
Abstract
Introduction
The importance of the tumor microenvironment in breast cancer has been increasingly recognized. Critical molecular changes in the tumor stroma accompanying cancer progression, however, remain largely unknown. We conducted a comparative analysis of global gene expression changes in the stromal and epithelial compartments during breast cancer progression from normal to preinvasive to invasive ductal carcinoma.
Methods
We combined laser capture microdissection and gene expression microarrays to analyze 14 patient-matched normal epithelium, normal stroma, tumor epithelium and tumor-associated stroma specimens. Differential gene expression and gene ontology analyses were performed.
Results
Tumor-associated stroma undergoes extensive gene expression changes during cancer progression, to a similar extent as that seen in the malignant epithelium. Highly upregulated genes in the tumor-associated stroma include constituents of the extracellular matrix and matrix metalloproteases, and cell-cycle-related genes. Decreased expression of cytoplasmic ribosomal proteins and increased expression of mitochondrial ribosomal proteins were observed in both the tumor epithelium and the stroma. The transition from preinvasive to invasive growth was accompanied by increased expression of several matrix metalloproteases (MMP2, MMP11 and MMP14). Furthermore, as observed in malignant epithelium, a gene expression signature of histological tumor grade also exists in the stroma, with high-grade tumors associated with increased expression of genes involved in immune response.
Conclusions
Our results suggest that the tumor microenvironment participates in tumorigenesis even before tumor cells invade into stroma, and that it may play important roles in the transition from preinvasive to invasive growth. The immune cells in the tumor stroma may be exploited by the malignant epithelial cells in high-grade tumors for aggressive invasive growth.
Similar content being viewed by others


Introduction
The tumor microenvironment or the stroma hosting the malignant breast epithelial cells is comprised of multiple cell types, including fibroblasts, myoepithelial cells, endothelial cells and various immune cells [1–4]. One prevailing view is that tumor-associated stroma is activated by the malignant epithelial cells to foster tumor growth – for example, by secreting growth factors, increasing angiogenesis, and facilitating cell migration, ultimately resulting in metastasis to remote organ sites [3]. For example, two chemokines (chemokine (C-X-C motif) ligand (CXCL) 12 and CXCL14) that bind to tumor epithelial cells to promote proliferation, migration and invasion have recently been shown to be overexpressed by the activated tumor fibroblasts and myoepithelial cells [5–7]. Genes involved in tumor-microenvironment interactions may therefore provide novel targets for diagnostic development and therapeutic intervention. Our understanding of the interactions between epithelial and stromal components of breast cancer, however, remains limited at the molecular level. Using the serial analysis of gene expression technique, Allinen and coworkers performed the first systematic profiling of the various stromal cell types isolated via cell-type-specific cell surface markers and magnetic beads [7]. They demonstrated gene expression alterations in all cell types within the tumor microenvironment accompanying progression from normal breast tissue to ductal carcinoma in situ (DCIS) to invasive ducal carcinoma (IDC) [8], providing evidence that these cell types all participate in tumorigenesis.
Using laser capture microdissection (LCM), we previously performed gene expression analysis of the epithelial compartment of the malignant lesions during breast cancer progression. We discovered that most of the gene expression changes take place prior to local invasion (even in atypical ductal hyperplasia) and that there are no major changes in gene expression accompanying the in situ to invasive growth transition [9]. In the present article we extend this analysis to the tumor stromal microenvironment and demonstrate that, like the tumor epithelium, the tumor stromal microenvironment undergoes extensive gene expression alterations even at the preinvasive stage of DCIS, supporting the view that cell-cell communication via paracrine mechanisms between the two compartments plays an important role in tumor progression.
Materials and methods
Clinical specimen
All breast cancer specimens were fresh-frozen biopsies obtained from the Massachusetts General Hospital between 1998 and 2001. The diagnostic criteria and tumor grading were described previously [9]. Patient and tumor characteristics of the 14 tumor specimens in this study are presented in Table 1. Patients were selected in which patient-matched normal and tumor samples were available and the normal breast lobules did not show fibrocystic change. The research was deemed exempt from informed consent as the samples are unidentifiable to the research team. The study was approved by the Massachusetts General Hospital human research committee in accordance with National Institutes of Health human research study guidelines.
Laser capture microdissection, RNA extraction and microarray analysis
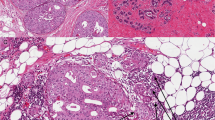
Highly enriched populations of patient-matched normal or malignant epithelial cells and of normal stroma or tumor-associated stroma from the different stages of breast cancer progression were procured by LCM using a PixCell IIe system (Molecular Devices, Mountain View, CA, USA) as previously described [9]. Enrichment for cells of interest was verified by microscopic examination of the LCM cap after microdissection. The microdissected normal stromal compartment consisted of the intralobular, rather than the extralobular, stromal compartment of normal breast tissue that was a minimum 0.3 cm from any premalignant or malignant lesion (Figure 1). The DCIS-associated stroma (DCIS-S) consisted of a 25 μm rim of cells that surrounded the DCIS; for cases in which synchronous DCIS and IDC were present, the DCIS-S was obtained from areas of DCIS that were at least 0.3 cm from the invasive component. The IDC-associated stroma (IDC-S) consists of stromal cells predominantly within the invasive tumor mass.

