
Abstract
Autoantibodies to neutrophil cytoplasmic antigen-associated vasculitis (AAV) is characterised by inflammation of blood vessels. The introduction of immunosuppressive therapy with glucocorticoids and cyclophosphamide transformed AAV from a fatal condition to a largely treatable condition. Over the past 30 years, considerable progress has been made refining immunosuppressive regimens with a focus on minimising toxicity. There is, however, a high unmet need in the treatment of AAV. A proportion of patients are refractory to current therapies; 50% experience a relapse within 5 years and treatment toxicity contributes to mortality and chronic disability. As knowledge of the pathogenesis of vasculitis grows, it is mirrored by the availability of biological agents, which herald a revolution in the treatment of vasculitis. Lymphocyte-targeted and cytokine-targeted agents have been evaluated for the treatment of AAV and are entering the routine therapeutic arena with the potential to improve patient outcomes. As rare diseases, treatment advances in vasculitis depend on international collaborative research networks both to establish an evidence base for newer agents and to develop recommendations for patient management.
Similar content being viewed by others


Introduction
The systemic vasculitides include a diverse range of conditions characterised by leucocyte infiltration of blood vessel walls, fibrinoid necrosis and vascular damage with occlusion or aneurysm formation. These conditions may be primary in origin, when they are classified according to the predominant size of vessel affected [1] (Table 1), or secondary to infection, malignancy, drugs or another autoimmune process, such as rheumatoid arthritis or systemic lupus erythematosus (SLE). Of the primary systemic vasculitides, the most prevalent syndrome is autoantibodies to neutrophil cytoplasmic antigens (ANCA)-associated vasculitis (AAV), which has attracted the most attention in the optimisation of existing therapies and the introduction of newer agents. AAV will be the focus of this review.
ANCA-associated vasculitis
AAV includes (Wegener's) granulomatosis with polyangiitis (GPA), microscopic polyangiitis (MPA) and Churg-Strauss syndrome (CSS). These conditions are usually associated with the presence of circulating ANCA [2]. The pathogenicity of ANCA has been confirmed in experimental models [3]. AAV predominantly involves microscopic blood vessels, with the respiratory tract and kidneys being the most common organs affected, although immune deposits are scanty [1, 4]. AAV has an annual incidence of 20 per million and a prevalence over 200 per million [5]. Across the spectrum of AAV, disease is sub-classified according to the severity and extent of organ involvement, ranging from localised to severe organ-threatening or life-threatening disease [6] (Table 2). This subclassification underlies current treatment guidelines, and has contributed to the design of clinical trials to ensure comparable patients are enrolled across multiple centres.
Current treatment outcomes
Prior to effective treatment, AAV had a mortality of 93% within 2 years, primarily due to renal and respiratory failure [7]. The introduction of glucocorticoids in 1948 and cyclophosphamide in the 1960s, together with adjunctive therapies such as antihypertensive drugs and renal replacement therapy, has transformed survival - with 5-year survival rates now approaching 80% [6]. This therapeutic revolution has converted vasculitis into a chronic relapsing disorder with progressive organ damage and disability, eventually affecting over 95% of patients. The cumulative exposure to glucocorticoids and immunosuppressive drugs contributes to organ damage, and there has been particular concern caused by cyclophosphamide-related toxicities of myelosuppression, infections, urothelial malignancy and infertility [8, 9].
Rates of cardiovascular disease and malignancy are increased and it is unclear to what extent these reflect the underlying disease process or its treatment [10]. Early treatment-related leucopaenia and infection, and later cardiovascular disease, are now the predominant causes of death, rather than uncontrolled vasculitis [11].
Treatment-related toxicity, together with the 10 to 30% of patients who do not respond to traditional immunosuppressive agents and pursue a refractory course and the 50% of patients who relapse within 5 years despite continued immunosuppression, has shifted the focus of treatment for AAV [12]. From learning how to best use traditional agents to minimise toxicity without losing efficacy, to the advent of new targeted biological agents, a second revolution in the treatment of AAV is on the horizon.
