycles, each cycle consisting of 30 seconds at 94°C, 30 seconds at 60°C, and 1 minute at 72°C. Following the RT-PCR procedure, the reaction products were electrophoresed on 1.6% TBE (Tris borate-ethylenediaminetetraacetic acid)-agarose gel containing 0.22 μg/mL ethidium bromide. The gel was then photographed and the band density was analyzed by a digital image system.
Transfection of RAW 264.7 cells with protein disulfide isomerase short interfering RNA
Silencer select predesigned PDI specific short interfering RNA (siRNA) (catalog number 4390771) previously annealed was obtained from Ambion, Inc., Austin, TX, USA. RAW 264.7 cells were plated at 5 × 105 cells in 12-well dishes and incubated overnight at 37°C and 5% CO2. Cells were then transfected with 100 nM PDI siRNA or negative control siRNA using Dharmafect Reagent 4 (Dharmacon RNAi Technologies, Chicago, IL, USA) in 1 mL media containing 10% serum according to the manufacturer's instructions. The transfected cells were incubated at 37°C for 48 hours. Afterward, cells were harvested for RNA isolation and the supernatant was collected for cytokine measurement. Total RNA isolated was reverse-transcribed to cDNA and used in real-time PCR with relative quantification analysis using primers specific for mouse PDI: forward 5'-TACCTGCTGGTGGAGTTCTATGC-3' and reverse 5'-TCGGGAGCCAGAGCTTTG-3'. The mouse β-actin primers were used as a control to quantitate the fold change in PDI gene expression. The supernatant collected from the transfected cells was used to measure TNF-α levels using ELISA kits specific for mouse TNF-α. The GPDH siRNA (100 nM) was used as a positive control for the transfection studies.
Statistical analysis
All data were expressed as mean ± standard error and compared by one-way analysis of variance and Tukey's test or Student t test. Differences in value were considered significant if the P value was less than 0.05.
Results
Protein disulfide isomerase gene expression in the liver after cecal ligation and puncture and in RAW 264.7 after lipopolysaccharide incubation
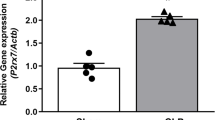
As shown in Figure 1, despite the fact that the PDI gene expression in hepatic tissues decreased by 19% at 5 hours after CLP, such a decrease was not statistically significant. In contrast, hepatic PDI gene expression decreased by 28% at 20 hours after CLP (P < 0.05, Figure 1). At 20 hours after the continuous infusion of LPS (1 μg/kg BW) in normal rats, the hepatic PDI gene expression markedly decreased by 69% (P < 0.05, Figure 2). This suggests that LPS may be responsible for the downregulation of the PDI gene expression observed 20 hours after the onset of sepsis. In cells of the cultured macrophage-like cell line, RAW 264.7, the PDI protein expression was also significantly reduced (by 33%) after incubation with LPS (100 ng/mL) for 4 hours (Figure 3).

Alterations in the protein disulfide isomerase (PDI) gene expression in hepatic tissues at 5 and 20 hours after cecal ligation and puncture (CLP). The ratio of PDI and the housekeeping gene glyceraldehyde 3-phosphate dehydrogenase (G3PDH) is calculated. Values (n = 4 to 5/group) are presented as mean ± standard error and are compared by one-way analysis of variance and Tukey's test: *P < 0.05 versus respective sham-operated animals.

Alterations in the protein disulfide isomerase (PDI) gene expression in hepatic tissues after continuous infusion of lipopolysaccharide (LPS) or normal saline (control). The ratio of PDI and the housekeeping gene glyceraldehyde 3-phosphate dehydrogenase (G3PDH) is calculated. Values (n = 4 to 6/group) are presented as mean ± standard error and are compared by Student t test: *P < 0.05 versus control.

Alterations in the protein disulfide isomerase (PDI) protein expression in RAW 264.7 cells after stimulation of lipopolysaccharide (LPS) (100 ng/mL) for 4 hours. The ratio of PDI and the housekeeping gene β-actin is calculated. Values (n = 4/group) are presented as mean ± standard error and are compared by Student t test: *P < 0.05 versus control.