Laser capture microdissection experimental design. Example of the tumor microenvironment compartments targeted by laser capture microdissection: epithelial (white asterisk) and stromal (black outlined areas with black asterisk) compartments of the normal terminal ductal lobular unit, of ductal carcinoma in situ (DCIS) and of invasive ductal carcinoma (IDC).
Total RNA was isolated from captured cells using the Picopure™ RNA isolation kit (Molecular Devices), amplified by T7 RNA amplification (RiboAmp™; Molecular Devices), labeled and hybridized to the whole genome array U133X3P (3'-biased design) according to the manufacturer's instructions (Affymetrix, Santa Clara, CA, USA). The hybridized microarrays were then washed, stained and scanned as per the manufacturer's protocols (Affymetrix).
Data analysis
Raw data from the U133X3P arrays were processed using the Bioconductor rma package with default parameters for background correction, quantile normalization and signal summation [10, 11]. Differential gene expression analyses were performed using linear regression models in the limma package [12]. For comparing normal and tumor samples, we used the patient identification as a blocking variable. For tumor grade comparison, we used the tumor stage (in situ or invasive) as the blocking variable. Statistical significance was corrected for multiple testing using the Benjamini-Hochberg procedure [13]. All procedures were performed in the R statistical environment [14]. For gene ontology analysis, ranked gene lists were first generated according to the moderated t statistics from linear models and then examined for enriched ontology terms using the Gene Set Enrichment Analysis software [15]. The data discussed in this publication have been deposited in the NCBI Gene Expression Omnibus [16] and are accessible [GEO:GSE14548] [17].
Quantitative real-time PCR and immunohistochemistry
TaqMan™ real-time PCR was performed on amplified RNA used for microarray analysis as previously described [9]. Briefly, amplified RNA was converted to double-stranded cDNA, and the cDNA was quantitated with PicoGreen (Molecular Probes, Eugene, OR, USA) using a spectrofluorometer (Molecular Devices). Each gene was analyzed in triplicate in a 96-well plate using ABI 7900 HT (Applied Biosystems, Foster City, CA., USA).
For each gene, the sequences of the PCR primer pairs and the fluorogenic MGB or TAMRA probe (5' to 3'), respectively, are as follows: ESR1, ATGATCAACTGGGCGAAGA, GGTGGACCTGATCATGGA and VIC-TGCCAGGCTTTGTGGA; RRM2, CCTTTAACCAGCACAGCCAGTT, TTATTTGTTTGTAAAGTGCCAGGTTT and VIC-TGCAGCCTCACTGCTTCAACGCA-TAMRA; gremlin 1 (GREM1), ACGGCAAAGAATTATATAGACTATGAGGTA, TTTTATGAGACTATCAACTCCCCTTTC and VIC-CTTGCTGTGTAGGAGGA; and WNT inhibitory factor 1(WIF1), CACTGTGGTAGTGGCATTTAAACAATA, GCCAATGCAAAAAGTTCATACATT and VIC-TTCTAAACACAATGAAATAGGGA.
Estrogen receptor and progesterone receptor immunohistochemistry staining was performed as previously described, using the rabbit monoclonal antibody (SP1) from Lab Vision (Fremont, CA, USA) for the estrogen receptor (1:50 dilution) and using the mouse monoclonal antibody (PgR 636) from Dako (Carpinteria, CA, USA) for the progesterone receptor (1:50 dilution) [18].
Results
Experimental design
The present study included 14 patients with primary ductal breast cancer (Table 1). These patients were primarily estrogen receptor positive (78.6%), lymph node positive (78.6%), and premenopausal (mean age 41 years). We used LCM to isolate the epithelial and stroma compartments separately from each of the 14 fresh-frozen biopsies. In the epithelial compartment, we captured normal and malignant epithelium from DCIS and/or IDC. In the stromal compartment, we captured normal stroma at least 3 mm from the malignant lesion and the DCIS-S and/or IDC-S whenever possible. An example of the microdissected compartments is shown in Figure 1. As shown in Table 2, in the epithelial compartment four cases had all three stages (normal breast epithelium, DCIS, and IDC) available, five cases had normal breast epithelium and IDC only, and five cases had normal breast epithelium and DCIS only; in the stroma, six cases had all three stages available, five cases had normal stromal compartment and DCIS-S, and three cases had the normal stromal compartment and IDC-S. RNA was isolated from the captured cells and interrogated with the Affymetrix whole-genome array U133X3P.
Gene expression changes in the stromal and epithelial compartments during breast cancer progression
We compared the gene expression patterns of the tumor epithelium and stroma at each stage of progression (DCIS or IDC) with their respective normal state using the limma (linear models of microarrays) software package [12]. The resulting P values for differential gene expression in each pair-wise comparison were adjusted for multiple testing [13], and the genes with a significant adjusted P value (P <0.05) were extracted.
