
Abstract
Introduction
Prevalence of insulin resistance and the metabolic syndrome has been reported to be high in rheumatoid arthritis (RA) patients. Tumor necrosis factor (TNF), a pro-inflammatory cytokine with a major pathogenetic role in RA, may promote insulin resistance by inducing Ser312 phosphorylation (p-Ser312) of insulin receptor substrate (IRS)-1 and downregulating phosphorylated (p-)AKT. We examined whether anti-TNF therapy improves insulin resistance in RA patients and assessed changes in the insulin signaling cascade.
Methods
Prospective study of RA patients receiving anti-TNF agents (infliximab, n = 49, adalimumab, n = 11, or etanercept, n = 1) due to high disease activity score in 28 joints (DAS28 > 5.1). A complete biochemical profile was obtained at weeks 0 and 12 of treatment. Insulin resistance, insulin sensitivity and pancreatic beta cell function were measured by the Homeostasis Model Assessment (HOMA-IR), the Quantitative Insulin Sensitivity Check Index (QUICKI) and the HOMA-B respectively. Protein extracts from peripheral blood mononuclear cells were assayed by western blot for p-Ser312 IRS-1 and p-AKT. RA patients treated with abatacept (CTLA4.Ig) were used as a control group for insulin signaling studies.
Results
At study entry, RA patients with high insulin resistance (HOMA-IR above median) had significantly higher mean DAS28 (P = 0.011), serum triglycerides (P = 0.015), and systolic blood pressure levels (P = 0.024) than patients with low insulin resistance. After 12 weeks of anti-TNF therapy, patients with high insulin resistance demonstrated significant reduction in HOMA-IR (P < 0.001), HOMA-B (P = 0.001), serum triglycerides (P = 0.039), and increase in QUICKI (P < 0.001) and serum HDL-C (P = 0.022). Western blot analysis in seven active RA patients with high insulin resistance showed reduction in p-Ser312 IRS-1 (P = 0.043) and increase in p-AKT (P = 0.001) over the study period. In contrast, the effect of CTLA4.Ig on p-Ser312 IRS-1 and p-AKT levels was variable.
Conclusions
Anti-TNF therapy improved insulin sensitivity and reversed defects in the insulin signaling cascade in RA patients with active disease and high insulin resistance. The impact of these biochemical changes in modifying cardiovascular disease burden in active RA patients remains to be seen.
Similar content being viewed by others


Introduction
Insulin resistance is a key feature of obesity, metabolic syndrome, and type 2 diabetes mellitus (T2DM). Insulin signaling is a complex process; binding of insulin to its receptor induces both auto-phosphorylation and phosphorylation of tyrosine residues on insulin receptor substrate (IRS) proteins, the most prominent being IRS-1 and IRS-2, thus initiating the intracellular signaling cascade [1, 2]. IRS-1 and IRS-2 mediate their metabolic effects through the phosphatidyl-inositol 3-kinase (PI-3K) pathway, which results in activation of AKT and other downstream effector molecules. IRS-1 may be more closely linked to glucose homeostasis, whereas IRS-2 is primarily involved in lipid metabolism [3]. Insulin signaling may also activate the mitogen activated protein kinase (MAPK) isoforms ERK1 and ERK2, through Grb/Sos and ras. This pathway mediates the mitogenic and pro-inflammatory responses of insulin signaling while it does not affect glucose homeostasis [4]. In obese patients with insulin resistance, the pathways leading to PI-3K activation are blocked, whereas the MAPK pathway remains active or even hypersensitive [5].
Inflammation and insulin resistance are closely linked and inflammatory cytokines such as tumor necrosis factor (TNF), interleukin (IL)-6, IL-1 and IL-8 may inhibit insulin signaling by multiple mechanisms [6]. TNF induces phosphorylation of IRS-1 at serine instead of tyrosine residues and promotes insulin resistance [7, 8]. Both IL-6 and TNF may inhibit the transcription of IRS-1 and glucose transporter (GLUT)-4 genes, thus reducing glucose transport and enhancing insulin resistance in obese patients [9].
Patients with rheumatoid arthritis (RA) are at increased risk for cardiovascular disease [10] independently of traditional vascular risk factors [11]. Cohort studies have demonstrated increased prevalence of metabolic syndrome in patients with RA, correlating with disease activity and markers of atherosclerosis [12–14]. RA patients are also at increased risk for T2DM compared with non-rheumatic controls (adjusted hazard ratio 1.5) [15], and pancreatic beta cell function is associated with disease activity and cumulative dose of glucocorticoids [14].
Observational studies suggest that anti-TNF therapy improves disease activity and may reduce cardiovascular events in RA patients (age-sex adjusted rate ratio 0.46) [16, 17]. This effect is thought to be mediated by reduction in insulin resistance and metabolic syndrome components demonstrated in patients treated with TNF blockade [18–22]. However, the results of the aforementioned studies are limited by the inclusion of a small number of RA patients and the lack of any mechanistic insights to the molecular effects of TNF blockade on insulin signaling.
To this end, we set out a 12-week prospective study in patients with RA, who were receiving anti-TNF agents due to active disease, to assess changes in insulin resistance/sensitivity, serum lipoproteins, and activation of the insulin receptor signaling cascade. Anti-TNF therapy resulted in significant improvement in insulin resistance/sensitivity and pancreatic beta cell function in RA patients with high baseline insulin resistance. Importantly, our studies in peripheral blood mononuclear cells from RA patients demonstrated reduction in IRS-1 serine phosphorylation and increase in AKT phosphorylation following anti-TNF therapy, both modifications linked to improved glucose homeostasis.