Pathogenesis of AAV
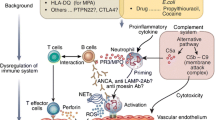
Both genetic susceptibility and environmental exposures contribute to the aetiology of AAV. ANCA have been demonstrated pathogenic in animal models but their contribution to human disease remains unclear. Transfer of murine myeloperoxidase-ANCA IgG to mice without functioning B cells or T cells results in a pauci immune, necrotising crescentic glomerulonephritis, similar to that seen in AAV in humans [3]. Human anti-PR3 antibodies from individuals with AAV were recently demonstrated to cause comparable renal and pulmonary lesions in mice with a humanised immune system [13]. Two lines of investigation have linked infections to ANCA formation through molecular mimicry [14, 15]. Fimbriated bacteria can induce novel ANCA, antibodies to human lysosome membrane protein-2, which can induce a crescentic glomerulonephritis in animal models [15]. Microbial superantigens are responsible for T-cell dysregulation in Kawasaki disease [16], and infection with Staphylococcus aureus is associated with relapse of GPA [17, 18]. In sensitive assays, the PR3-ANCA binding level is predictive of outcome and rises in ANCA precede relapse. The clinical correlation with ANCA is closest for alveolar and glomerular capillaritis, both lesions capable of being induced by ANCA in experimental models. Patients who are consistently ANCA-negative, however, may fit the clinical phenotype of AAV, and the efficacy of B-cell depletion with rituximab is not associated with ANCA status. Despite the pauci immune nature of the histology in ANCA vasculitis, there is evidence of a role for immune complexes and complement activation in renal vasculitis.
There is activation of circulating T lymphocytes and B lymphocytes and infiltration of plasmoblasts into affected tissues [19]. Autoreactive B cells are necessary for the development of autoantibody-producing cells, but may play a more important role in supporting autoreactive T-cell activity through antigen presentation, co-stimulation and the direct production of proinflammatory cytokines, such as IL-6 and TNFα. In view of their role as precursors of ANCA-secreting plasma cells, B cells are a therapeutic target in AAV. T cells also play an important role in the pathogenesis of AAV [20]. The autoantibodies are class switched (that is, mainly IgG), which means the autoreactive B cell has received cognate T-cell help. Also, in biopsy specimens T cells are seen to infiltrate tissues, causing damage via direct cytotoxicity and by the recruitment and activation of macrophages [21]. A CD8 T-cell gene expression signature has been associated with relapse frequency in AAV [22], and T-cell targeted therapies including cyclosporin [23] and alemtuzumab have been effective [24].
Treatment
After making a diagnosis of systemic vasculitis, treatment is divided into remission induction, then 3 to 6 months of intensive therapy, followed by a longer period of remission maintenance therapy. Disease state definitions have been agreed for active disease, remission, low disease activity state and refractory disease [25]. Further key management concepts are the subgrouping of patients by severity, the minimisation of adverse event risk and proactive management of increased cardiovascular and malignancy risks.
GPA has been subdivided into limited or nonsevere versus generalised or severe - the most important determinant being the presence or absence of nephritis. An analogous system was developed by the European Vasculitis Study Group to include both GPA and MPA (Table 2) [25] in order to define eligibility for a sequence of randomised controlled trials aimed at defining the optimal standard of care.
Standard induction remission therapy
Glucocorticoids
Despite the introduction of glucocorticoids into treatment strategies for vasculitis over 50 years ago, there are no randomised controlled trials to support their use. Evidence is also lacking to guide dosage. Current regimens start oral prednisone/prednisolone at 1 mg/kg/day, reducing to 10 to 20 mg daily by 12 weeks, with up to 3 g intravenous (i.v.) methylprednisolone for generalised or severe presentations. Although they contribute to disease control, there is increasing evidence that high-dose steroids contribute to morbidity [26]. The PEXIVAS trial aims to address the efficacy of a rapidly reducing glucocorticoid regimen, as well as the place of plasma exchange in severe AAV (ClinicalTrials.gov: NCT00987389).
Cyclophosphamide
Cyclophosphamide, together with high-dose glucocorticoids, was introduced empirically for induction treatment of AAV over 40 years ago, as a lymphocyte-depleting therapy following its use in lymphoproliferative diseases. Together with glucocorticoids, cyclophosphamide remains the standard of care in consensus guidelines for the treatment of generalised disease [27]. The use of cyclophosphamide has been refined, however, employing strategies to reduce exposure and cumulative toxicity.