The DCIS and IDC stages were each associated with thousands of gene expression alterations relative to their respective normal state in both the tumor epithelium and the stroma (Figure 2). Furthermore, within each compartment, the expression patterns of DCIS-associated and IDC-associated genes were highly similar to each other (Figure 3).

Comparative analysis of gene expression changes in tumor and stroma. Gene expression changes in normal breast epithelium (N), ductal carcinoma in situ (DCIS), invasive ductal carcinoma (IDC), normal stromal compartment (N-S), ductal carcinoma in situ-associated stroma (DCIS-S) and invasive ductal carcinoma-associated stroma (IDC-S). ↑, upregulated genes; ↓, downregulated genes.

Heatmap of expression patterns of ductal carcinoma in situ-associated and invasive ductal carcinoma-associated genes. (a) Heatmap of 849 genes with >3-fold differential expression in either ductal carcinoma in situ (DCIS) versus normal breast or invasive ductal carcinoma (IDC) versus normal breast in the epithelium. (b) Heatmap of 557 genes with >3-fold differential expression in either ductal carcinoma in situ-associated stroma (DCIS-S) versus normal stromal compartment or invasive ductal carcinoma-associated stroma (IDC-S) versus normal stromal compartment. Data shown are log2(fold change) relative to the average expression in normal controls (normal breast epithelium or normal stromal compartment). In each heatmap, genes (rows) are hierarchically clustered using 1 – Pearson correlation as the distance metric. IS, ductal carcinoma in situ; INV, invasive ductal carcinoma; ISS, ductal carcinoma in situ-associated stroma; INVS, invasive ductal carcinoma-associated stroma.
To gain an overview of the biological processes in which these differentially expressed genes are involved, we performed gene set enrichment analysis [19] using the gene ontology database [20]. Table 3 presents the top 20 gene ontology terms significantly enriched within genes upregulated in the invasive stage in the epithelium and the stroma. In the epithelium, the genes were dominated by those associated with the cell cycle (mitosis in particular). In the stroma, the genes prominently featured the components of the extracellular matrix and the matrix metalloproteases responsible for remodeling the extracellular matrix. Additionally, the stromal genes also included those related to the cell cycle, indicating increased proliferation as a common feature in both the tumor epithelium and the stroma.
In both compartments, the single gene ontology term STRUCTURAL_CONSTITUENT_OF_RIBOSOME was significantly enriched within the downregulated genes (Table 3). To examine this further, we extracted all ribosomal protein-encoding genes that were differentially expressed between DCIS or IDC versus the normal breast in the epithelium and visualized their expression patterns in both compartments. Interestingly, there was an almost complete bipartite partitioning of these genes (Figure 4): while the downregulated genes were all those encoding for the cytoplasmic ribosomal proteins, the upregulated genes were mostly those encoding for the mitochondrial ribosomal proteins.

Heatmap of differential expression of ribosomal protein genes in the malignant epithelium and tumor stroma. Differential expression of ribosomal protein genes in ductal carcinoma in situ (DCIS), invasive ductal carcinoma (IDC), ductal carcinoma in situ-associated stroma (DCIS-S) and invasive ductal carcinoma-associated stroma (IDC-S). Data shown are log2(fold change) relative to the average expression level in the normal controls (normal breast epithelium or normal stromal compartment). Expression measurements for multiple probe sets representing the same gene were collapsed to the single representative probe set with the largest differential gene expression. All genes shown were significant at adjusted P < 0.05. IS, ductal carcinoma in situ; INV, invasive ductal carcinoma; ISS, ductal carcinoma in situ-associated stroma; INVS, invasive ductal carcinoma-associated stroma.
In addition to these global patterns, Tables 4 and 5 present the top 50 differentially expressed genes in the epithelium and the stroma, respectively. In these tables, besides the dominant features of cell-cycle-related genes in the epithelium and extracellular matrix genes in the stroma discussed earlier, we note several additional genes important in cell signaling pathways. Two antagonists of WNT receptor signaling, WIF1 and secreted frizzled-related protein 1 (SFRP1), were downregulated in both the tumor epithelium and the stroma. In addition, two members of the transforming growth factor beta superfamily, GREM1 and inhibin beta A (INHBA), showed markedly increased expression specifically in the tumor stroma (Table 5).
Stromal gene expression signature associated with tumor invasion
We next compared the gene expression patterns associated with the DCIS to IDC transition within each compartment. In the tumor epithelium, there were only three genes (POSTN, periostin; SPARC, osteoconectin; SPARCL1, SPARC-like 1) that were significantly upregulated in IDC relative to DCIS. All three genes are known to be specifically expressed in the stroma [21–23] and were indeed strongly expressed in the stroma samples in our dataset. Their apparent overexpression in IDC relative to DCIS might therefore be due to contaminating stromal cells in the procured epithelial cell populations in the IDC samples but not in DCIS samples. In the stroma, however, there were more significant changes in comparing IDC-S with DCIS-S, with 76 upregulated genes and 229 downregulated genes (Figure 2). The lack of significant changes in gene expression in the epithelium associated with the DCIS-IDC transition seen here was consistent with that in our previous study [9].