Effects of protein disulfide isomerase inhibition on tumor necrosis factor-alpha gene expression and production in RAW 264.7 cells
To investigate the role of PDI in the regulation of proinflammatory cytokine TNF-α, we incubated RAW 264.7 cells with a specific PDI inhibitor, bacitracin (24-hour culture). Figure 4 shows the effect of bacitracin on the TNF-α gene expression in RAW 264.7 cells. Bacitracin significantly increased TNF-α gene expression in a dose-dependent manner. The TNF-α gene expression was increased by 33%, 84%, and 93% at 0.25, 1.25, and 3.75 mM bacitracin, respectively (Figure 4). Alterations in the supernatant and cellular TNF-α levels in cells cultured with bacitracin are shown in Figures 5 and 6. As shown in Figure 5, the supernatant levels of TNF-α significantly increased (by 55%) at 0.25 mM bacitracin and further increased by 317% and 327% at the higher concentrations, 1.25 and 3.75 mM, respectively (Figure 5). Similarly, cellular concentrations of TNF-α were markedly elevated by bacitracin in the range of 12- to 54-fold in a dose-response fashion (Figure 6).

Alterations in tumor necrosis factor-alpha (TNF-α) gene expression in RAW 264.7 cells after culture with bacitracin (0.25, 1.25, and 3.75 mM) for 24 hours. The ratio of TNF-α and the housekeeping gene β-actin is calculated. Values (n = 4 to 5/group) are presented as mean ± standard error and are compared by one-way analysis of variance and Tukey's test: *P < 0.05 versus control; #P < 0.05 versus 0.25 mM bacitracin.

Alterations in supernatant tumor necrosis factor-alpha (TNF-α) levels in RAW 264.7 cells after culture with bacitracin (0.25, 1.25, and 3.75 mM) for 24 hours. TNF-α levels were determined by enzyme-linked immunosorbent assay. Values (n = 7 to 8/group) are presented as mean ± standard error and are compared by one-way analysis of variance and Tukey's test: *P < 0.05 versus control; #P < 0.05 versus 0.25 mM bacitracin.

Alterations in cellular tumor necrosis factor-alpha (TNF-α) levels in RAW 264.7 cells cultured with bacitracin (0.25, 1.25, and 3.75 mM) for 24 hours. TNF-α levels were determined by enzyme-linked immunosorbent assay. Values (n = 7 to 8/group) are presented as mean ± standard error and are compared by one-way analysis of variance and Tukey's test: *P < 0.05 versus control; #P < 0.05 versus 0.25 mM bacitracin.
Effect of protein disulfide isomerase inhibition by short interfering RNA on tumor necrosis factor-alpha gene expression and release in RAW 264.7 cells
To further confirm the role of PDI in the regulation of proinflammatory cytokine TNF-α, RAW 264.7 cells were transfected with PDI siRNA for 48 hours and TNF-α release into the cell supernatant was assessed. Transfection with 100 nM PDI siRNA produced an average 36.8% inhibition of the PDI gene expression (Figure 7a, P < 0.001). Interestingly, the PDI downregulation by siRNA caused a 3.19-fold increase in TNF-α release (Figure 7b, P < 0.001).

Alterations in the protein disulfide isomerase (PDI) gene expression and supernatant tumor necrosis factor-alpha (TNF-α) levels in RAW 264.7 cells transfected with PDI short interfering RNA (siRNA) for 48 hours. (a) PDI gene expression was determined by real-time polymerase chain reaction using specific PDI primers. (b) The TNF-α release into the cell supernatant was measured by enzyme-linked immunosorbent assay. Values (n = 3 to 6/group) are presented as mean ± standard error and are compared by paired Student t test. *P < 0.05 versus control.
Discussion
The notion that reduced/denatured proteins would spontaneously reoxidize and refold to form their native conformation led to the search for a physiological catalyst of this process. An enzyme was found that catalyzed the formation of native proteins from the reduced/denatured state and has been termed as PDI [38]. PDI is widely distributed and has been detected in most vertebrate tissues, although detailed studies have been confined to the enzyme from the liver. In the mammalian liver homogenates, PDI is found in crude microsomal membrane fractions [39]. In the rat liver, the enzyme co-sediments with markers of the endoplasmic reticulum [38]. PDI is a membrane-associated enzyme of the endoplasmic reticulum and its function, in part, is translational modification of proteins [40]. PDI may also catalyze the covalent crosslinking of native proteins or the covalent immobilization of biologically active molecules to the extracellular matrix.