Materials and methods
Patients and treatment
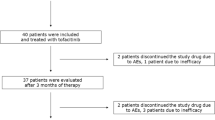
We prospectively studied patients with RA who were started on anti-TNF agents due to high disease activity, defined by a disease activity score in 28 joints (DAS28) > 5.1 despite treatment with disease-modifying anti-rheumatic drugs (DMARDs) [23]. Patients fulfilled the American College of Rheumatology 1987 classification criteria for RA [24]. Patients aged < 18 years or with history of DM were excluded. Anti-TNF agents (infliximab, adalimumab or etanercept) were given at approved standard doses. The dosage of background DMARDs and glucocorticoids, as well as of anti-hypertensive and lipid-lowering treatments remained stable during the study period. Detailed medical and drug history was obtained, and the use of tobacco was recorded. Disease activity (DAS28) was assessed at baseline and after 12 weeks of treatment [25]. Coronary heart disease was defined as presence of any of the following: angina pectoris, myocardial infarction or coronary revascularization procedures (bypass grafting, percutaneous coronary intervention). As a control group, we consecutively enrolled seven patients with RA who were started on abatacept (CTLA4.Ig), another class of biologic therapy which targets T-cell costimulation rather than TNF, due to high disease activity (DAS28 > 5.1). All patients signed informed consent forms, and the study protocol was approved by the University Hospital of Heraklion Ethics Committee (13/12/99).
Assessment of insulin resistance/sensitivity and other metabolic parameters
Blood was drawn after 12-hour overnight fasting. Serum concentrations of total cholesterol (TC), high-density lipoprotein cholesterol (HDL-C), and triglycerides (TG) were measured using an automated chemistry analyzer (Olympus AU-600, Tokyo, Japan). Low-density lipoprotein cholesterol (LDL-C) was calculated according to the Friedewald formula except for samples with serum TG > 400 mg/dl (4.5 mmol/L). Apolipoprotein (apo)-A, apo-B and lipoprotein a (Lpa) were analyzed by immunonephelometry (BN II analyzer, Eschborn, Germany). Plasma glucose concentration was determined by standard enzymatic colorimetric assay (Roche Diagnostics, Mannheim, Germany). C-reactive protein (CRP) was measured by immune nephelometry (BN II analyzer, Eschborn, Germany) and erythrocyte sedimentation rate (ESR) by an automated analyzer (Ves-matic 20, Florence, Italy). Serum insulin was determined by chemiluminescent microparticle immunoassay (ARCHITECT Insulin Reagent Kit, 8K41, Abbott Diagnostics, Lake Forest, Illinois, USA)
While the hyperinsulinemic euglycemic clamp technique is the gold standard for evaluating insulin sensitivity, the Homeostasis Model Assessment (HOMA) for insulin resistance and the Quantitative Insulin Sensitivity Check Index (QUICKI) are widely used as noninvasive surrogate markers of insulin resistance and sensitivity, respectively [26, 27]. Pancreatic beta cell function was assessed by the HOMA for beta cell function (HOMA-B) [28]. We used the National Cholesterol Education Program (NCEP) Adult Treatment Panel (ATP) III criteria for metabolic syndrome [29].
Isolation of peripheral blood mononuclear cells (PBMCs) and protein extracts
PBMCs were isolated by Ficoll-Histopaque (Sigma-Aldrich, St. Louis, MO, USA) density-gradient centrifugation of heparinized venous blood. PBMCs were lysed with RIPA buffer containing 50 mM Tris-HCl, pH 7.2, 150 mM sodium chloride, 1% NP-40, 12 mM sodium deoxycholate, 3 mM sodium dodecyl sulfate, 4 mM sodium azide, 0.57 mM phenylmethysulfonyl fluoride and complete protease inhibitor cocktail. Protein concentrations were determined by Bradford protein assay (Bio-Rad laboratories, Inc, Hercules, CA, USA).
Western blot analysis
Proteins were separated by 8% sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose membrane (Whatman, Schwerte, Germany). Membranes were probed overnight with anti-phospho-IRS1-Ser312 (Upstate, Lake Placid, NY, USA) and anti-phospho-Akt-Ser473 (Cell Signalling, Frankfurt, Germany) monoclonal antibodies. Blots were also probed with anti-β-actin (Santa Cruz, CA, USA) to control for total protein content. The secondary antibodies horseradish peroxidase (HRP)-conjugated anti-mouse IgG and anti-rabbit IgG were used (Santa Cruz, CA, USA). Enhanced chemiluminescence (ECL) from Thermo Scientific (Vantaa, Finland) was used for detection, and densitometry analysis was performed with Image J program.
Statistical analysis
Results are expressed as the mean and 95% confidence interval (95% CI) or as proportions. Parameters showing abnormal distribution were logarithmically transformed. In a post-hoc analysis, we used the median HOMA-IR value (3.18) to define high insulin resistance, and we compared the recorded variables in RA patients with high versus low insulin resistance using the independent samples t-test (continuous variables) or the chi-squared test (categorical variables). The Pearson's test was used for bivariate correlation analysis. Logistic regression (backward selection) was further performed to calculate odds ratios (ORs) for high versus low HOMA-IR according to demographic and lifestyle (gender, age, tobacco use), metabolic (obesity, hypertension, use of statins, metabolic syndrome), and clinical parameters, including the DAS28, health assessment questionnaire (HAQ), CRP, and RA treatment modality (glucocorticoids, methotrexate). Changes in recorded parameters between baseline and the end of the 12-week study period were analyzed by paired t-test or the Wilcoxon signed ranks test as appropriate. To address whether the effect of anti-TNF treatment on insulin resistance is confounded by changes in disease activity, we performed repeated measures analysis of variance (ANOVA) entering HOMA-IR values at baseline and at 12 weeks after treatment as dependent variables (within-subject variables), the insulin resistance group (high versus low baseline HOMA-IR) as grouping factor (between-subject variable), and the change in DAS28 score as a covariate. P values (two-tailed) < 0.05 were considered statistically significant. SPSS software version 18.0 (SPSS, Inc) was used for the statistical calculations.