Table 6 presents the top 50 differentially expressed genes between DICS-S and IDC-S (see Additional data file 1). Among genes with increased expression in IDC-S, three matrix metalloproteases (MMP11, MMP2 and MMP14) were notable. In fact, one additional matrix metalloprotease (MMP13) had higher expression in IDC-S than in DCIS-S, with adjusted P = 0.06. These genes have been known to be involved in tumor invasion [3]. On the other hand, genes with decreased expression in IDC-S included many genes involved in vasculature development (for example, EMCN, FLT1, KDR, SELE, MYH11, EDNRB and PODXL), a process expected to increase in invasive cancer. This paradoxical result might reflect the decreased vascular density in the leading invasive front where we microdissected the stroma relative to the stroma surrounding DCIS.
Stromal gene expression signature associated with tumor grade
We have previously shown that tumor grade is associated with a strong gene expression signature in malignant breast epithelial cells [9]. We therefore examined whether a similar signature also exists in the tumor stroma. Comparing grade I (n = 8) and grade III (n = 7) tumor-associated stroma samples (DCIS-S and IDC-S), we identified 526 upregulated genes and 94 downregulated genes in grade III samples (Figure 5; see also Additional data file 2). The gene set enrichment analysis indicated that the tumor stroma in grade III tumors were associated with a strong immune response signature (interferon signaling, activation of leukocytes and T cells) and with increased mitotic activity (Table 7).

Heatmap of gene expression signature correlated with tumor grade in the stroma. Comparison of grade III tumors with grade I tumors identified 526 upregulated genes and 94 downregulated genes in grade III stroma. Data shown are log2(fold change) relative to the median expression level across all samples. Genes in rows were hierarchically clustered, and samples in columns were arranged by sample type. E, epithelium; S, stroma.
Validation of selected differentially expressed genes
We next used quantitative real-time PCR to validate selected genes differentially expressed in the various comparisons presented above. Quantitative real-time PCR analysis of the same samples as used in the microarray analysis confirmed the marked downregulation of WIF1 in both neoplastic epithelium and tumor stroma (Figure 6a) and the marked upregulation of GREM1 in both DCIS-associated and IDC-associated stroma (Figure 6b). In addition, two representative genes (ESR1, estrogen receptor alpha; and RRM2, ribonucleotide reductase M2 subunit) differentially expressed in the stroma between grade III and grade I tumors (see Additional data file 2) were also confirmed by quantitative real-time PCR. In both the epithelium and stroma, RRM2, a cell proliferation marker, was more highly expressed in grade III tumors (Figure 6c), whereas ESR1 was more highly expressed in grade I tumors (Figure 6d). Although expression of estrogen receptor alpha is thought to be restricted to the tumor epithelial cells in human breast cancer [24], we confirmed the low but detectable levels of estrogen receptor alpha expression in stromal fibroblasts by immunohistochemical staining (Figure 6e).

Validation of selected genes. (a) to (d) Boxplots of relative gene expression by quantitative real-time PCR in ductal carcinoma in situ (DCIS), invasive ductal carcinoma (IDC), ductal carcinoma in situ-associated stroma (DCIS-S) and invasive ductal carcinoma-associated stroma (IDC-S). (a) and (b) Reference groups were the normal components (N, normal breast epithelium; N-S, normal stromal compartment). (c) and (d) Reference groups were grade I (EI, epithelium; SI, stroma). y axis, cycling threshold values relative to the median value for the entire series. Statistically significant differences by Wilcoxon rank sum test: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.(e) Immunostaining of an estrogen-receptor-positive breast cancer. Arrows point to positive staining in stromal fibroblasts.
Discussion
Exploratory genome-wide analysis of the tumor microenvironment in breast cancer has been limited to date. Using serial analysis of gene expression coupled with antibody-based ex vivo tissue fractionation, Allinen and colleagues identified a limited set of 417 cell-type-specific genes among the most prominent cell types in breast cancer (epithelial, myoepithelial, and endothelial cells, fibroblasts, and leukocytes) [7]. Finak and colleagues more recently obtained gene expression profiles of both epithelial and stromal compartments from the same tumor biopsy via LCM [25]. These workers only analyzed the morphologically normal epithelium and normal stroma, however, leaving the gene expression changes in the tumor-activated stroma unexplored. Our work therefore provides the first comprehensive comparative analysis of in vivo gene expression changes in the tumor epithelium and its stromal microenvironment during breast cancer progression from normal to DCIS to IDC.
We observed extensive gene expression changes in the stroma associated with DCIS and IDC, suggesting that tumor-adjacent stroma coevolves with the tumor epithelium, even before tumor invasion occurs. These alterations included many components of the extracellular matrix and the extracellular-matrix-remodeling matrix metalloproteases. Increased mitotic gene expression occurred both in the malignant epithelium and adjacent stroma, which may reflect the often observed desmoplastic reaction around the tumor cells. Expression of cytoplasmic ribosomal proteins was generally decreased in both compartments during cancer progression. While this result may seem paradoxical in that increased protein synthesis is considered a hallmark of cancer, it is supported by several different lines of studies. First, decreased expression of many ribosomal proteins has also been observed in colorectal cancer compared with normal mucosal epithelium [26]. Secondly, many ribosomal protein genes have been found to be haploinsufficient tumor suppressors in zebrafish [27]. Thirdly, the oncogenic activity of c-Myc is inhibited by the ribosomal protein L11, and inactivation of the L11 gene by small interfering RNA increases c-Myc-induced transcription and cell proliferation [28].
