
Abstract
Introduction
Arginine vasopressin (AVP) is increasingly used to restore mean arterial pressure (MAP) in low-pressure shock states unresponsive to conventional inotropes. This is potentially deleterious since AVP is also known to reduce cardiac output by increasing vascular resistance. The effects of AVP on blood flow to vital organs and cardiac performance in a circulation altered by cardiac ischemia are still not sufficiently clarified. We hypothesised that restoring MAP by low dose, therapeutic level AVP would reduce vital organ blood flow in a setting of experimental acute left ventricular dysfunction.
Methods
Cardiac output (CO) and arterial blood flow to the brain, heart, kidney and liver were measured in nine pigs using transit-time flow probes. Left ventricular pressure-volume catheter and central arterial and venous catheters were used for haemodynamic recordings and blood sampling. Transient left ventricular ischemia was induced by intermittent left coronary occlusions resulting in a 17% reduction in cardiac output and a drop in MAP from 87 ± 3 to 67 ± 4 mmHg (p < 0.001). A low-dose therapeutic level of AVP (0.005 U/kg/min) was used to restore MAP to pre-ischemic values (93 ± 4 mmHg).
Results
AVP further impaired systemic perfusion (CO and brain, heart and kidney blood flow reduced by 29, 18, 23 and 34%, respectively) due to a 2.0-, 2.2-, 1.9- and 2.1-fold increase in systemic, brain, heart and kidney specific vascular resistances. The hypoperfusion induced by AVP was associated with an increased systemic oxygen extraction. Oxygen saturation in blood drawn from the great cardiac vein fell from 29 ± 1 to 21 ± 3% (p = 0.01). Finally, these effects were reversed 40 min after AVP was withdrawn.
Conclusion
Low dose AVP induced a pronounced reduction in vital organ blood flow in pigs after transient cardiac ischemia. This indicates a potentially deleterious effect of AVP in patients with heart failure or cardiogenic shock due to impaired coronary perfusion.
Similar content being viewed by others


Introduction
Synthetic arginine vasopressin (AVP) has been increasingly used to restore systemic blood pressure in low pressure shock states unresponsive to conventional inotropes [1, 2]. The association found between low plasma AVP levels and advanced vasodilatory shock has given a rationale for administrating this hormone in septic shock and in systemic inflammatory response syndrome (SIRS) [1–4]. AVP has also been given to patients in post-cardiotomy cardiac failure [1] and in cardiogenic shock complicating myocardial infarction [5]. Interestingly, the modulating effect of AVP on cardiac index (CI) seems to lower CI in hyperdynamic patients whereas an elevation of CI has been found in hypodynamic patients [6]. AVP treatment in patients with low or normal CI is however controversial, since experimental studies [7–9] and small clinical trials [10–13] have shown that the hormone may compromise cardiac output and organ perfusion due to its vasoconstrictive effect.
The response in vital organ perfusion, that is, pulmonary, coronary and cerebral circulation, to AVP-infusions is still insufficiently clarified [14]. From its basic physiological effect on V1-receptor stimulation in vascular smooth muscle [14], one would predict an overall vasoconstrictive effect after AVP infusion. However, infusion of the drug has been found to dilate the coronary, pulmonary and vertebrobasilary arteries [14, 15].
The aim of the present study was to asses whether infusion of AVP in a dose at therapeutic level in pigs after transient cardiac ischemia would reduce coronary, cerebral, renal and total organ blood flow (cardiac output). As the treatment with AVP after cardiac ischemia is potentially the most deleterious of conditions if vasoconstrictive in vital organs, we elected to assess the vascular response after reperfusion of an ischemic condition in the pig heart.
Materials and methods
Animals
The experimental protocol was approved by the local steering committee of the National Animal Research Authority (NARA). Nine castrated male domestic pigs (cross breed of Norwegian Landrace and Yorkshire pigs) weighing 30 ± 1 kg were adapted to the animal department for 5–7 days and fasted overnight before the experiment with free access to water.