In the present study, by using animal models of sepsis or endotoxemia, we have shown that the PDI gene expression is decreased at 20 hours after CLP or LPS infusion. Similarly, PDI gene expression is downregulated in a macrophage-like cell line after stimulation by LPS for 4 hours. These results indicate that PDI gene expression is downregulated under inflammatory conditions and that LPS plays an important role in the downregulation of PDI. In addition, to evaluate the role of PDI on TNF-α gene expression, we have used bacitracin, a specific inhibitor of PDI, on the TNF-α release and the expression in 24-hour-cultured RAW 264.7 cells. TNF-α levels in the supernatant and cellular TNF-α in RAW 264.7 cells cultured with bacitracin were significantly increased. In addition, we further confirmed that downregulation of PDI using PDI siRNA significantly increased TNF-α release from cells. These results suggest that PDI plays an important role in the production of proinflammatory cytokine TNF-α.
PDI has been found to be secreted from a variety of cell types, including hepatocytes [41], pancreatic exocrine cells [42], endothelial cells [43], and activated platelets [44]. While the biological importance of these secreted proteins remains in most cases obscure, the function of PDI secreted by thyrocytes into the lumen of the thyroid follicles has been identified [45]. It has been shown that the enzyme is involved in the control of thyroglobulin folding and multimerization, probably by reducing the intermolecular disulfide bridges and thus limiting the extent of multimer formation. While the full biological importance of the protein disulfide activity must still be understood, some interesting examples of PDI in pathological events such as Sindbis virus [46] and HIV [47] have been demonstrated. It has been suggested that PDI is specifically upregulated in response to hypoxia/ischemia in astrocytes [48]. In addition, the overexpression of this gene into neurons protects against apoptopic cell death induced by hypoxia/brain ischemia. Further studies by the same group indicate that ubiquilin, an endoplasmic reticulum-associated protein, together with PDI, has critical functions as a regulatory protein for cell death and therefore that upregulation of these proteins may result in the acquisition of tolerance against ischemic stress in glial cells [48]. A recent report also indicates that the transcriptional activity of NF-κB is negatively regulated by PDI [32]. Overexpression of PDI in RAW 264.7 cells strongly suppressed the LPS-induced production of inflammatory cytokines as well as NF-κB-dependent luciferase activity. This negative regulation of NF-κB was reversed by bacitracin, a PDI inhibitor. Finally, PDI expression was induced by the anti-inflammatory cytokine IL-10, and IL-10-mediated inhibition of LPS-induced IL-6 expression was reduced by bacitracin. These findings clearly demonstrate that PDI is a negative regulator of NF-κB and may act downstream of IL-10 in this signal pathway [32].
Our present study with septic rats, in which immunomodulation is known, also indicates that PDI is a regulator of inflammatory cytokines. Previous studies have demonstrated that proinflammatory cytokines play a critical role in the initiation and progression of sepsis syndrome and that TNF-α, IL-1β, and IL-6 are important mediators of hemodynamic, metabolic, and immunologic alterations in the host during sepsis [27–31]. Studies have also shown that circulating concentrations of TNF-α, IL-1β, and IL-6 increase significantly in the early, hyperdynamic stage of sepsis and remain elevated in the late, hypodynamic stage of sepsis [27, 49]. In the present study, we have provided a clue that TNF-α release increased significantly in RAW 264.7 cells treated with bacitracin, which is an inhibitor of PDI. This result indicates the important role of PDI in TNF-α release in sepsis.
Conclusion
In summary, our results indicate that PDI gene expression is downregulated in sepsis or endotoxemia. In addition, PDI gene expression is attenuated in a macrophage-like cell line after stimulation with LPS. Since the PDI inhibitor bacitracin significantly increases TNF-α release in a macrophage cell line, it appears that prevention of PDI downregulation may be a novel approach to reduce proinflammatory cytokine release in sepsis. Further studies are necessary in this direction.
Key messages
-
Protein disulfide isomerase (PDI), an important factor for the protein modification step in the post-translational event, plays an essential role in cell survival under stress conditions.
-
In an experimental model, PDI gene and protein expressions were significantly downregulated in late sepsis.
-
Similar downregulation was also observed in lipopolysaccharide-treated RAW 264.7 cells, a macrophage-like cell line.
-
Bacitracin, a specific PDI inhibitor, significantly increased tumor necrosis factor-alpha (TNF-α) gene expression and TNF-α release as well as its cellular levels in a dose-dependent manner.
-
Collectively, the data suggest that prevention of downregulation of PDI in sepsis attenuates hyperinflammation and reduces tissue injury.