The efficacy of a sequential design whereby cyclophosphamide was replaced by an alternative at the time of remission was explored in the CYCAZAREM trial [28]. In this study, 155 patients were randomised to receive either 1 year of oral cyclophosphamide or 3 months (maximum 6 months) of oral cyclophosphamide followed by azathioprine. Both groups followed a similar steroid taper. There was no difference in relapse rates between the two groups at 18 months, demonstrating that the duration of exposure to cyclophosphamide may be safely reduced.
An i.v. pulsed administration has potential advantages over daily oral cyclophosphamide of reduced exposure, bladder protection and improved compliance. A meta-analysis of three studies of i.v. cyclophosphamide, including 143 patients, demonstrated that pulsed regimens reduced cumulative cyclophosphamide exposure by 50%, and were at least as effective at inducing remission, with fewer infective and myelosuppressive side effects but possibly with an increased risk of relapse [29].
The CYCLOPS study randomised 149 patients with generalised AAV to receive either i.v. cyclophosphamide, 15 mg/kg at 2-week intervals for the first three doses and every 3 weeks thereafter, or daily oral cyclophosphamide 2 mg/kg/day [30]. Induction therapy was continued for 3 months after remission was achieved in both groups prior to a switch to azathioprine for maintenance therapy until the end of the 18-month trial. There was no difference in time to achieve remission or the proportion of patients in remission at 9 months between the groups, despite the i.v. cyclophosphamide group receiving 8.2 g as compared with 15.9 g cyclophosphamide for the daily oral route. The reduced dose resulted in fewer leucopaenic episodes, but this did not translate to fewer infections. Long-term follow-up (median 4.3 years), however, has shown that pulsed i.v. cyclophosphamide is associated with a higher relapse rate, but this does not seem to impact survival or long-term morbidity [31]. The CYCLOPS protocol also standardised cyclophosphamide dose reductions for age over 60 years and for renal impairment, which improved the safety of these regimens.
Methotrexate
Following nonrandomised studies of methotrexate in limited GPA, 100 patients with a new diagnosis of early systemic AAV, without critical organ manifestations and with creatinine <150 μmol/l were randomised to receive either methotrexate targeting 25 mg weekly or oral cyclophosphamide in the NORAM study [32]. Both groups followed the same glucocorticoid regimen, and treatment was withdrawn at 12 months. Methotrexate was not inferior at inducing remission, but remission was slower than with cyclophosphamide in those patients with more extensive disease or pulmonary involvement. There was less leucopaenia with methotrexate, but more liver dysfunction and a higher relapse rate.
Adjunctive therapies for severe disease
For those presenting with severe vasculitis when vital organ function is already compromised, adjunctive therapies have been considered that include, plasma exchange, i.v. methylprednisolone, intravenous immunoglobulin (IVIg) and TNFα blockade. Despite an absence of randomised evidence there is widespread use of i.v. methylprednisolone in doses between 1,000 and 3,000 mg for generalised and severe presentations.
Advanced renal failure at presentation correlates with an increased risk of end-stage renal failure and death, and the aims of therapy are both to control disease and recover organ function [6]. Several small trials have produced conflicting results regarding the merits of plasma exchange, although improvements in recovery rates for renal vasculitis presenting with advanced renal failure were found [33]. The MEPEX trial compared adjunctive therapy with plasma exchange or i.v. methylprednisolone in 137 patients with AAV and serum creatinine >500 μmol/l (5.8 mg/dl) at presentation [34]. Both groups received oral cyclophosphamide and steroids. Plasma exchange decreased the risk of progression to end-stage renal failure by 24% at 12 months, but had no effect on longer-term renal function or survival [35].
The mode of action of plasma exchange is unclear, but removal of complement and coagulation factors, ANCA and other circulating factors is likely to be important. A smaller trial found benefit in terms of renal outcome for patients presenting with less severe nephritis [36]. There remains uncertainty regarding the long-term benefits of plasma exchange, and a recent meta-analysis failed to find convincing evidence that there was an influence on the composite endpoint of death or end-stage renal disease after 1 year [37].
Severe alveolar haemorrhage is the most common vasculitic cause of early death, and, in view of similarities in pathogenesis with renal vasculitis, plasma exchange has been recommended. Single-centre reviews have suggested a role for plasma exchange, but one study of ventilator-dependent haemorrhage still found a 50% mortality despite this intervention [38, 39].