Instrumentation
The animals were pre-medicated with intramuscular injections of 20 mg/kg ketalar (Pfizer AS, Norway) and 1 mg atropine (Nycomed Pharma, Norway). Anaesthesia was induced by intravenous injection of 10 mg/kg pentobarbital-sodium (Abbott, Sweden) and 0.01 mg/kg fentanyl (Hameln Pharmaceuticals, Germany), and the animals were normoventilated after tracheotomy. A central venous catheter was placed through the left internal jugular vein, and anaesthesia was maintained throughout the experiment using a continuous infusion of 4.0 mg/kg/h pentobarbital-sodium, 0.02 mg/kg/h fentanyl and 0.3 mg/kg/h midazolam (B. Braun, Melsungen, Germany). The circulating volume was maintained by a 20 ml/kg/h continuous infusion of 0.9% NaCl supplemented with 1.25 g/l glucose. The animals received 2 500 IU heparin, and 5 mg/kg amiodarone (Sanofi-Synthelabo, Sweden) to avoid blood clotting of catheters and cardiac arrhythmias. Central venous pressure (CVP) was monitored through the right jugular vein. Measurement of mean arterial pressure (MAP) and arterial blood sampling in the abdominal aorta was performed through a catheter inserted through the left femoral artery. After sternotomy, the left hemiazygos vein was ligated to avoid return of systemic blood to the coronary sinus. Transit time flow probes (CardioMed CM-4000, Medi-Stim AS, Horten, Norway) were placed on the left anterior descending coronary, the right carotid artery and the pulmonary artery in order to measure changes in coronary blood flow, cerebral blood flow and cardiac output, respectively. A 7 Fr balloon catheter was introduced to the inferior caval vein for preload reduction. Also, a 7 Fr dual field, combined pressure-conductance catheter (CD Leycom, Zoetermeer, the Netherlands) was inserted into the left ventricular cavity via the left carotid artery for measurements of left ventricular pressure and volume (P-V). Assessment of the individual segments of the pressure-volume loops confirmed the proper placement of the catheter. Myocardial venous blood was drawn from a catheter placed in the great cardiac vein via the coronary sinus. A catheter was inserted into the main pulmonary trunk through the right ventricular wall for measurement of mean pulmonary artery pressure (MPAP) and central venous oxygen saturation (sVO2). Thereafter, a midline laparotomy was performed and the urine bladder was catheterised via a cystotomy. Transit time flow probes were placed on the main hepatic artery and left renal artery in order to measure liver and kidney blood flow, respectively. The animals were allowed to rest for 1 h before baseline measurements.
Experimental protocol
Full data sets were collected five times during a complete experiment. Baseline values were recorded 1 h after the end of instrumentation. The ischemia/reperfusion protocol was then implemented during the next 30–50 min (see below). Post-ischemic values were recorded 40 min after the end of the ischemia/reperfusion protocol. The pigs were subsequently given 0.005 U/kg/min AVP for 40 min as an initial dose (in a series of pilot experiments this was found to be the lowest dose necessary to restore blood pressure to pre-ischemic values). The third recordings were then performed (AVP-initial). Thereafter, the AVP infusion rate was individually adjusted to keep the mean arterial pressure stable at approximately 90 mmHg. The fourth measurements (AVP-90 mmHg) were carried out 40 min into the infusions of this individual AVP dose. Finally, the AVP was withdrawn and the last measurements (Withdrawal) were performed 40 min thereafter. Obtaining each data set required approximately 3 min and the samples were carried out in the following order: 1, blood sampling from femoral artery, pulmonary artery and great cardiac vein; 2, blood oxygen levels from femoral artery (arterial or artO2 sat), pulmonary artery (central venous or SVO2) and great cardiac vein (sinus coronarius or sin cor O2 sat); 3 the respirator was then disconnected for 10 s to avoid pressure influence on haemodynamics, and steady state P-V data, MAP, MPAP, CVP, carotid artery flow, renal artery flow, LAD flow, hepatic and pulmonary artery flows were recorded. After a further period of 1 min, the respirator was disconnected for another 10 s and the balloon catheter in the inferior caval vein was inflated to obtain P-V data during vena cava occlusions (VCO) in accordance with previous protocols from our laboratory [16].
Ischemia reperfusion protocol
The ischemia reperfusion protocol has in principle been described previously [17]. The present modified protocol was based on repeated coronary occlusions to induce an acute impairment of cardiac performance, and aimed to reversibly reduce the flow in the larger part of the left ventricle but to avoid the arrhythmias and pump failure connected to prolonged coronary occlusions. To create coronary flow perturbations, rubber bands were looped around the LAD and circumflex coronary arteries. The bands were tightened sufficiently to demonstrate zero flow in the LAD signal during occlusions. The protocol was initiated by two occlusion periods of 60 s followed by 120 s of reperfusion. The following occlusions were guided by a maximum occlusion time of 120 s or were terminated if MAP fell below 30 mmHg during occlusions. The reperfusion time between occlusions were 30–120 s with the next occlusion starting at the onset of maximal hyperaemia. The cumulative ischemic time was 20 ± 4 min, and was terminated when MAP recovered to only 90% of baseline. This protocol resulted in a slightly more pronounced post-ischemic hypotension compared to our previous study [17]. In four pigs, defibrillation was necessary to resuscitate ventricular arrhythmias occurring during the LAD/CX occlusions.