The mechanism by which ribosomal proteins contribute to tumorigenesis is unknown. Decreased expression of ribosomal proteins in cancer may reflect a qualitative change in ribosomal structure, which may allow differential translation of gene products required for rapid tumor growth. Alternatively, it may reflect some unknown nonribosomal functions by these proteins. In contrast to the decreased expression of these cytoplasmic ribosomal protein genes, we observed increased expression of a number of mitochondrial ribosomal protein genes in both the tumor epithelium and the stroma. The human mitochondrial ribosomes are responsible for the production of several key proteins in bioenergetics including subunits of the ATP synthase. Given the importance of mitochondria in cancer [29, 30], our novel finding suggests that the mitochondrial ribosome may be a potential therapeutic target and thus warrants further study.
The top differentially expressed genes between tumor-associated stroma and the adjacent normal stroma included several signaling molecules known to be important for tumorigenesis. Two antagonists of WNT receptor signaling, WIF1 and SFRP1, were consistently downregulated both in the tumor epithelium and stroma. The WNT signaling pathway plays an important role in development and tissue homeostasis, and its aberrant activation by loss of expression WIF1 or SFRP1 has been shown to be an important early event in breast cancer progression [31–33]. Two transforming growth factor beta superfamily members (GREM1 and INHBA) are strongly induced in the tumor-associated stroma. GREM1 is a bone morphogenetic protein antagonist, and it is overexpressed in cancer-associated stromal cells in many solid tumors [34]. It has been hypothesized that bone morphogenetic proteins and bone morphogenetic protein antagonists may play opposing roles in the maintenance of a niche of self-renewing stem cells, with bone morphogenetic protein antagonists such as GREM1 blocking cell differentiation [34]. WNT3A was recently demonstrated in human fibroblasts to markedly increase the expression of GREM2, a close paralog of GREM1 – raising the possibility that the significant downregulation of WNT antagonists (WIF1 and SFRP1) and upregulation of GREM1 in the stroma [35] we observed here may be functionally linked.
INHBA is the gene for the beta A subunit of inhibin and activin, which are pleiotropic growth factors regulating the growth and differentiation of many cell types via autocrine and paracrine mechanisms [36]. Although its role in breast cancer remains unclear, circulating levels of INHBA has been shown to be higher in breast cancer patients with bone metastasis [37]. These signaling molecules could serve as key messengers between the tumor and its microenvironment, as shown for CXCL12 and CXCL14, which are overexpressed in tumor-associated myoepithelial cells and myofibroblasts [6, 7, 38]. We note that in our dataset, however, CXCL12 and CXCL14 were also expressed in normal stroma. This discrepancy could be due to the fact that Allinen and colleagues used purified stromal cell types [7] and we used the whole stroma compartment in our study.
A watershed event in breast cancer progression is the invasion of tumor cells into the stromal compartment. The only morphological diagnostic criterion distinguishing DCIS from IDC is the association of DCIS with a complete basement membrane. Understanding the molecular events that drive the DCIS-IDC transition has been of great interest. We have previously shown [9], and confirm in the present study, that the malignant epithelium of DCIS and IDC are very similar without significant differences at the transcriptome level. This conclusion is supported by the recent demonstration that MCFDCIS cells, a cell line model for DCIS, make the DCIS-IDC transition spontaneously without further molecular changes in the malignant epithelial cells themselves [39]. Instead, this transition is driven by fibroblasts and blocked by myoepithelial cells.
In the present article we demonstrated that the stromal compartment is associated with a relatively small number of significant changes accompanying the DCIS-IDC transition. In particular, several matrix metalloproteases (MMP2, MMP11 and MMP14) showed significantly increased expression in IDC-associated stroma. MMP14, a membrane-type matrix metalloprotease, can activate MMP2 protease activity, which degrades type IV collagen, the major structural component of the basement membrane [40, 41]. MMP11 has recently been shown to exhibit protease activity towards type VI collagen and to promote tumor progression [42]. MMP11 has been shown to be differentially expressed in IDC relative to DCIS in two other studies. Schuetz and colleagues conducted a study similar to ours, using LCM and microarrays to profile the epithelium of patient-matched DCIS and IDC, and found MMP11 to be upregulated in IDC relative to DCIS [43]. Their result differs from ours, however, in that we observed upregulation of MMP11 in the IDC-associated stroma but not in the epithelium. A stromal origin of MMP11 expression had been established previously [44]. The result by Schuetz and coworkers might be due to contaminating nonepithelial cells in their LCM samples, a possibility acknowledged by these authors [43]. In another study, Hannemann and colleagues identified a gene expression signature including MMP11 to be able to distinguish IDC from DCIS [45]. Since no microdissection was performed in that study, the gene expression profiles they obtained were from mixtures of tumor epithelium and stroma. Nevertheless, our results together with these other studies support the notion that stroma-produced matrix metalloproteases may be key players driving the DCIS-IDC transition.