Newer induction agents
B-cell depletion therapies (rituximab)
Following the successful use of rituximab in rheumatoid arthritis and increasing appreciation of a role for B cells in the pathogenesis of AAV, several studies have reported responses to ritxumab for patients with relapsing or refractory disease [40]. The largest multicentre series of 65 patients with refractory disease reported complete remission in 75%, partial remission in 23%, and only one nonresponder. However, 57% of those who achieved remission relapsed after a median of 12 months [41].
Two randomised control studies - RITUXVAS [42], a randomised trial of a rituximab-based regimen versus cyclophosphamide in ANCA-associated renal vasculitis; and RAVE [43], rituximab for ANCA-associated vasculitis - have demonstrated that rituximab has similar efficacy to cyclophosphamide for the induction of remission in AAV. Table 3 summarises the key features of these studies.
Neither study reported a reduction in severe adverse events with rituximab, which suggested that high-dose glucocorticoids and the disease itself are the main contributors to these events, and that current cyclophosphamide regimens are, in expert centres, reasonably safe. Although two doses of cyclophosphamide were administered in addition to rituximab in the RITUXVAS study, the RAVE study used no immunosuppressive in the rituximab group. Response rates were comparable, so it is now justified to withdraw immunosuppression at the time of rituximab treatment. The response rates in both the rituximab and cyclophosphamide limbs of the RAVE study were lower in those patients who had glucocorticoids withdrawn by 6 months, highlighting their importance in remission induction strategies. However, this observation must be balanced against their longer-term toxicity, and the optimal time for withdrawal is yet to be established. For the subgroup presenting with relapsing disease as compared with new-onset disease, rituximab was more effective in obtaining remission than cyclophosphamide and can be recommended for this indication. The role of rituximab for new-onset disease is more controversial; both trials had a short duration of 12 and 6 months, respectively, and longer-term results are awaited.
Meanwhile rituximab can be recommended as an alternative to cyclophosphamide in generalised, severe or refractory AAV when the latter is contraindicated. However, controlled data for the use of rituximab in localised and early systemic disease are lacking at present.
Mycophenolate mofetil
Two observational studies reported remissions after mycophenolate mofetil (MMF) 2 g/day and glucocorticoids in 25/32 (78%) patients with active, mostly relapsing, AAV and in 13/17 (76%) patients with MPA [44, 45]. However, the former study found high rates of relapse [44].
Thirty-five Chinese patients with MPA and serum creatinine <500 μmol/l (5.8 mg/dl) were randomised to either receive MMF 2 g/day or i.v. cyclophosphamide [46]. At 6 months, 14/18 (77.8%) of the MMF group compared with 8/17 (47.1%) in the cyclophosphamide group were in complete remission. In addition, 44.4% of patients in the MMF group recovered renal function compared with only 16.7% in the cyclophosphamide group. Adverse events were comparable between the two groups. A larger induction trial to compare MMF and cyclophosphamide, involving 140 patients - the MYCYC trial - is being conducted by the European Vasculitis Study Group [47].
Remission maintenance therapy
AAV is a relapsing condition, with disease and treatment factors influencing relapse risk. In a retrospective cohort study of 246 patients with AAV and renal involvement, the relapse rate was 34% with a median time to relapse of 13 months after induction of remission [12]. Aggregated data from four European Vasculitis Study Group trials, encompassing the spectrum of disease severity, reported a 50% relapse rate at 5 years (unpublished data), with a higher rate for GPA (70%) than for MPA (30%). Relapse risk is increased by upper respiratory tract involvement, nasal carriage of S. aureus infection and the absence of renal disease. Persistent ANCA positivity at the time of remission and a subsequent rise in ANCA level can be predictive of relapse [48–50]. The utility of ANCA monitoring varies with disease type, however, and ongoing treatment dissociates changes in ANCA from changes in disease activity, which accounts for some negative reports [51].
Withdrawal of immunosuppression or glucocorticoids is associated with a higher relapse rate [52]. The cyclophosphamide group had a relapse rate of 46.5% at 18 months in the NORAM trial [32], when therapy was stopped at 12 months, compared with 13.7% at the same time point in the cyclophosphamide group in the CYCAZAREM trial [28], when azathioprine was substituted for cyclophosphamide at 12 months and continued until the end of the study. Consensus guidelines advocate continuation of maintenance immunosuppression for at least 18 to 24 months [27, 53]. The REMAIN study is comparing the relapse rate between 2 years and 4 years of maintenance therapy [54].