Results
Patients' clinical characteristics at baseline
We prospectively studied 61 patients with RA (43 women) of mean age 60 years and mean disease duration 7.2 years (Table 1). All patients had active disease with mean DAS28 score 5.8 and they were started on one of the following anti-TNF agents: infliximab (n = 49), adalimumab (n = 11), or etanercept (n = 1). Fifty six patients (92%) were on single (n = 36) or combination (n = 20) therapy with DMARDs, including methotrexate (n = 42), leflunomide (n = 15), and hydroxychloroquine (n = 17). Thirteen patients were on glucocorticoids. Patients treated with abatacept had comparable demographic and clinical characteristics and metabolic profiles to patients treated with anti-TNFagents (data not shown).
Association of clinical and metabolic parameters with insulin resistance in RA patients
We used the median HOMA-IR at baseline to assign patients to the low (HOMA-IR ≤ 3.18, n = 31) and high (HOMA-IR > 3.18, n = 30) insulin resistance group. Patients in the high insulin resistance group had higher mean disease activity (P = 0.011), fasting serum insulin (P < 0.001), plasma glucose (P < 0.001), triglycerides (P = 0.015), and systolic blood pressure levels (P = 0.024) than patients with low insulin resistance (Table 1). Metabolic syndrome was more frequent in RA patients with high insulin resistance (63% versus 19% in those with low insulin resistance, P = 0.001). In accordance with previous reports [30], we found significantly increased pancreatic beta cell function, assessed by the HOMA-B index, in RA patients with high insulin resistance.
By using a cut-off of HOMA-IR > 2.0 [31], 74% (n = 45) of RA patients were classified as having insulin resistance. Patients with HOMA-IR > 2.0 at baseline had higher serum insulin (P < 0.001), HOMA-B (P < 0.001), and triglycerides (P = 0.014), and lower QUICKI (P < 0.001) than those with HOMA-IR ≤ 2.0. RA patients with insulin resistance had a trend for higher DAS28 (P = 0.105) and significantly higher HAQ (P = 0.037). Fasting plasma glucose levels were not significantly different between the two groups, although 61% in patients with insulin resistance versus 6% in those without insulin resistance had metabolic syndrome (P = 0.001) (data not shown).
HOMA-IR correlated with DAS28 (r = 0.31, P = 0.016) and fasting triglycerides (r = 0.38, P = 0.003), and HOMA-B correlated with body mass index (BMI) (r = 0.28, P = 0.035) (data not shown). In multivariate regression analysis, presence of the metabolic syndrome and DAS28 score > 6.86 (75th percentile) were independent predictors for high insulin resistance with ORs of 10.8 (95% CI 2.8 to 42.6, P = 0.001) and 6.4 (95% CI 1.2 to 32.9, P = 0.027), respectively. BMI > 30 kg/m2 was associated with an OR of 3.8 (95% CI 1.1 to 12.4, P = 0.030) for increased pancreatic beta cell function.
Anti-TNF therapy improves insulin resistance/sensitivity, pancreatic beta cell function, and metabolic parameters in patients with active RA and high insulin resistance
Patients with high baseline insulin resistance demonstrated significant improvement in DAS28 (P = 0.013), HAQ (P = 0.012), and CRP (P = 0.007) after 12 weeks of treatment with anti-TNF agents (Table 2). A similar trend was observed in patients with lower baseline insulin resistance, yet only reduction in HAQ was statistically significant (P = 0.018). Anti-TNF therapy resulted in significant improvement in HOMA-IR (P < 0.001), QUICKI (P < 0.001), and HOMA-B (P = 0.001) only in RA patients with high baseline insulin resistance (Table 2). In these patients, significant decreases in fasting plasma glucose (P = 0.016), serum insulin (P < 0.001), and serum triglycerides (P = 0.039), and increase in serum HDL-C (P = 0.022) levels were observed.
We repeated the analysis in RA patients with baseline HOMA-IR ≤ 2.0 (n = 16) or > 2.0 (n = 45). Anti-TNF treatment reduced DAS28 (P = 0.007), HAQ (P = 0.002), and CRP (P = 0.002) in patients with insulin resistance but not in those with HOMA-IR ≤ 2.0 (data not shown). TNF blockade resulted in significant reduction in serum insulin (P < 0.001) and improvements in HOMA-IR (P = 0.001), HOMA-B (P = 0.001), and QUICKI (P = 0.001) in patients with HOMA-IR > 2.0. In contrast, there were no significant changes in serum triglycerides (P = 0.058), HDL-C (P = 0.101), homocysteine (P = 0.312) and blood glucose (P = 0.166). These findings suggest that the metabolic effects of anti-TNF treatment may be more pronounced in RA patients with very high insulin resistance levels compared to those with low-level or no insulin resistance.
We next examined whether changes in insulin resistance/sensitivity and pancreatic beta cell function correlated with changes in RA disease activity. Significant improvements in the HOMA-IR, QUICKI, and HOMA-B indices were found in RA patients with high baseline insulin resistance irrespective of their clinical response according to the European League Against Rheumatism (EULAR) criteria (data not shown). In addition, repeated measures ANOVA revealed a significant effect of anti-TNF treatment (F test = 11.89, P = 0.001) and of the interaction of anti-TNF treatment with baseline insulin resistance group (F test = 24.68, P < 0.001) on HOMA-IR. Conversely, the interaction of anti-TNF treatment with the change in DAS28 had no significant impact on HOMA-IR (F test = 0.06, P = 0.810), suggesting that the effect of anti-TNF treatment on insulin resistance is not confounded by improvement in disease activity.