Abbreviations
- BW:
-
body weight
- CLP:
-
cecal ligation and puncture
- ELISA:
-
enzyme-linked immunosorbent assay
- G3PDH:
-
glyceraldehyde 3-phosphate dehydrogenase
- IL:
-
interleukin
- LPS:
-
lipopolysaccharide
- NF-κB:
-
nuclear factor-kappa-B
- PCR:
-
polymerase chain reaction
- PDI:
-
protein disulfide isomerase
- RAW 264.7:
-
murine macrophage-like cell line
- RT-PCR:
-
reverse transcription-polymerase chain reaction
- siRNA:
-
short interfering RNA
- TNF-α:
-
tumor necrosis factor-alpha.
References
Bernard GR, Vincent JL, Laterre PF, LaRosa SP, Dhainaut JF, Lopez-Rodriguez A, Steingrub JS, Garber GE, Helterbrand JD, Ely EW, Fisher CJ Jr: Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med 2001, 344: 699-709. 10.1056/NEJM200103083441001
Martin GS, Mannino DM, Eaton S, Moss M: The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med 2003, 348: 1546-1554. 10.1056/NEJMoa022139
Hotchkiss RS, Karl IE: The pathophysiology and treatment of sepsis. N Engl J Med 2003, 348: 138-150. 10.1056/NEJMra021333
Martin GS, Mannino DM, Moss M: The effect of age on the development and outcome of adult sepsis. Crit Care Med 2006, 34: 15-21. 10.1097/01.CCM.0000194535.82812.BA
Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR: Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med 2001, 29: 1303-1310. 10.1097/00003246-200107000-00002
Deitch EA: Animal models of sepsis and shock: a review and lessons learned. Shock 1998, 9: 1-11. 10.1097/00024382-199801000-00001
Remick DG, Bolgos GR, Siddiqui J, Shin J, Nemzek JA: Six at six: interleukin-6 measured 6 h after the initiation of sepsis predicts mortality over 3 days. Shock 2002, 17: 463-467. 10.1097/00024382-200206000-00004
Zingarelli B, Sheehan M, Hake PW, O'Connor M, Denenberg A, Cook JA: Peroxisome proliferator activator receptor-gamma ligands, 15-deoxy-delta(12,14)-prostaglandin J2 and ciglitazone, reduce systemic inflammation in polymicrobial sepsis by modulation of signal transduction pathways. J Immunol 2003, 171: 6827-6837.
Chung CS, Song GY, Lomas J, Simms HH, Chaudry IH, Ayala A: Inhibition of Fas/Fas ligand signaling improves septic survival: differential effects on macrophage apoptotic and functional capacity. J Leukoc Biol 2003, 74: 344-351. 10.1189/jlb.0102006
Chaudry IH: Sepsis: lessons learned in the last century and future directions. Arch Surg 1999, 134: 922-929. 10.1001/archsurg.134.9.922
Wang P, Chaudry IH: Mechanism of hepatocellular dysfunction during hyperdynamic sepsis. Am J Physiol 1996, 270: R927-R938.
Wang H, Liao H, Ochani M, Justiniani M, Lin X, Yang L, Al Abed Y, Wang H, Metz C, Miller EJ, Tracey KJ, Ulloa L: Cholinergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nat Med 2004, 10: 1216-1221. 10.1038/nm1124
Wichterman KA, Baue AE, Chaudry IH: Sepsis and septic shock: a review of laboratory models and a proposal. J Surg Res 1980, 29: 189-201. 10.1016/0022-4804(80)90037-2
Yang S, Cioffi WG, Bland KI, Chaudry IH, Wang P: Differential alterations in systemic and regional oxygen delivery and consumption during the early and late stages of sepsis. J Trauma 1999, 47: 706-712.
Wang P, Ba ZF, Chaudry IH: Hepatocellular dysfunction occurs earlier than the onset of hyperdynamic circulation during sepsis. Shock 1995, 3: 21-26. 10.1097/00024382-199503000-00008
Wang P, Ba ZF, Chaudry IH: Hepatic extraction of indocyanine green is depressed early in sepsis despite increased hepatic blood flow and cardiac output. Arch Surg 1991, 126: 219-224.
Wang P, Zhou M, Rana MW, Ba ZF, Chaudry IH: Differential alterations in microvascular perfusion in various organs during early and late sepsis. Am J Physiol 1992, 263: G38-G43.