Standard remission maintenance therapy
Azathioprine
Cyclophosphamide is effective at inducing remission in AAV, and has been continued for prolonged periods to maintain remission. However, the dangers of high cumulative toxicity, particularly bladder cancer and myeloproliferative disease, have led to arguments for safer therapies [9]. The CYCAZAREM trial compared continued cyclophosphamide 1.5 mg/kg/day after remission induction to a switch to azathioprine 2 mg/kg/day [28]. No difference in subsequent relapse rate or other end-points was seen at 18 months, and this study has validated a sequential approach to treatment with cyclophosphamide withdrawal at the time of remission.
Methotrexate
The WEGENT study randomised 126 patients to receive either methotrexate or azathioprine as maintenance therapy after successful induction of remission with cyclophosphamide [55]. There were no differences between groups in either relapse or toxicity. Use of methotrexate is usually restricted to those with creatinine <150 μmol/l (1.8 mg/dl).
Glucocorticoids
There is variance in current practice regarding the use of prednisolone for remission maintenance. A review of published randomised trials in which the glucocorticoid protocol was defined found by multivariate analysis that glucocorticoid withdrawal was the strongest predictor for relapse [52].
Alternative remission maintenance therapies
A study involving 64 patients with GPA, comparing methotrexate (20 mg weekly) with leflunomide (30 mg daily) as a maintenance therapy, was terminated early due to a higher major relapse rate in the methotrexate group (46%) compared with the leflunomide group (23%) [56]. However, this was balanced by an increased rate of severe adverse events in the leflunomide group. The position of leflunomide as a remission maintenance agent remains uncertain.
Long-term treatment with sulfamethoxazole/trimethoprim is based on the assumption that controlling bacterial infection will reduce relapse risk [18]. When dosed at sulfamethoxazole/trimethoprim 960 mg twice a day in GPA there were reduced respiratory tract infections and respiratory tract relapses in a double-blind trial [57]. It is unclear whether this antibiotic has intrinsic antivasculitis activity separate from its antibiotic role, but the study supports the need for careful microbiological evaluation and treatment in GPA, especially in patients with respiratory tract involvement pursuing a relapsing course.
Mycophenolate mofetil
Following its success in the field of transplantation, MMF has been considered as an alternative to azathioprine for remission maintenance therapy in AAV. Results from a small pilot study in 11 patients were promising, with only one relapse occurring after 14 months of follow-up [58]. The IMPROVE randomised study that compared MMF 2 g/day and azathioprine 2 mg/kg/day as maintenance agents after cyclophosphamide induction in 174 patients, however, found an increased hazard ratio of 1.7 for relapse in the MMF group, and a shorter time to relapse [59]. MMF cannot be recommended as a first-line remission maintenance agent in AAV but it may have a role for patients intolerant of azathioprine for whom methotrexate is contraindicated by renal failure.
Refractory disease
Patients with refractory disease encompass those who have disease progression or inadequate disease control despite induction therapy; those who relapse on maintenance immunosuppression, and those who are intolerant of standard therapy, most commonly cyclophosphamide. Refractory disease has been defined in a European League Against Rheumatism statement to facilitate further clinical investigation [25]. In such challenging patients, it is important to identify underlying drives such as infection or malignancy, and then to consider whether further immunosuppressive agents or alternative agents are indicated.
Rituximab is now the preferred agent for refractory disease [41]. Rituximab was more effective than cyclophosphamide in the relapsing subgroup of the RAVE trial [40], although a retrospective series suggests it is less effective for granulomatous lesions, especially orbital masses than for vasculitic manifestations [60].
Additional agents
Intravenous immunoglobulin
Small studies have demonstrated a benefit of IVIg in persistent disease [61, 62]. Postulated mechanisms for action of IVIg include the interference of binding of ANCA to their antigens through idiotypic mechanisms and the inhibition of ANCA-induced neutrophil activation [63, 64]. Thirty-four patients with persistent disease activity were randomised to receive either IVIg (a single course at 2 g/kg) or placebo in a double-blind trial. Fourteen of 17 patients in the IVIg group as compared with 6/17 patients who received placebo had a reduction in disease activity, but this effect was not sustained beyond 3 months [65]. IVIg has also been used to maintain remission in patients with refractory disease [66]. Cost and limited availability have been barriers to the use of IVIg; however, it remains an alternative to consider in sepsis or pregnancy in order to minimise or avoid the use of standard agents.