Biochemical analyses
Blood gases and base excess were immediately analysed on a blood gas analyser (Rapid lab, Chiron Diagnostics, Emeryville, CA, USA). Blood samples for the other analyses were put on ice, quickly centrifuged and plasma was then frozen. Plasma lactate levels were determined on a biosensor (ABL 800, Radiometer, Bergman Diagnostica, Lillestrom, Norway), while troponin T was measured by electrochemiluminiscense (Modular E, Roche, Diagnostics, Basel, Switzerland). Plasma AVP was analysed using an immunoassay kit (Assay Designs, Ann Arbor, MI, USA). Sampling, preparation and analysis of plasma catecholamines were carried out as previously described [18, 19] with minor modifications. Noradrenaline and adrenaline were separated by HPLC (Dionex P680, Dionex ASI-100, Chromsystems analytical column and eluent) and their concentrations determined with an electrochemical detector (ESA Coulochem III).
Calculation of haemodynamic indices
Vascular resistance in various systemic beds were calculated as the pressure drop (MAP-CVP) divided by the arterial blood flow to the respective organs. Data from the pressure-volume catheter was analysed on CircLab (GTX Medical Software, Zoetermeer, the Netherlands) to obtain various indices of ventricular function. Calibration and practical use of the left ventricular P-V catheter has been described in detail previously [16]. Absolute volume assessment by the conductance technology requires an estimate of parallel conductance. This is performed by hypertonic saline infusion. In the present study, injection of hypertonic NaCl was not employed since a hypertonic coronary flush could influence the coronary response to AVP in the experimental protocol. Calculations of absolute ventricular volumes were therefore not performed. Such calculations would have demanded an alternate technique such as echocardiography.
The first derivative of ventricular pressure (dP/dt) was recorded as maximum and minimum values. The time-constant of isovolumic relaxation (Tau) was calculated according to Mirsky [20]. The slope of EDPVR and ESPVR denotes the line drawn through the end diastolic and end systolic pressure volume relationship for a family of PV-loops obtained during an abrupt vena cava occlusion [21]. The preload recruitable stroke work index (PRSWi) integrates the systolic and diastolic performances as the relation between stroke work (SW) and end diastolic volume (EDV).
Statistics
The data are expressed as mean ± SEM. A one-way repeated measures analysis of variance (ANOVA), followed by the Holm-Sidak test, were used to determine differences in the series of interventions. Data obtained 40 min after the ischemia/reperfusion protocol were regarded as reference value. We labelled significant differences between all data points as compared to this reference value. In addition, we labelled significant differences between (AVP-90 mmHg) and (Withdrawal). Differences between means were regarded as statistically significant when p values were less than 0.05.
Results
Post-ischemic circulation
The repetitive ischemia/reperfusion (IR) protocol was characterised by a 17% reduction in cardiac output (Figure 1). This was solely a consequence of a pronounced reduction in stroke volume since heart rate increased from 89 ± 7 to 114 ± 6 (p = 0.013) (Figure 2). The fall in systemic blood pressure from 87 ± 3 mmHg at baseline to 67 ± 4 mmHg (p = 0.001) after IR, was meditated by this reduction in CO since vascular resistance and central venous pressures were unaffected (Figures 2 and 3). Furthermore, LAD, carotid and hepatic arterial blood flow dropped from 47 ± 5, 257 ± 23 and 65 ± 12 ml to 42 ± 4, 195 ± 34 and 47 ± 11 ml. This flow reduction was only significant for carotid flow (p = 0.013). Arterial blood flow to the kidney remained constant despite the reduced CO following ischemia (Figure 1). In addition, systemic hypoperfusion was evident from an increased oxygen extraction (art O2 sat-SVO2) and a reduced base excess (Figure 4). Lactate increased significantly only in the coronary sinus (Table 1). No significant troponin T release occurred. Myocardial contractility was depressed by the ischemia/reperfusion protocol shown by a drop in dP/dt max from 1 566 ± 85 mmHg/sec to 985 ± 74 mmHg/sec (p < 0.001) (Figure 5). Neither end systolic elastance (Ees), nor pre-load recruitable stroke work index (PRSWi) were significantly altered by ischemia (p = 0.31, p = 0.16). The reduced left ventricular function was associated with a trend towards increase in mean pulmonary artery pressure (20 ± 1 mmHg to 23 ± 1 after ischemia (p = 0.074) (Figure 2)). Finally, the diastolic function was modestly affected by the ischemia/reperfusion protocol, as seen by a reduction in dP/dt min from -1 704 ± 82 mmHg/sec to -1 172 ± 78 mmHg/sec (p < 0.001). However, Tau and ventricular compliance (EDP and EDPVR) were unaffected (Figure 5).