Anti-TNF therapy reduces IRS-1 phosphorylation at Ser312
TNF may compromise insulin signaling by induction of serine 312 (Ser312) phosphorylation of IRS-1 [7, 32]. Based on our findings of improved insulin resistance in patients with active RA following treatment with anti-TNF agents, we examined whether this effect is associated with altered IRS-1 phosphorylation. We used PBMCs, which provide a useful tool to determine alterations in the insulin signaling cascade in vivo as they include all of the signaling proteins present in insulin target tissues [33, 34]. We purified total protein extracts from PBMCs of seven randomly selected RA patients at weeks 0 and 12 of anti-TNF therapy, and we quantified IRS-1 Ser312 phosphorylation by western blot. At study entry, all patients had active disease with median DAS28 7.4 (range 5.6 to 9.0), and high insulin resistance with median HOMA-IR 7.6 (range 3.1 to 10.3). At week 12, all but one patient demonstrated reduction in IRS-1 phosphorylation (Figure 1A). Phosphorylated IRS-1 normalized for β-actin protein expression was significantly reduced from 0.96 (arbitrary units) at week 0 to 0.57 at week 12 (Wilcoxon signed ranks test, P = 0.043) (Figure 1B). Both DAS28 and HOMA-IR improved over the study period (Figures 1C, D). We found no correlation between longitudinal changes in HOMA-IR and IRS-1 Ser312 phosphorylation levels.

Anti-TNF treatment decreases insulin receptor substrate (IRS)-1 Ser312 phosphorylation and increases AKT phosphorylation in patients with active rheumatoid arthritis (RA) and high insulin resistance. (A) Seven patients with active RA received anti-TNF treatment for 12 weeks. Protein extracts from PBMCs were analyzed by western blotting for the Ser312-phosphorylated form of IRS-1 at weeks 0 and 12 of treatment. All but one patient demonstrated decrease in IRS-1 phosphorylation at Ser312. (B) Quantitative analysis (protein densitometry) showed that median phosphorylated IRS-1 normalized for β-actin levels were reduced from 0.96 to 0.57 (arbitrary units) (Wilcoxon signed ranks test, P = 0.043). (C) Decrease in disease activity following anti-TNF treatment (from median DAS28 7.4 to 6.3, P = 0.049). (D) Improvement in insulin resistance following anti-TNF treatment (from median HOMA-IR 7.6 to 3.6, P = 0.028). (E) Protein extracts from peripheral blood mononuclear cells (n = 7 RA patients) were analyzed by western blotting for Ser473- phosphorylated AKT at weeks 0 and 12 of anti-TNF therapy. (F) Phosphorylated AKT levels, normalized for β-actin, increased in all RA patients from a median 0.24 (arbitrary units) at week 0 to 0.94 at week 12 (P = 0.001).
Anti-TNF therapy increases AKT phosphorylation
IRS-1 mediates its metabolic effects through the PI-3K pathway which results in phosphorylation of AKT and other downstream molecules [32]. TNF-induced insulin resistance involves inhibition of AKT through a ceramide-activated protein-phosphatase (PP) 2A [35]. Individuals with insulin resistance display reduced insulin stimulation of the PI-3K pathway [5], and insulin-stimulated AKT kinase activity is reduced in patients with T2DM [36, 37]. Therefore, we measured phosphorylated AKT levels in protein extracts from PBMCs of RA patients at weeks 0 and 12 of anti-TNF therapy. Phosphorylated AKT levels, normalized for β-actin, increased in all seven RA patients (Figure 1E) from a median 0.24 at week 0 to 0.94 at week 12 (P = 0.001) (Figure 1F). We found no correlation between longitudinal changes in HOMA-IR or IRS-1 Ser312 phosphorylation and AKT phosphorylation levels.
Improvement in RA disease activity following therapy with CTLA4.Ig does not correlate with changes in insulin resistance, IRS-1 and AKT phosphorylation
To assess the specificity of the effect of TNF blockade on insulin resistance, we prospectively studied seven RA patients (median age 63 years, median disease duration 13 years) with high disease activity (median DAS28 5.92) treated with abatacept (CTLA4.Ig), a biologic agent that targets T-cell co-stimulation. All patients were receiving DMARDs (five on methotrexate, two on leflunomide), and three patients were receiving low-dose glucocorticoids. At baseline, three patients had low insulin resistance (median HOMA-IR 1.16, range 0.87 to 1.91), and four patients had high insulin resistance (median HOMA-IR 5.34, range 3.45 to 8.85).
At week 12 of abatacept treatment, disease activity was reduced in all seven RA patients (Wilcoxon signed ranks test, P = 0.046) (Figure 2A). Insulin resistance followed a variable trend, and HOMA-IR increased in three patients (two with low baseline insulin resistance), remained stable in one patient, and decreased in three patients (two with high baseline insulin resistance) (Figure 2B). Similarly, HOMA-B was not significantly altered during the study period (data not shown).

Variable effect of CTLA4.Ig (abatacept) treatment on insulin receptor substrate (IRS)-1 and AKT phosphorylation in patients with rheumatoid arthritis (RA). (A) Improvement in disease activity in RA patients (n = 7) following treatment with abatacept for 12 weeks, from median disease activity score in 28 joints (DAS28) 5.9 to 4.8 (P = 0.046, Wilcoxon signed ranks test). (B) Treatment with abatacept had variable effects on the Homeostasis Model Assessment for insulin resistance (HOMA-IR) in RA patients with low or high baseline insulin resistance. (C) Protein extracts from periperhal blood mononuclear cells of the RA patients were analyzed by western blotting for the Ser312 and Ser473- phosphorylated forms of insulin resistance substrate (IRS)-1 and AKT, respectively, at baseline and after 12 weeks treatment with abatacept. (D) Quantitative analysis of p-Ser321 IRS-1 normalized to β-actin expression showed no significant change following treatment with abatacept. (E) Variable trend in p-AKT levels from baseline to week 12 of treatment with abatacept in RA patients with low or high baseline insulin resistance.