Wang P, Ba ZF, Chaudry IH: Mechanism of hepatocellular dysfunction during early sepsis: key role of increased gene expression and release of proinflammatory cytokines tumor necrosis factor and interleukin-6. Arch Surg 1997, 132: 364-370.
Koo DJ, Chaudry IH, Wang P: Kupffer cells are responsible for producing inflammatory cytokines and hepatocellular dysfunction during early sepsis. J Surg Res 1999, 83: 151-157. 10.1006/jsre.1999.5584
Yang S, Zhou M, Fowler DE, Wang P: Mechanisms of the beneficial effect of adrenomedullin and adrenomedullin-binding protein-1 in sepsis: down-regulation of proinflammatory cytokines. Crit Care Med 2002, 30: 2729-2735. 10.1097/00003246-200212000-00018
Darby NJ, Creighton TE: Functional properties of the individual thioredoxin-like domains of protein disulfide isomerase. Biochem 1995, 34: 11725-11735. 10.1021/bi00037a009
Freedman RB, Hirst TR, Tuite MF: Protein disulphide isomerase: building bridges in protein folding. Trends Biochem Sci 1994, 19: 331-336. 10.1016/0968-0004(94)90072-8
Frand AR, Kaiser CA: Ero1p oxidizes protein disulfide isomerase in a pathway for disulfide bond formation in the endoplasmic reticulum. Mol Cell 1999, 4: 469-477. 10.1016/S1097-2765(00)80198-7
Cabibbo A, Pagani M, Fabbri M, Rocchi M, Farmery MR, Bulleid NJ, Sitia R: ERO1-L, a human protein that favors disulfide bond formation in the endoplasmic reticulum. J Biol Chem 2000, 275: 4827-4833. 10.1074/jbc.275.7.4827
Mikami T, Genma R, Nishiyama K, Ando S, Kitahara A, Natsume H, Yoshimi T, Horiuchi R, Nakamura H: Alterations in the enzyme activity and protein contents of protein disulfide isomerase in rat tissues during fasting and refeeding. Metabolism 1998, 47: 1083-1088. 10.1016/S0026-0495(98)90282-7
Turano C, Coppari S, Altieri F, Ferraro A: Proteins of the PDI family: unpredicted non-ER locations and functions. J Cell Physiol 2002, 193: 154-163. 10.1002/jcp.10172
Ertel W, Morrison MH, Wang P, Ba ZF, Ayala A, Chaudry IH: The complex pattern of cytokines in sepsis. Association between prostaglandins, cachectin, and interleukins. Ann Surg 1991, 214: 141-148. 10.1097/00000658-199108000-00008
Waage A, Halstensen A, Espevik T: Association between tumour necrosis factor in serum and fatal outcome in patients with meningococcal disease. Lancet 1987, 1: 355-357. 10.1016/S0140-6736(87)91728-4
Debets JM, Kampmeijer R, Linden MP, Buurman WA, Linden CJ: Plasma tumor necrosis factor and mortality in critically ill septic patients. Crit Care Med 1989, 17: 489-494. 10.1097/00003246-198906000-00001
Marano MA, Fong Y, Moldawer LL, Wei H, Calvano SE, Tracey KJ, Barie PS, Manogue K, Cerami A, Shires GT: Serum cachectin/tumor necrosis factor in critically ill patients with burns correlates with infection and mortality. Surg Gynecol Obstet 1990, 170: 32-38.
Busund R, Lindsetmo RO, Rasmussen LT, Rokke O, Rekvig OP, Revhaug A: Tumor necrosis factor and interleukin 1 appearance in experimental gram-negative septic shock. The effects of plasma exchange with albumin and plasma infusion. Arch Surg 1991, 126: 591-597.