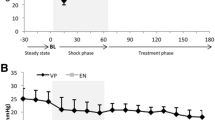
Flow recordings. Data in mL/min (L/min for CO) obtained at baseline, after ischemia/reperfusion (IR) protocol, initial AVP infusions (AVP-initial), mean pressure of 90 mmHg (AVP-90 mmHg) and after drug withdrawal (Withdrawal) (n = 9). CO, cardiac output; carotid, right carotid artery; LAD, left anterior descending; Renal, left renal artery; Hepatic, main liver artery. *Significant difference from (AHF); †significant difference (AVP-90 mmHg) and (Withdrawal).

Heart rate (HR) and pressure recordings. Data obtained in mmHg at baseline, after ischemia/reperfusion (IR) protocol, initial AVP infusions (AVP-initial), mean pressure of 90 mmHg (AVP-90 mmHg) and after drug withdrawal (Withdrawal) (see text for details) (n = 9). MAP, mean arterial pressure; MPAP, mean pulmonary arterial pressure; CVP, central venous pressure. *Significant difference from (IR); †significant difference between (AVP-90 mmHg) and (Withdrawal).

Vascular resistance. Data in dynes/sec/cm5 are obtained at baseline, after ischemia/reperfusion (IR) protocol, initial AVP infusions (AVP-initial), mean pressure of 90 mmHg (AVP-90 mmHg) and after drug withdrawal (Withdrawal) (n = 9). The values are calculated as (MAP-CVP)/(specific organ flow). SVR, systemic vascular resistance; Carotid, specific resistance in right carotid artery; LAD, specific resistance in left anterior descending artery supplying left ventricle; Renal, specific resistance in main artery supplying left kidney; Hepatic, specific resistance in main liver artery. *Significant difference from (IR); †significant difference between (AVP-90 mmHg) and (Withdrawal).

Blood gas analysis. Data obtained at baseline, after ischemia/reperfusion (IR) protocol, initial AVP infusions (AVP-initial), mean pressure of 90 mmHg (AVP-90 mmHg) and after drug withdrawal (Withdrawal) (n = 9). Cor sinus, Venous and Arterial are (%) oxygen saturation in blood obtained from the main heart vein, pulmonary artery and femoral artery, respectively. Base excess was calculated from systemic arterial blood. *Significant difference from (IR); †significant difference between (AVP-90 mmHg) and (Withdrawal).

Indices of left ventricular function. Data obtained at baseline, after ischemia/reperfusion (IR) protocol, initial AVP infusions (AVP-initial), mean pressure of 90 mmHg (AVP-90 mmHg) and after drug withdrawal (Withdrawal) (n = 9). dP/dt max and min (mmHg/sec), maximal acceleration and declaration of pressure in the cardiac cycle; PRSWi (mmHg), slope of preload recruitable stroke work index; Tau (ms), time constant of isovolumetric ventricular relaxation; SW (ml/mmHg), stroke work; EDP (mmHg), end diastolic pressure; Ees (mmHg/ml), end systolic elastance; EDPVR (mmHg/ml), end diastolic pressure volume relationship. All values are obtained by left ventricle pressure-volume catheter at steady state or vena cava occlusions (PRSWi, Ees and EDPVR). *Significant difference from (IR); †significant difference between (AVP-90 mmHg) and (Withdrawal).
Effect of AVP treatment
All haemodynamic responses were evident within the first minutes of AVP infusion and levelled of to the new steady state within the first 20 min. These indices remained stable during the 20 min prior to the recording. The only delayed vasoconstrictive effect was observed in the carotid artery (delayed for approximately 5 min; unpublished results).