To examine the effect of CTLA4.Ig treatment on the insulin signaling cascade, we measured the levels of Ser312-phosphorylated IRS-1 and phosphorylated AKT (p-AKT) in protein extracts from PBMCs (Figure 2C). We found no significant change in p-Ser321 IRS-1 or in p-AKT levels in RA patients with either low or high baseline insulin resistance (Figure 2D, E). We found no correlation between changes in HOMA-IR and IRS-1 or AKT phosphorylation levels.
Discussion
We evaluated the outcome on insulin resistance and other metabolic parameters among 61 patients with active RA disease, after 12 weeks of treatment with anti-TNF agents. In a subset of patients, we also examined the effect of TNF blockade on the phosphorylation status of IRS-1 and AKT, which are major mediators of insulin signaling. Our findings suggest that treatment with anti-TNF agents may improve insulin resistance and sensitivity in RA patients with active disease and high insulin resistance, and this effect correlates with reduced levels of the insulin-resistant Ser312-phosphorylated form of IRS-1 and increased levels of activated/phosphorylated AKT.
Insulin resistance constitutes a major mechanism in the pathophysiology of metabolic syndrome [38]. In our cohort of patients with active RA, those with high insulin resistance had significantly higher fasting plasma glucose, serum triglycerides, and systolic blood pressure levels than their counterparts in the low insulin resistance group. Accordingly, patients with metabolic syndrome had an OR of 10.8 for high insulin resistance, independent of the effect of other parameters.
In patients with RA, insulin resistance has been shown to correlate both with anthropometric parameters (abdominal obesity) and high disease activity [13, 14]. We found that RA patients with high insulin resistance had significantly higher DAS28 values than those with low insulin resistance, and in multivariate analysis, patients in the upper quartile of disease activity (DAS28 > 6.86) had more than six times increased risk (OR 6.4) for high insulin resistance. This is in agreement with our previous result of increased prevalence of metabolic syndrome in RA patients with moderate or high disease activity (OR 9.2 for DAS28 > 3.2) [12], and corroborates the link between inflammation and insulin resistance in chronic inflammatory disorders.
Previous studies have demonstrated that treatment with TNF antagonists improves insulin sensitivity in RA [18–22]. We further corroborate these results by examining the metabolic effects of anti-TNF therapy in a larger cohort of 61 patients with active RA. Among patients with high insulin resistance at baseline, treatment with anti-TNF agents for 12 weeks resulted in significant improvement in insulin sensitivity as evidenced by reduction in the HOMA-IR and increase in the QUICKI indices. Of note, serum triglycerides were reduced by an average 24.5 mg/dl, and HDL-C levels were increased by an average 4.2 mg/dl. Our findings agree with those of Kiortsis, et al. [20] who reported significant decrease in HOMA-IR and increase in QUICKI among patients with RA and ankylosing spondylitis in the highest tertile of insulin resistance, suggesting that this subset of patients may benefit most from anti-TNF therapy. In accordance with the results of previous studies [18–20, 22], we found no correlation between changes in HOMA-IR and fasting insulin levels with improvements in disease activity or CRP level.
We found that treatment with anti-TNF agents resulted in significant reduction in the HOMA-B index of pancreatic beta cell function in RA patients with high baseline insulin resistance. This could be explained by the therapy-driven reduction in the inflammatory burden of the disease and the insulin resistance levels, thus resulting in reduced compensatory beta cell hyperfunction. Whether this effect could lead to preservation of beta cell function in the long term is currently unknown. Intriguingly, a recent epidemiological study found that among patients with RA or psoriasis, the adjusted risk of new-onset DM was lower for individuals on TNF inhibitor (adjusted hazard ratio 0.62) compared with initiation of other non-biologic DMARDs [39].
TNF may impair insulin signaling at the level of the IRS proteins, and the Ser307 (or Ser312 in humans) residue in IRS-1 has been identified as a site for the inhibitory effects of TNF, with p38 MAPK and inhibitor kB kinase being involved in the phosphorylation of this residue [7, 8, 32]. TNF mediates insulin resistance by inhibition of AKT activity through a ceramide-activated protein-phosphatase (PP) 2A [35]. Since anti-TNF therapy significantly reduced HOMA-IR in patients with active RA and high insulin resistance, we examined the extent to which the aforementioned biochemical effects are affected by TNF inhibition. Our western blot assays demonstrated significant reduction in p-Ser312 IRS-1 and increase in p-AKT levels, associated with reduction in HOMA-IR in patients with active RA and high baseline insulin resistance. These alterations were consistent in all but one patient in our study and their magnitude did not correlate with the degree of improvement in disease activity or insulin resistance. To our knowledge, this is the first demonstration that anti-TNF therapy may improve insulin resistance in patients with active RA by reversing defects in the phosphorylation/activation status of the insulin signaling pathway.
To determine whether the observed improvements in insulin resistance and phosphorylation status of IRS-1 and AKT are directly linked to TNF blockade or are secondary to reduction in disease activity and inflammation, we studied seven patients with RA, who received abatacept (CTLA4.Ig) due to high disease activity. Patients treated with abatacept did not demonstrate reduction in HOMA-IR levels over the 12-week study period despite improvement in disease activity, nor did they display any consistent alterations in p-Ser312 IRS-1 and p-AKT levels. It should be noted, however, that these observations are limited to a small number of patients and further documentation in a larger cohort will be required. Moreover, it may be argued that the duration of the study was too short to capture the metabolic effects of abatacept treatment, or that we did not evaluate changes in other molecules implicated in insulin resistance such as expression of the protein-tyrosine phosphatase (PTP)-1B or the glucose transporter (GLUT)-4 [32].