Finally, we showed that – like the epithelial compartment [9] – tumor stroma also exhibited a robust gene expression signature correlating with the histological tumor grade. These genes are primarily involved in immune response and cell-cycle progression. The association of an immune response signature with the more aggressive high-grade tumors is seemingly paradoxical. The interactions between tumor cells and the various immune cells are complex, however, ranging from tumor growth-suppressing effects to tumor growth-promoting effects [46–48]. Perhaps the immune response signature associated with high-grade tumors represents the escape phase [48], when the cancer cells become resistant to immune attack and hijack the abundant cytokines and chemokines made by the immune cells to grow, invade and spread to distant organs.
Conclusions
The present study provides the first comparative analysis of the in situ gene expression profiles of patient-matched normal and neoplastic breast epithelial and stromal compartments of both preinvasive and invasive stages of human breast cancer progression. This study of the breast cancer microenvironment at the transcriptome level and previous studies at the genomic [49, 50] and epigenetic [51, 52] levels support the view that the tumor microenvironment is an important co-conspirator rather than a passive bystander during tumorigenesis. Molecular alterations within the stroma offer novel avenues for therapeutic interventions and disease prognosis [53]. This gene expression dataset of carefully procured in situ tumor epithelium and stroma should be a timely and valuable addition to the resources for the breast cancer research community.
Abbreviations
- CXCL:
-
chemokine (C-X-C motif) ligand
- DCIS:
-
ductal carcinoma in situ
- DCIS-S:
-
DCIS-associated stroma
- GREM1:
-
gremlin 1
- IDC:
-
invasive ductal carcinoma
- IDC-S:
-
IDC-associated stroma
- INHBA:
-
inhibin beta A
- LCM:
-
laser capture microdissection
- PCR:
-
polymerase chain reaction
- SFRP1:
-
secreted frizzled-related protein 1
- WIF1:
-
WNT inhibitory factor 1.
References
Bissell MJ, Radisky D: Putting tumours in context. Nat Rev Cancer. 2001, 1: 46-54.
de Visser KE, Coussens LM: The inflammatory tumor microenvironment and its impact on cancer development. Contrib Microbiol. 2006, 13: 118-137.
Liotta LA, Kohn EC: The microenvironment of the tumour-host interface. Nature. 2001, 411: 375-379.
Egeblad M, Littlepage LE, Werb Z: The fibroblastic coconspirator in cancer progression. Cold Spring Harb Symp Quant Biol. 2005, 70: 383-388.
Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, Barrera JL, Mohar A, Verastegui E, Zlotnik A: Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001, 410: 50-56.
Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, Carey VJ, Richardson AL, Weinberg RA: Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005, 121: 335-348.
Allinen M, Beroukhim R, Cai L, Brennan C, Lahti-Domenici J, Huang H, Porter D, Hu M, Chin L, Richardson A, Schnitt S, Sellers WR, Polyak K: Molecular characterization of the tumor microenvironment in breast cancer. Cancer Cell. 2004, 6: 17-32.
Allred DC, Mohsin SK, Fuqua SA: Histological and biological evolution of human premalignant breast disease. Endocr Relat Cancer. 2001, 8: 47-61.
Ma XJ, Salunga R, Tuggle JT, Gaudet J, Enright E, McQuary P, Payette T, Pistone M, Stecker K, Zhang BM, Zhou YX, Varnholt H, Smith B, Gadd M, Chatfield E, Kessler J, Baer TM, Erlander MG, Sgroi DC: Gene expression profiles of human breast cancer progression. Proc Natl Acad Sci USA. 2003, 100: 5974-5979.
Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JY, Zhang J: Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004, 5: R80-
Bolstad BM, Irizarry RA, Astrand M, Speed TP: A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003, 19: 185-193.
Wettenhall JM, Smyth GK: limmaGUI: a graphical user interface for linear modeling of microarray data. Bioinformatics. 2004, 20: 3705-3706.
Benjamin RS, Hochberg Y: Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B. 1995, 57: 289-300.
R Project for Statistical Computing. [http://www.r-project.org]
Subramanian A, Kuehn H, Gould J, Tamayo P, Mesirov JP: GSEA-P: a desktop application for Gene Set Enrichment Analysis. Bioinformatics. 2007, 23: 3251-3253.
Edgar R, Domrachev M, Lash AE: Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30: 207-210.
Gene Expression Omnibus. [http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE14548]
Ma XJ, Wang Z, Ryan PD, Isakoff SJ, Barmettler A, Fuller A, Muir B, Mohapatra G, Salunga R, Tuggle JT, Tran Y, Tran D, Tassin A, Amon P, Wang W, Enright E, Stecker K, Estepa-Sabal E, Smith B, Younger J, Balis U, Michaelson J, Bhan A, Habin K, Baer TM, Brugge J, Haber DA, Erlander MG, Sgroi DC: A two-gene expression ratio predicts clinical outcome in breast cancer patients treated with tamoxifen. Cancer Cell. 2004, 5: 607-616.
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP: Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005, 102: 15545-15550.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G: Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000, 25: 25-29.