Higuchi T, Watanabe Y, Waga I: Protein disulfide isomerase suppresses the transcriptional activity of NF-kappaB. Biochem Biophys Res Commun 2004, 318: 46-52. 10.1016/j.bbrc.2004.04.002
Kaiser BK, Yim D, Chow IT, Gonzalez S, Dai Z, Mann HH, Strong RK, Groh V, Spies T: Disulphide-isomerase-enabled shedding of tumour-associated NKG2D ligands. Nature 2007, 447: 482-486. 10.1038/nature05768
Short DM, Heron ID, Birse-Archbold JL, Kerr LE, Sharkey J, McCulloch J: Apoptosis induced by staurosporine alters chaperone and endoplasmic reticulum proteins: identification by quantitative proteomics. Proteomics 2007, 7: 3085-3096. 10.1002/pmic.200600964
Wang P, Chaudry IH: A single hit model of polymicrobial sepsis: cecal ligation and puncture. Sepsis 1998, 2: 227-233. 10.1023/A:1009833813546
Zhou M, Simms HH, Wang P: Adrenomedullin and adrenomedullin binding protein-1 attenuate vascular endothelial cell apoptosis in sepsis. Ann Surg 2004, 240: 321-330. 10.1097/01.sla.0000133253.45591.5b
Zhou M, Wu R, Dong W, Jacob A, Wang P: Endotoxin downregulates peroxisome proliferator-activated receptor-{gamma} via the increase in TNF-{alpha} release. Am J Physiol Regul Integr Comp Physiol 2008, 294: R84-R92.
Ibbetson AL, Freedman RB: Thiol-protein disulphide oxidoreductases. Assay of microsomal membrane-bound glutathione-insulin transhydrogenase and comparison with protein disulphide-isomerase. Biochem J 1976, 159: 377-384.
Goldberger RF, Epstein CJ, Anfinsen CB: Acceleration of reactivation of reduced bovine pancreatic ribonuclease by a microsomal system from rat liver. J Biol Chem 1963, 238: 628-635.
Freedman RB: Protein disulfide isomerase: multiple roles in the modification of nascent secretory proteins. Cell 1989, 57: 1069-1072. 10.1016/0092-8674(89)90043-3
Terada K, Manchikalapudi P, Noiva R, Jauregui HO, Stockert RJ, Schilsky ML: Secretion, surface localization, turnover, and steady state expression of protein disulfide isomerase in rat hepatocytes. J Biol Chem 1995, 270: 20410-20416. 10.1074/jbc.270.35.20410
Yoshimori T, Semba T, Takemoto H, Akagi S, Yamamoto A, Tashiro Y: Protein disulfide-isomerase in rat exocrine pancreatic cells is exported from the endoplasmic reticulum despite possessing the retention signal. J Biol Chem 1990, 265: 15984-15990.
Hotchkiss KA, Matthias LJ, Hogg PJ: Exposure of the cryptic Arg-Gly-Asp sequence in thrombospondin-1 by protein disulfide isomerase. Biochim Biophys Acta 1998, 1388: 478-488.
Chen K, Lin Y, Detwiler TC: Protein disulfide isomerase activity is released by activated platelets. Blood 1992, 79: 2226-2228.
Delom F, Mallet B, Carayon P, Lejeune PJ: Role of extracellular molecular chaperones in the folding of oxidized proteins. Refolding of colloidal thyroglobulin by protein disulfide isomerase and immunoglobulin heavy chain-binding protein. J Biol Chem 2001, 276: 21337-21342. 10.1074/jbc.M101086200
Abell BA, Brown DT: Sindbis virus membrane fusion is mediated by reduction of glycoprotein disulfide bridges at the cell surface. J Virol 1993, 67: 5496-5501.
Ryser HJ, Levy EM, Mandel R, DiSciullo GJ: Inhibition of human immunodeficiency virus infection by agents that interfere with thiol-disulfide interchange upon virus-receptor interaction. Proc Natl Acad Sci USA 1994, 91: 4559-4563. 10.1073/pnas.91.10.4559
Ko HS, Uehara T, Nomura Y: Role of ubiquilin associated with protein-disulfide isomerase in the endoplasmic reticulum in stress-induced apoptotic cell death. J Biol Chem 2002, 277: 35386-35392. 10.1074/jbc.M203412200
Blackwell TS, Christman JW: Sepsis and cytokines: current status. Br J Anaesth 1996, 77: 110-117.
Acknowledgements
This study was supported by National Institutes of Health grants R01 GM053008 and R01 GM057468 (PW).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
MZ designed the study, collected data, interpreted the data, performed statistical analysis, and drafted the manuscript. NH is a summer student who helped MZ to collect the data. MM and RW participated in the design of the study. SRM and AJ participated in the critical revision of the manuscript. PW conceived of the study, participated in its design and interpretation, and helped to draft the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Zhou, M., Jacob, A., Ho, N. et al. Downregulation of protein disulfide isomerase in sepsis and its role in tumor necrosis factor-alpha release. Crit Care 12, R100 (2008). https://doi.org/10.1186/cc6977
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/cc6977