Although our results suggest a beneficial effect of anti-TNF treatment on insulin signaling in RA patients with high insulin resistance, the clinical importance of such treatment in reducing risk of cardiovascular disease in the long term remains to be determined. In the interim, clinical decisions for initiation of anti-TNF or other biologic agents in RA should be based on disease activity and severity indices [40]. To this end, according to the existing EULAR recommendations, 'adequate control of disease activity is necessary to lower the cardiovascular disease risk' in RA patients [41]. Consequently, anti-TNF agents may be preferred as first-line biologic treatment in patients with active RA and high inflammatory burden, and metabolic abnormalities or insulin resistance.
Conclusions
In summary, we found that 12 weeks of treatment with anti-TNF agents may improve insulin resistance in patients with active RA and high insulin resistance. Treatment with anti-TNF was shown to restore the phosphorylation status of Ser312-IRS-1 and AKT, which are important mediators in the insulin signaling cascade. The impact of these biochemical changes in modifying the burden of cardiovascular disease in patients with chronic inflammatory arthritis remains to be seen.
Abbreviations
- ANOVA:
-
analysis of variance
- apo-A:
-
apolipoprotein
- ATP:
-
Adult Treatment Panel
- BMI:
-
body mass index
- CI:
-
confidence interval
- CRP:
-
C-reactive protein
- DAS28:
-
disease activity score of 28 joints
- DBP:
-
diastolic blood pressure
- DMARD:
-
disease-modifying anti-rheumatic drug
- ESR:
-
erythrocyte sedimentation rate
- EULAR:
-
European League Against Rheumatism
- FBG:
-
fasting blood glucose
- GC:
-
glucocorticoids
- HAQ:
-
health assessment questionnaire
- HDL-C:
-
high-density lipoprotein cholesterol
- HOMA-IR:
-
Homeostasis Model Assessment for insulin resistance
- HRP:
-
horseradish peroxidase
- IL:
-
interleukin
- IRS:
-
insulin receptor substrate
- LDL-C:
-
low-density lipoprotein cholesterol
- Lpa:
-
lipoprotein a
- MAPK:
-
mitogen activated protein kinase
- NCEP:
-
National Cholesterol Education Program
- OR:
-
odds ratio
- PBMC:
-
peripheral blood mononuclear cell
- PI-3K:
-
phosphatidyl-inositol 3-kinase
- PP:
-
protein-phosphatase
- QUICKI:
-
Quantitative Insulin Sensitivity Check Index
- RA:
-
rheumatoid arthritis
- SBP:
-
systolic blood pressure
- SDS-PAGE:
-
sodium dodecyl sulphate-polyacrylamide gel electrophoresis
- T2DM:
-
type 2 diabetes mellitus
- TC:
-
total cholesterol
- TG:
-
triglycerides
- TNF:
-
tumor necrosis factor.
References
Tamemoto H, Kadowaki T, Tobe K, Yagi T, Sakura H, Hayakawa T, Terauchi Y, Ueki K, Kaburagi Y, Satoh S, Sekihara H, Yoshioka S, Horikoshi H, Furuta Y, Ikawa Y, Kasuga M, Yazaki Y, Aizawa S: Insulin resistance and growth retardation in mice lacking insulin receptor substrate-1. Nature. 1994, 372: 182-186. 10.1038/372182a0.
Withers DJ, Gutierrez JS, Towery H, Burks DJ, Ren JM, Previs S, Zhang Y, Bernal D, Pons S, Shulman GI, Bonner-Weir S, White MF: Disruption of IRS-2 causes type 2 diabetes in mice. Nature. 1998, 391: 900-904. 10.1038/36116.
Taniguchi CM, Ueki K, Kahn R: Complementary roles of IRS-1 and IRS-2 in the hepatic regulation of metabolism. J Clin Invest. 2005, 115: 718-727.
Lazar DF, Wiese RJ, Brady MJ, Mastick CC, Waters SB, Yamauchi K, Pessin JE, Cuatrecasas P, Saltiel AR: Mitogen-activated protein kinase kinase inhibition does not block the stimulation of glucose utilization by insulin. J Biol Chem. 1995, 270: 20801-20807. 10.1074/jbc.270.35.20801.
Cusi K, Maezono K, Osman A, Pendergrass M, Patti ME, Pratipanawatr T, DeFronzo RA, Kahn CR, Mandarino LJ: Insulin resistance differentially affects the PI 3-kinase- and MAP kinase-mediated signaling in human muscle. J Clin Invest. 2000, 105: 311-320. 10.1172/JCI7535.
Wellen KE, Hotamisligil GS: Inflammation, stress, and diabetes. J Clin Invest. 2005, 115: 1111-1119.
Hotamisligil GS, Murray DL, Choy LN, Spiegelman BM: Tumor necrosis factor alpha inhibits signaling from the insulin receptor. Proc Natl Acad Sci USA. 1994, 91: 4854-4858. 10.1073/pnas.91.11.4854.
Hotamisligil GS, Peraldi P, Budavari A, Ellis R, White MF, Spiegelman BM: IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science. 1996, 271: 665-668. 10.1126/science.271.5249.665.
Rotter V, Nagaev I, Smith U: Interleukin-6 (IL-6) induces insulin resistance in 3T3-L1 adipocytes and is, like IL-8 and tumor necrosis factor-alpha, overexpressed in human fat cells from insulin-resistant subjects. J Biol Chem. 2003, 278: 45777-45784. 10.1074/jbc.M301977200.