Intravenous infusion of AVP caused a further drop in cardiac output from 2.3 ± 0.2 L to 1.7 ± 0.2 L (p < 0.001). This was an effect of the induced bradycardia since HR simultaneously dropped from 114 ± 6 beats/min to 88 ± 6 beats/min (p = 0.004) whereas stroke volume remained unaffected (22 ± 2 ml IR versus 19 ± 2 ml AVP-initial, not significant). AVP had a pronounced effect on systemic vascular resistance, which increased twofold during drug administration. In fact, the vasoconstriction caused by AVP resulted in a normalisation of systemic pressure from 67 ± 4 mmHg to 93 ± 4 mmHg despite the reduction in CO (Figure 2). AVP administration caused vasoconstriction in several vascular beds, that is, a reduction of flow in LAD, carotid, and renal arteries of 23, 18 and 34%, respectively (p = 0.008, p = 0.033, p = 0.001) (Figure 1). This was not observed in the hepatic artery as the blood flow increased from 47 ± 11 ml to 112 ± 19 ml by AVP infusion (p = 0.002). The reduced CO and peripheral vasoconstriction escalated the tissue hypoperfusion shown by a further reduction in SVO2 and sin cor O2 sat from 49 ± 4 and 29 ± 1% to 42 ± 4 and 21 ± 3% (p = 0.05, p = 0.012), respectively (Figure 4). The lowest measured sin cor O2 sat measured was 10% during AVP infusion. Plasma AVP increased after exogenous administration of the hormone confirming an adequate dosage to reach pharmacological levels (Table 1). Concomitantly, the circulation levels of catecholamines fell in the same timeframe. AVP had no effect on contractile function as seen by an unaffected dP/dt max and PRSWi. The Ees value increased from 2.8 ± 0.5 mmHg/ml to 4.3 ± 0.8 mmHg/ml (p = 0.007). Furthermore, there was an impairment of diastolic function induced by AVP infusion. Isovolumetric relaxation time (Tau) increased by 48% (p < 0.001), whereas ventricular compliance was reduced as evident by an increased EDP and EDPVR (Figure 5). An individual adjustment of AVP infusion rate guided by a MAP of 90 mmHg did not principally alter any of the parameters.
Withdrawal of AVP
By 40 min after AVP infusion was stopped, most of the indices had returned to pre-AVP levels (Tau, EDP, EDPVR, HR, CO, carotid flow, LAD flow, renal flow, hepatic flow, MAP, SVO2, sin cor O2 sat and SVR). However, base excess remained at values comparable to AVP intervention.
Discussion
The most striking observation in the present study was an immediate drop in coronary and renal blood flow after administering AVP. Only minutes following the onset of AVP infusion, normal blood pressure was re-established due to a twofold increase in SVR. This was consistent with a twofold increase in organ specific vascular resistance in the brain, heart and kidney. The increased coronary resistance induced by AVP was similar to previous observations in vivo [7, 22] and ex vivo [23, 24]. Interestingly, AVP caused a reduction in coronary blood flow, despite an increased perfusion pressure, to such extent that it seems to override metabolic flow regulation in the heart. This was supported by a drop in oxygen saturation in blood drawn from the great cardiac vein. This potentially deleterious effect of AVP on myocardial oxygen delivery is in line with a previous case report [25] and studies of felypressin infusions in dogs [7]. A partly compensatory response to the vasoconstriction could be observed in the pigs, as the heart rate and systemic catecholamines fell after AVP infusions.
AVP at this therapeutic level further impaired post-ischemic blood flow to the brain and kidney by vasoconstriction of the respective vascular beds, which is a matter of some concern. This is somewhat in contrast to observations of similar AVP doses in another pig model including septic shock [26]. Malay and co-workers [26] reported a heterogenic vasoconstriction with increased CO and carotid flow and dilated coronary arteries whereas renal flow dropped compatible with our observations. The explanation for the divergent findings of AVP effects on various systemic vascular beds is not known, and whether the vasoactive response to AVP is fundamentally different in sepsis and cardiac ischemia is also not clear. In our study, LAD flow, renal flow and cardiac output fell abruptly, whereas carotid flow had a transient increase during the course of several minutes before the flow dropped below pre-AVP values. This trend (a non sustained cerebral blood flow increase with AVP) was also seen in a previous study during cardiopulmonary resuscitation (CPR) in pigs [27]. The authors did not, however, measure the pre-CPR blood flow in their study, making comparisons difficult. Blood flow in the hepatic artery increased during AVP treatment consistent with a reduction in vascular resistance. However, portal vein blood flow was not measured, and the total hepatic blood flow could therefore have been reduced by the AVP infusion [28, 26, 29].
There has been some debate regarding the effect of AVP on cardiac contractility. Studies in isolated hearts [30], papillary muscle [31] and myocytes [32, 33] have reported a possibly positive inotropic effect. By contrast, studies in intact animals have shown a general trend towards negative effects on contractility [34–36]. However, a major criticism against earlier in vivo studies has been that the use of unphysiologically high doses of AVP may cause myocardial ischemia by coronary vasoconstriction [36]. In the present study, with therapeutically AVP levels, AVP had no effect on cardiac contractility, evident from unchanged dP/dt max and PRSWi. Interestingly however, there was a diastolic dysfunction following AVP infusions, evident from reduced isovolumetric relaxation (Tau) and ventricular compliance (EDPVR and EDP). Also, the increased EES indicate an excessive load dependence in the left ventricle and a manifestation of early contractile failure [36]. These effects are probably due to the increased afterload induced by AVP.