Kanno A, Satoh K, Masamune A, Hirota M, Kimura K, Umino J, Hamada S, Satoh A, Egawa S, Motoi F, Unno M, Shimosegawa T: Periostin, secreted from stromal cells, has biphasic effect on cell migration and correlates with the epithelial to mesenchymal transition of human pancreatic cancer cells. Int J Cancer. 2008, 122: 2707-2718.
Coutu DL, Wu JH, Monette A, Rivard GE, Blostein MD, Galipeau J: Periostin, a member of a novel family of vitamin K-dependent proteins, is expressed by mesenchymal stromal cells. J Biol Chem. 2008, 283: 17991-18001.
Framson PE, Sage EH: SPARC and tumor growth: where the seed meets the soil?. J Cell Biochem. 2004, 92: 679-690.
Jensen EV, Cheng G, Palmieri C, Saji S, Makela S, Van Noorden S, Wahlstrom T, Warner M, Coombes RC, Gustafsson JA: Estrogen receptors and proliferation markers in primary and recurrent breast cancer. Proc Natl Acad Sci USA. 2001, 98: 15197-15202.
Finak G, Sadekova S, Pepin F, Hallett M, Meterissian S, Halwani F, Khetani K, Souleimanova M, Zabolotny B, Omeroglu A, Park M: Gene expression signatures of morphologically normal breast tissue identify basal-like tumors. Breast Cancer Res. 2006, 8: R58-
Kasai H, Nadano D, Hidaka E, Higuchi K, Kawakubo M, Sato TA, Nakayama J: Differential expression of ribosomal proteins in human normal and neoplastic colorectum. J Histochem Cytochem. 2003, 51: 567-574.
Amsterdam A, Sadler KC, Lai K, Farrington S, Bronson RT, Lees JA, Hopkins N: Many ribosomal protein genes are cancer genes in zebrafish. PLoS Biol. 2004, 2: E139-
Dai MS, Arnold H, Sun XX, Sears R, Lu H: Inhibition of c-Myc activity by ribosomal protein L11. EMBO J. 2007, 26: 3332-3345.
Carew JS, Huang P: Mitochondrial defects in cancer. Mol Cancer. 2002, 1: 9-
Hagland H, Nikolaisen J, Hodneland LI, Gjertsen BT, Bruserud O, Tronstad KJ: Targeting mitochondria in the treatment of human cancer: a coordinated attack against cancer cell energy metabolism and signalling. Expert Opin Ther Targets. 2007, 11: 1055-1069.
Ugolini F, Charafe-Jauffret E, Bardou VJ, Geneix J, Adelaide J, Labat-Moleur F, Penault-Llorca F, Longy M, Jacquemier J, Birnbaum D, Pebusque MJ: WNT pathway and mammary carcinogenesis: loss of expression of candidate tumor suppressor gene SFRP1 in most invasive carcinomas except of the medullary type. Oncogene. 2001, 20: 5810-5817.
Wissmann C, Wild PJ, Kaiser S, Roepcke S, Stoehr R, Woenckhaus M, Kristiansen G, Hsieh JC, Hofstaedter F, Hartmann A, Knuechel R, Rosenthal A, Pilarsky C: WIF1, a component of the Wnt pathway, is down-regulated in prostate, breast, lung, and bladder cancer. J Pathol. 2003, 201: 204-212.
Klopocki E, Kristiansen G, Wild PJ, Klaman I, Castanos-Velez E, Singer G, Stohr R, Simon R, Sauter G, Leibiger H, Essers L, Weber B, Hermann K, Rosenthal A, Hartmann A, Dahl E: Loss of SFRP1 is associated with breast cancer progression and poor prognosis in early stage tumors. Int J Oncol. 2004, 25: 641-649.
Sneddon JB, Zhen HH, Montgomery K, Rijn van de M, Tward AD, West R, Gladstone H, Chang HY, Morganroth GS, Oro AE, Brown PO: Bone morphogenetic protein antagonist gremlin 1 is widely expressed by cancer-associated stromal cells and can promote tumor cell proliferation. Proc Natl Acad Sci USA. 2006, 103: 14842-14847.
Klapholz-Brown Z, Walmsley GG, Nusse YM, Nusse R, Brown PO: Transcriptional program induced by Wnt protein in human fibroblasts suggests mechanisms for cell cooperativity in defining tissue microenvironments. PLoS ONE. 2007, 2: e945-
Reis FM, Luisi S, Carneiro MM, Cobellis L, Federico M, Camargos AF, Petraglia F: Activin, inhibin and the human breast. Mol Cell Endocrinol. 2004, 225: 77-82.
Mylonas I, Jeschke U, Shabani N, Kuhn C, Friese K, Gerber B: Inhibin/activin subunits (inhibin-alpha, -betaA and -betaB) are differentially expressed in human breast cancer and their metastasis. Oncol Rep. 2005, 13: 81-88.
Burger JA, Kipps TJ: CXCR4: a key receptor in the crosstalk between tumor cells and their microenvironment. Blood. 2006, 107: 1761-1767.