Boers M, Dijkmans B, Gabriel S, Maradit-Kremers H, O'Dell J, Pincus T: Making an impact on mortality in rheumatoid arthritis: targeting cardiovascular comorbidity. Arthritis Rheum. 2004, 50: 1734-1739. 10.1002/art.20306.
del Rincon ID, Williams K, Stern MP, Freeman GL, Escalante A: High incidence of cardiovascular events in a rheumatoid arthritis cohort not explained by traditional cardiac risk factors. Arthritis Rheum. 2001, 44: 2737-2745. 10.1002/1529-0131(200112)44:12<2737::AID-ART460>3.0.CO;2-#.
Karvounaris SA, Sidiropoulos PI, Papadakis JA, Spanakis EK, Bertsias GK, Kritikos HD, Ganotakis ES, Boumpas DT: Metabolic syndrome is common among middle-to-older aged Mediterranean patients with rheumatoid arthritis and correlates with disease activity: a retrospective, cross-sectional, controlled, study. Ann Rheum Dis. 2007, 66: 28-33.
Chung CP, Oeser A, Solus JF, Avalos I, Gebretsadik T, Shintani A, Raggi P, Sokka T, Pincus T, Stein CM: Prevalence of the metabolic syndrome is increased in rheumatoid arthritis and is associated with coronary atherosclerosis. Atherosclerosis. 2008, 196: 756-763. 10.1016/j.atherosclerosis.2007.01.004.
Dessein PH, Joffe BI: Insulin resistance and impaired beta cell function in rheumatoid arthritis. Arthritis Rheum. 2006, 54: 2765-2775. 10.1002/art.22053.
Solomon DH, Love TJ, Canning C, Schneeweiss S: Risk of diabetes among patients with rheumatoid arthritis, psoriatic arthritis and psoriasis. Ann Rheum Dis. 2010, 69: 2114-2117. 10.1136/ard.2009.125476.
Jacobsson LT, Turesson C, Gulfe A, Kapetanovic MC, Petersson IF, Saxne T, Geborek P: Treatment with tumor necrosis factor blockers is associated with a lower incidence of first cardiovascular events in patients with rheumatoid arthritis. J Rheumatol. 2005, 32: 1213-1218.
Dixon WG, Watson KD, Lunt M, Hyrich KL, Silman AJ, Symmons DP: Reduction in the incidence of myocardial infarction in patients with rheumatoid arthritis who respond to anti-tumor necrosis factor alpha therapy: results from the British Society for Rheumatology Biologics Register. Arthritis Rheum. 2007, 56: 2905-2912. 10.1002/art.22809.
Gonzalez-Gay MA, De Matias JM, Gonzalez-Juanatey C, Garcia-Porrua C, Sanchez-Andrade A, Martin J, Llorca J: Anti-tumor necrosis factor-alpha blockade improves insulin resistance in patients with rheumatoid arthritis. Clin Exp Rheumatol. 2006, 24: 83-86.
Gonzalez-Gay MA, Gonzalez-Juanatey C, Vazquez-Rodriguez TR, Miranda-Filloy JA, Llorca J: Insulin resistance in rheumatoid arthritis: the impact of the anti-TNF-alpha therapy. Ann N Y Acad Sci. 2010, 1193: 153-159. 10.1111/j.1749-6632.2009.05287.x.
Kiortsis DN, Mavridis AK, Vasakos S, Nikas SN, Drosos AA: Effects of infliximab treatment on insulin resistance in patients with rheumatoid arthritis and ankylosing spondylitis. Ann Rheum Dis. 2005, 64: 765-766. 10.1136/ard.2004.026534.
Seriolo B, Paolino S, Ferrone C, Cutolo M: Impact of long-term anti-TNF-alpha treatment on insulin resistance in patients with rheumatoid arthritis. Clin Exp Rheumatol. 2008, 26: 159-160. author reply 160
Tam LS, Tomlinson B, Chu TT, Li TK, Li EK: Impact of TNF inhibition on insulin resistance and lipids levels in patients with rheumatoid arthritis. Clin Rheumatol. 2007, 26: 1495-1498. 10.1007/s10067-007-0539-8.
Smolen JS, Landewe R, Breedveld FC, Dougados M, Emery P, Gaujoux-Viala C, Gorter S, Knevel R, Nam J, Schoels M, Aletaha D, Buch M, Gossec L, Huizinga T, Bijlsma JW, Burmester G, Combe B, Cutolo M, Gabay C, Gomez-Reino J, Kouloumas M, Kvien TK, Martin-Mola E, McInnes I, Pavelka K, van Riel P, Scholte M, Scott DL, Sokka T, Valesini G, et al: EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs. Ann Rheum Dis. 2010, 69: 964-975. 10.1136/ard.2009.126532.
Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, Healey LA, Kaplan SR, Liang MH, Luthra HS, Medsger TA, Mitchell DM, Neustadt DH, Pinals RS, Schaller JG, Sharp JT, Wilder RL, Hunder GG: The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988, 31: 315-324. 10.1002/art.1780310302.
van Gestel AM, Haagsma CJ, van Riel PL: Validation of rheumatoid arthritis improvement criteria that include simplified joint counts. Arthritis Rheum. 1998, 41: 1845-1850. 10.1002/1529-0131(199810)41:10<1845::AID-ART17>3.0.CO;2-K.
Bonora E, Kiechl S, Willeit J, Oberhollenzer F, Egger G, Targher G, Alberiche M, Bonadonna RC, Muggeo M: Prevalence of insulin resistance in metabolic disorders: the Bruneck Study. Diabetes. 1998, 47: 1643-1649. 10.2337/diabetes.47.10.1643.
Katz A, Nambi SS, Mather K, Baron AD, Follmann DA, Sullivan G, Quon MJ: Quantitative insulin sensitivity check index: a simple, accurate method for assessing insulin sensitivity in humans. J Clin Endocrinol Metab. 2000, 85: 2402-2410. 10.1210/jc.85.7.2402.
Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC: Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985, 28: 412-419. 10.1007/BF00280883.
Executive Summary of The Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III). JAMA. 2001, 285: 2486-2497. 10.1001/jama.285.19.2486.
Festa A, Williams K, D'Agostino R, Wagenknecht LE, Haffner SM: The natural course of beta-cell function in nondiabetic and diabetic individuals: the Insulin Resistance Atherosclerosis Study. Diabetes. 2006, 55: 1114-1120. 10.2337/diabetes.55.04.06.db05-1100.
Balkau B, Charles MA: Comment on the provisional report from the WHO consultation. European Group for the Study of Insulin Resistance (EGIR). Diabet Med. 1999, 16: 442-443.
Nieto-Vazquez I, Fernandez-Veledo S, Kramer DK, Vila-Bedmar R, Garcia-Guerra L, Lorenzo M: Insulin resistance associated to obesity: the link TNF-alpha. Arch Physiol Biochem. 2008, 114: 183-194. 10.1080/13813450802181047.
Hennige AM, Stefan N, Kapp K, Lehmann R, Weigert C, Beck A, Moeschel K, Mushack J, Schleicher E, Haring HU: Leptin down-regulates insulin action through phosphorylation of serine-318 in insulin receptor substrate 1. FASEB J. 2006, 20: 1206-1208. 10.1096/fj.05-4635fje.
McElduff A, Hedo JA, Taylor SI, Roth J, Gorden P: Insulin receptor degradation is accelerated in cultured lymphocytes from patients with genetic syndromes of extreme insulin resistance. J Clin Invest. 1984, 74: 1366-1374. 10.1172/JCI111547.
Teruel T, Hernandez R, Lorenzo M: Ceramide mediates insulin resistance by tumor necrosis factor-alpha in brown adipocytes by maintaining Akt in an inactive dephosphorylated state. Diabetes. 2001, 50: 2563-2571. 10.2337/diabetes.50.11.2563.
Krook A, Roth RA, Jiang XJ, Zierath JR, Wallberg-Henriksson H: Insulin-stimulated Akt kinase activity is reduced in skeletal muscle from NIDDM subjects. Diabetes. 1998, 47: 1281-1286. 10.2337/diabetes.47.8.1281.
Smith U, Axelsen M, Carvalho E, Eliasson B, Jansson PA, Wesslau C: Insulin signaling and action in fat cells: associations with insulin resistance and type 2 diabetes. Ann N Y Acad Sci. 1999, 892: 119-126. 10.1111/j.1749-6632.1999.tb07790.x.
Mottillo S, Filion KB, Genest J, Joseph L, Pilote L, Poirier P, Rinfret S, Schiffrin EL, Eisenberg MJ: The metabolic syndrome and cardiovascular risk a systematic review and meta-analysis. J Am Coll Cardiol. 2010, 56: 1113-1132. 10.1016/j.jacc.2010.05.034.
Solomon DH, Massarotti E, Garg R, Liu J, Canning C, Schneeweiss S: Association between disease-modifying antirheumatic drugs and diabetes risk in patients with rheumatoid arthritis and psoriasis. JAMA. 2011, 305: 2525-2531. 10.1001/jama.2011.878.
Singh JA, Furst DE, Bharat A, Curtis JR, Kavanaugh AF, Kremer JM, Moreland LW, O'Dell J, Winthrop KL, Beukelman T, Bridges SL, Chatham WW, Paulus HE, Suarez-Almazor M, Bombardier C, Dougados M, Khanna D, King CM, Leong AL, Matteson EL, Schousboe JT, Moynihan E, Kolba KS, Jain A, Volkmann ER, Agrawal H, Bae S, Mudano AS, Patkar NM, Saag KG: 2012 update of the 2008 american college of rheumatology recommendations for the use of disease-modifying antirheumatic drugs and biologic agents in the treatment of rheumatoid arthritis. Arthritis Care Res (Hoboken). 2012, 64: 625-639. 10.1002/acr.21641.
Peters MJ, Symmons DP, McCarey D, Dijkmans BA, Nicola P, Kvien TK, McInnes IB, Haentzschel H, Gonzalez-Gay MA, Provan S, Semb A, Sidiropoulos P, Kitas G, Smulders YM, Soubrier M, Szekanecz Z, Sattar N, Nurmohamed MT: EULAR evidence-based recommendations for cardiovascular risk management in patients with rheumatoid arthritis and other forms of inflammatory arthritis. Ann Rheum Dis. 2010, 69: 325-331. 10.1136/ard.2009.113696.
Acknowledgements
We thank Eva Choustoulaki, RN, and Chara Misirlaki, RN, for sample collection and patient care; Aggeliki Klisarxaki, MD, for technical assistance. The paper is dedicated to the memory of Heraklis Kritikos, a devoted physician and inspired clinical researcher. This study was supported by funds from the Hellenic Society of Rheumatology, Pancretan Health Association and the European Union (AutoCure program).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
IS performed PBMC separation and western blotting. GB contributed to study design, statistical analysis, and drafting the manuscript. SK performed patients' data acquisition and took care of the patients. MK and DV helped with blood sampling and performed western blotting. AR took care of the patients and also helped with patients' data acquisition. DK contributed to the design of the experimental part of the study. DTB and PIS supervised the study, drafted and revised the manuscript. All authors read and approved the final manuscript.
Ilias Stagakis, George Bertsias contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Stagakis, I., Bertsias, G., Karvounaris, S. et al. Anti-tumor necrosis factor therapy improves insulin resistance, beta cell function and insulin signaling in active rheumatoid arthritis patients with high insulin resistance . Arthritis Res Ther 14, R141 (2012). https://doi.org/10.1186/ar3874
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/ar3874