We infused AVP in the lowest dose a giving a significant response in blood pressure. This approach led to plasma levels similar to what have been observed in experimental haemorrhagic or endotoxemic shock [14]. Also, these levels have been observed in patients on extracorporeal circulation [37] with no infusion of AVP. Unfortunately, in the clinical studies on patients in cardiogenic shock indicating a beneficial effect of AVP [6, 5], no AVP levels after drug infusion were reported. The drug levels used in these studies (3.6 U/h and 4 U/h) were approximately five times lower compared to AVP levels used in our experimental study. The discrepancy between the experimental and clinical studies indicates that much is still unknown about AVP levels and drug effects in different types of cardiac related shock states [38].
Limitations
The dose of AVP used in this study was derived from a dose-response study (0.0001 U/kg/min to 0.1 U/kg/min), and the lowest dose giving a significantly increased MAP was chosen for the actual protocol. Comparing this dose (0.005 U/kg/min) to previous clinical studies, it seems that the pigs in our study have a dose-response profile different from humans. It is therefore evident that the dose used in our study should not be applied in clinical practice.
The ischemia reperfusion protocol in our study creates an ischemic cardiac dysfunction where all haemodynamic parameters remain stable for several hours [17]. We used this model to investigate the effects of AVP under stable conditions. With regards to the clinical application of AVP in cardiogenic shock, it would be of great interest to examine the effects of AVP in more advanced acute heart failure. However, haemodynamic studies in an unstable and deteriorating circulation such as cardiogenic shock are difficult to conduct and the data equally hard to interpret when large animal variations occur. Further studies should be undertaken to address the role of AVP in treatment of advanced circulatory shock though our study concurs with previous studies showing pronounced vasoconstrictive properties of AVP [7–13]. It should be mentioned that in contrast to our experimental study, AVP is never administered as monotherapy in patients with acute heart failure, and the aspects of drug interactions and synergy might alter the AVP response in multidrug treatments.
Clinical implications
The haemodynamic effects of therapeutic level AVP raise serious concerns about the therapeutic potential in patients with attenuated organ perfusion due to acute ischemic heart failure. The negative spiral in cardiogenic shock could possibly be hastened by applying AVP. Therefore, the drug should as a rule not be applied in such patients [39]. Of particular importance, the drug should be used with extreme caution in cardiac surgical patients due to the pronounced vasoconstrictive effect on vascular grafts [37].
Conclusion
The main observations in this study were that AVP, at therapeutic levels, reduces cardiac output, carotid-, renal- and myocardial blood flow by increased vascular resistance after experimental acute cardiac ischemia. This contributes to an aggravation of systemic hypoperfusion reflected by a reduced oxygen saturation of venous blood from systemic and heart veins. Furthermore, infusing AVP in these dosages resulted in an impairment of the diastolic left ventricular function, whereas myocardial contractility was unaffected by the drug. Finally, these effects were abolished and the pigs returned to pre-AVP values 40 min after withdrawal of the drug.
Key messages
Vasopressin in therapeutic level impairs blood flow to the brain, heart and kidney in pigs with post-ischemic cardiac dysfunction. The vasoconstriction induced by vasopressin leads to an increased oxygen extraction in the coronary circulation. This indicates that the constrictive effect overrides the myocardial metabolic regulation of the coronary circulation.
Vasopressin has no effect on cardiac contractility, and vasopressin infusion hampers left ventricular diastolic function, probably related to an elevated afterload.
References
Argenziano M, Chen JM, Choudhri AF, Cullinane S, Garfein E, Weinberg AD, Smith CR Jr, Rose EA, Landry DW, Oz MC: Management of vasodilatory shock after cardiac surgery: identification of predisposing factors and use of a novel pressor agent. J Thorac Cardiovasc Surg 1998, 116: 973-980. 10.1016/S0022-5223(98)70049-2
Sharshar T, Blanchard A, Paillard M, Raphael JC, Gajdos P, Annane D: Circulating vasopressin levels in septic shock. Crit Care Med 2003, 31: 1752-1758. 10.1097/01.CCM.0000063046.82359.4A
Landry DW, Levin HR, Gallant EM, Ashton RC Jr, Seo S, D'Alessandro D, Oz MC, Oliver JA: Vasopressin deficiency contributes to the vasodilation of septic shock. Circulation 1997, 95: 1122-1125.