Hu M, Yao J, Carroll DK, Weremowicz S, Chen H, Carrasco D, Richardson A, Violette S, Nikolskaya T, Nikolsky Y, Bauerlein EL, Hahn WC, Gelman RS, Allred C, Bissell MJ, Schnitt S, Polyak K: Regulation of in situ to invasive breast carcinoma transition. Cancer Cell. 2008, 13: 394-406.
Rozanov DV, Savinov AY, Williams R, Liu K, Golubkov VS, Krajewski S, Strongin AY: Molecular signature of MT1-MMP: transactivation of the downstream universal gene network in cancer. Cancer Res. 2008, 68: 4086-4096.
Egeblad M, Werb Z: New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002, 2: 161-174.
Motrescu ER, Blaise S, Etique N, Messaddeq N, Chenard MP, Stoll I, Tomasetto C, Rio MC: Matrix metalloproteinase-11/stromelysin-3 exhibits collagenolytic function against collagen VI under normal and malignant conditions. Oncogene. 2008, 27: 6347-55.
Schuetz CS, Bonin M, Clare SE, Nieselt K, Sotlar K, Walter M, Fehm T, Solomayer E, Riess O, Wallwiener D, Kurek R, Neubauer HJ: Progression-specific genes identified by expression profiling of matched ductal carcinomas in situ and invasive breast tumors, combining laser capture microdissection and oligonucleotide microarray analysis. Cancer Res. 2006, 66: 5278-5286.
Basset P, Bellocq JP, Wolf C, Stoll I, Hutin P, Limacher JM, Podhajcer OL, Chenard MP, Rio MC, Chambon P: A novel metalloproteinase gene specifically expressed in stromal cells of breast carcinomas. Nature. 1990, 348: 699-704.
Hannemann J, Velds A, Halfwerk JB, Kreike B, Peterse JL, Vijver van de MJ: Classification of ductal carcinoma in situ by gene expression profiling. Breast Cancer Res. 2006, 8: R61-
de Visser KE, Eichten A, Coussens LM: Paradoxical roles of the immune system during cancer development. Nat Rev Cancer. 2006, 6: 24-37.
Clement JF, Meloche S, Servant MJ: The IKK-related kinases: from innate immunity to oncogenesis. Cell Res. 2008, 18: 889-899.
Strausberg RL: Tumor microenvironments, the immune system and cancer survival. Genome Biol. 2005, 6: 211-
Fukino K, Shen L, Matsumoto S, Morrison CD, Mutter GL, Eng C: Combined total genome loss of heterozygosity scan of breast cancer stroma and epithelium reveals multiplicity of stromal targets. Cancer Res. 2004, 64: 7231-7236.
Patocs A, Zhang L, Xu Y, Weber F, Caldes T, Mutter GL, Platzer P, Eng C: Breast-cancer stromal cells with TP53 mutations and nodal metastases. N Engl J Med. 2007, 357: 2543-2551.
Fiegl H, Millinger S, Goebel G, Muller-Holzner E, Marth C, Laird PW, Widschwendter M: Breast cancer DNA methylation profiles in cancer cells and tumor stroma: association with HER-2/neu status in primary breast cancer. Cancer Res. 2006, 66: 29-33.
Hu M, Yao J, Cai L, Bachman KE, Brule van den F, Velculescu V, Polyak K: Distinct epigenetic changes in the stromal cells of breast cancers. Nat Genet. 2005, 37: 899-905.
Finak G, Bertos N, Pepin F, Sadekova S, Souleimanova M, Zhao H, Chen H, Omeroglu G, Meterissian S, Omeroglu A, Hallett M, Park M: Stromal gene expression predicts clinical outcome in breast cancer. Nat Med. 2008, 14: 518-527.
Acknowledgements
The present study was supported by NIH RO1-1CA112021-01 (to DCS), the Department of Defense grants W81XWH-04-1-0606 and DAMD170310428 (to DCS), Susan G. Komen Breast Cancer Foundation grant BCTR0402932 (to DCS), and the Avon Foundation (to DCS). The authors thank Erica Chatfield for technical assistance in microdissection and RNA extraction, and they thank Sridhar Ramaswamy and E. Schmidt for helpful discussions and reading of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
XJM and ME are employees of BioTheranostics, Inc. ME, X-JM and DCS are named inventors on a patent application relating to the contents of the manuscript.
Authors' contributions
DCS, XJM and ME conceived of the study, participated in its design and coordination, and helped draft the manuscript. DCS and SD performed microdissection and RNA extraction. XJM and ME performed the microarray experiments and microarray data analysis. ER performed the real-time PCR assays. All authors have read and approved the final manuscript.
Electronic supplementary material
13058_2008_2186_MOESM1_ESM.xls
Additional file 1: An Excel file containing a table that lists the genes differentially expressed between DCIS-S and IDC-S. (XLS 74 KB)
13058_2008_2186_MOESM2_ESM.xls
Additional file 2: An Excel file containing a table that lists the genes differentially expressed between grade III and grade I samples. (XLS 114 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Ma, XJ., Dahiya, S., Richardson, E. et al. Gene expression profiling of the tumor microenvironment during breast cancer progression. Breast Cancer Res 11, R7 (2009). https://doi.org/10.1186/bcr2222
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/bcr2222