Patel BM, Chittock DR, Russell JA, Walley KR: Beneficial effects of short-term vasopressin infusion during severe septic shock. Anesthesiology 2002, 96: 576-582. 10.1097/00000542-200203000-00011
Jolly S, Newton G, Horlick E, Seidelin PH, Ross HJ, Husain M, Dzavik V: Effect of vasopressin on hemodynamics in patients with refractory cardiogenic shock complicating acute myocardial infarction. Am J Cardiol 2005, 96: 1617-1620. 10.1016/j.amjcard.2005.07.076
Luckner G, Dunser MW, Jochberger S, Mayr VD, Wenzel V, Ulmer H, Schmid S, Knotzer H, Pajk W, Hasibeder W, Mayr AJ, Friesenecker B: Arginine vasopressin in 316 patients with advanced vasodilatory shock. Crit Care Med 2005, 33: 2659-2666. 10.1097/01.CCM.0000186749.34028.40
Miyachi K, Ichinohe T, Kaneko Y: Effects of local injection of prilocaine-felypressin on the myocardial oxygen balance in dogs. Eur J Oral Sci 2003, 111: 339-345. 10.1034/j.1600-0722.2003.00050.x
Ertmer C, Morelli A, Bone HG, Stubbe HD, Schepers R, Van AH, Lange M, Broking K, Lucke M, Traber DL, Westphal M: Dobutamine reverses the vasopressin-associated impairment in cardiac index and systemic oxygen supply in ovine endotoxemia. Crit Care 2006, 10: R144. 10.1186/cc5065
Johnson KB, Pearce FJ, Jeffreys N, McJames SW, Cluff M: Impact of vasopressin on hemodynamic and metabolic function in the decompensatory phase of hemorrhagic shock. J Cardiothorac Vasc Anesth 2006, 20: 167-172. 10.1053/j.jvca.2005.11.015
Shelly MP, Greatorex R, Calne RY, Park GR: The physiological effects of vasopressin when used to control intra-abdominal bleeding. Intensive Care Med 1988, 14: 526-531. 10.1007/BF00263525
Medel J, Boccara G, Van de SE, Bertrand M, Godet G, Coriat P: Terlipressin for treating intraoperative hypotension: can it unmask myocardial ischemia? Anesth Analg 2001, 93: 53-55. 10.1097/00000539-200107000-00012
O'Brien A, Clapp L, Singer M: Terlipressin for norepinephrine-resistant septic shock. Lancet 2002, 359: 1209-1210. 10.1016/S0140-6736(02)08225-9
Lauzier F, Levy B, Lamarre P, Lesur O: Vasopressin or norepinephrine in early hyperdynamic septic shock: a randomized clinical trial. Intensive Care Med 2006, 32: 1782-1789. 10.1007/s00134-006-0378-0
Holmes CL, Patel BM, Russell JA, Walley KR: Physiology of vasopressin relevant to management of septic shock. Chest 2001, 120: 989-1002. 10.1378/chest.120.3.989
Mayr VD, Wenzel V, Wagner-Berger HG, Stadlbauer KH, Cavus E, Raab H, Muller TH, Jochberger S, Dunser MW, Krismer AC, Schwarzacher S, Lindner KH: Arginine vasopressin during sinus rhythm: effects on haemodynamic variables, left anterior descending coronary artery cross sectional area and cardiac index, before and after inhibition of NO-synthase, in pigs. Resuscitation 2007, 74: 366-371. 10.1016/j.resuscitation.2006.07.030
Korvald C, Elvenes OP, Ytrebo LM, Sorlie DG, Myrmel T: Oxygen-wasting effect of inotropy in the "virtual work model". Am J Physiol 1999, 276: H1339-H1345.
Korvald C, Elvenes OP, Aghajani E, Myhre ES, Myrmel T: Postischemic mechanoenergetic inefficiency is related to contractile dysfunction and not altered metabolism. Am J Physiol Heart Circ Physiol 2001, 281: H2645-H2653.
Sager G, Trovik T, Slordal L, Jaeger R, Prytz PS, Brox J, Reikeras O: Catecholamine binding and concentrations in acute phase plasma after surgery. Scand J Clin Lab Invest 1988, 48: 419-424. 10.3109/00365518809085751
Sager G, Slordal L, Huseby NE, Florholmen J: Beta-adrenoceptor regulation in insulin-dependent diabetes mellitus. Scand J Clin Lab Invest 1991, 51: 283-288. 10.3109/00365519109091616
Mirsky I: Assessment of diastolic function: suggested methods and future considerations. Circulation 1984, 69: 836-841.
Aghajani E, Muller S, Kjorstad KE, Korvald C, Nordhaug D, Revhaug A, Myrmel T: The pressure-volume loop revisited: is the search for a cardiac contractility index a futile cycle? Shock 2006, 25: 370-376. 10.1097/01.shk.0000209521.20496.7a
Michel JB, Tedgui A, Bardou A, Levy B: Effect of vasopressin on phasic coronary blood flow. Basic Res Cardiol 1985, 80: 221-230. 10.1007/BF01907898
Graf BM, Fischer B, Martin E, Bosnjak ZJ, Stowe DF: Differential effects of arginine vasopressin on isolated guinea pig heart function during perfusion at constant flow and constant pressure. J Cardiovasc Pharmacol 1997, 29: 1-7. 10.1097/00005344-199701000-00001
Bax WA, Van der Graaf PH, Stam WB, Bos E, Nisato D, Saxena PR: [Arg8]vasopressin-induced responses of the human isolated coronary artery: effects of non-peptide receptor antagonists. Eur J Pharmacol 1995, 285: 199-202. 10.1016/0014-2999(95)00503-D
Medel J, Boccara G, Van de SE, Bertrand M, Godet G, Coriat P: Terlipressin for treating intraoperative hypotension: can it unmask myocardial ischemia? Anesth Analg 2001, 93: 53-5. TOC 10.1097/00000539-200107000-00012
Malay MB, Ashton JL, Dahl K, Savage EB, Burchell SA, Ashton RC Jr, Sciacca RR, Oliver JA, Landry DW: Heterogeneity of the vasoconstrictor effect of vasopressin in septic shock. Crit Care Med 2004, 32: 1327-1331. 10.1097/01.CCM.0000128578.37822.F1
Lindner KH, Prengel AW, Pfenninger EG, Lindner IM, Strohmenger HU, Georgieff M, Lurie KG: Vasopressin improves vital organ blood flow during closed-chest cardiopulmonary resuscitation in pigs. Circulation 1995, 91: 215-221.
Ericsson BF: Hemodynamic effects of vasopressin. An experimental study in normovolemic and hypovolemic anesthetized dogs. Acta Chir Scand Suppl 1971, 414: 1-29.
Martikainen TJ, Uusaro A, Tenhunen JJ, Ruokonen E: Dobutamine compensates deleterious hemodynamic and metabolic effects of vasopressin in the splanchnic region in endotoxin shock. Acta Anaesthesiol Scand 2004, 48: 935-943. 10.1111/j.0001-5172.2004.00435.x
Walker BR, Childs ME, Adams EM: Direct cardiac effects of vasopressin: role of V1- and V2-vasopressinergic receptors. Am J Physiol 1988, 255: H261-H265.
Fujisawa S, Iijima T: On the inotropic actions of arginine vasopressin in ventricular muscle of the guinea pig heart. Jpn J Pharmacol 1999, 81: 309-312. 10.1254/jjp.81.309
Xu YJ, Gopalakrishnan V: Vasopressin increases cytosolic free [Ca2+] in the neonatal rat cardiomyocyte. Evidence for V1 subtype receptors. Circ Res 1991, 69: 239-245.
Chandrashekhar Y, Prahash AJ, Sen S, Gupta S, Roy S, Anand IS: The role of arginine vasopressin and its receptors in the normal and failing rat heart. J Mol Cell Cardiol 2003, 35: 495-504. 10.1016/S0022-2828(03)00053-1
Khayyal MA, Eng C, Franzen D, Breall JA, Kirk ES: Effects of vasopressin on the coronary circulation: reserve and regulation during ischemia. Am J Physiol 1985, 248: H516-H522.
Tipayamontri U, Young DB, Nuwayhid BS, Scott RE: Analysis of the cardiovascular effects of arginine vasopressin in conscious dogs. Hypertension 1987, 9: 371-378.
Share L: Role of vasopressin in cardiovascular regulation. Physiol Rev 1988, 68: 1248-1284.
Novella S, Martinez AC, Pagan RM, Hernandez M, Garcia-Sacristan A, Gonzalez-Pinto A, Gonzalez-Santos JM, Benedito S: Plasma levels and vascular effects of vasopressin in patients undergoing coronary artery bypass grafting. Eur J Cardiothorac Surg 2007, 32: 69-76. 10.1016/j.ejcts.2007.03.047
Luckner G, Mayr VD, Jochberger S, Wenzel V, Ulmer H, Hasibeder WR, Dunser MW: Comparison of two dose regimens of arginine vasopressin in advanced vasodilatory shock. Crit Care Med 2007, 35: 2280-2285.
Dunser MW, Hasibeder WR: Vasopressin in vasodilatory shock: ensure organ blood flow, but take care of the heart! Crit Care 2006, 10: 172. 10.1186/cc5089
Acknowledgements
The study was financially supported by the health authorities of north Norway ("Helse Nord").
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
SM, OJH and TM played a pivotal role in planning and carrying out the experiments as well as writing the manuscript. SEH and TAS contributed in scientific discussions as well as preparing the manuscript. GS planned and carried out the catecholamine analysis.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Müller, S., How, OJ., Hermansen, S.E. et al. Vasopressin impairs brain, heart and kidney perfusion: an experimental study in pigs after transient myocardial ischemia. Crit Care 12, R20 (2008). https://doi.org/10.1186/cc6794
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/cc6794