TNF blockade
TNF blockade ameliorates disease in experimental models of AAV [67]. However, a beneficial effect of TNF blockade in human disease has been more difficult to demonstrate. Etanercept did not improve the rates of stable remission in GPA when added to glucocorticoids and cyclophosphamide or methotrexate [68]. Rates of serious adverse events exceeded 50% in both groups and a possible increase in solid malignancies was recorded in the etanercept group [69]. All cases of malignancy occurred in patients with prior cyclophosphamide exposure, however, and TNF blockade should therefore be avoided in these patients [70]. Both infliximab and adalimumab have been proposed as adjunctive agents in renal vasculitis and a potential for steroid sparing has been demonstrated [71–73]. Prolonged infliximab for refractory AAV has been complicated by infection and relapse.
Alternative strategies
T-cell-targeted agents
T-cell depletion with anti-thymocyte globulin led to remissions in refractory AAV in the SOLUTION trial but was poorly tolerated [74]. Alemtuzumab, an anti-CD52 humanised monoclonal antibody, causes similar profound lymphocyte depletion. A retrospective cohort study found remission of vasculitis in 60/71 (85%) refractory AAV patients after 134 mg alemtuzumab. A minority had sustained treatment-free remissions for many years, but 43/60 (72%) patients relapsed after a median of 9 months [24]. Retreatment was effective. Infective morbidity was high in those over 50 years old and with fulminant disease or vital organ failure. These results have confirmed a pathogenic role for T cells and the potential for T-cell depletion to produce sustained remissions. A trial of abatacept that blocks co-stimulation via CD28 in GPA is ongoing (ClinicalTrials.gov: NCT00468208).
Gusperimus
Gusperimus (previously termed 15-deoxyspergualin) is a synthetic analogue of the antibiotic spergualin, and suppresses lymphocyte and macrophage function and impairs neutrophil production. Forty-two of 44 (95%) patients with relapsing GPA responded to six cycles, each lasting 21 days with a 7-day washout period, of 0.5 mg/kg gusperimus, followed by azathioprine for 6 months [75]. Twenty of 44 (45%) patients achieved complete remission, although 43% of patients relapsed a median of 6 months after achieving remission. Severe adverse events occurred in 53% of patients and were predominantly leucopaenias, although they rarely led to treatment discontinuation. Extended courses of gusperimus as a maintenance agent have been shown to be effective and relatively safe [76, 77].
Autologous stem cell transplantation
There is limited experience of stem cell transplantation in vasculitis. In the 15 cases recorded in the European Blood and Marrow Transplantation database there was a response rate of over 90%, although relapses still occurred [78]. This approach is yet to be evaluated prospectively, and appropriate patient selection - avoiding those with high prior cyclophosphamide exposure and irreversible end organ damage - is crucial in view of the high risk of complications.
Churg-Strauss syndrome
Although a form of AAV, the treatment of CSS has a number of distinct features. Glucocorticoid therapy alone may be sufficient, and patients are stratified on the basis of the Five Factor Score. Cardiac, gastrointestinal and central nervous system involvement are adverse prognostic features, as is creatinine >140 μmol/l and proteinuria >1 g in 24 hours [79]. A total 93% of 72 patients with a Five Factor Score of 0 achieved remission with glucocorticoids alone, although 35% relapsed on prednisolone taper, and many required long-term maintenance glucocorticoids in order to control asthma [80].
Cyclophosphamide is the first-line therapy for those with any adverse prognostic features. This treatment is highly effective when administered at a dose of 600 mg/m2 every 2 weeks for a month and 4-weekly thereafter, with 87.5% of 48 patients achieving remission. Cumulative cyclophosphamide exposure influences relapse risk, however, with 85.7% of patients relapsing after six doses compared with 61.9% after 12 doses [81].
Regardless of disease severity, relapses are common, and current maintenance strategies for CSS are as for GPA and MPA. Rituximab has been shown to be efficacious in refractory CSS. This observation is based on only 20 reported cases, however, and there were two reports of severe bronchospasm during rituximab infusion, which have raised concerns over possible hypersensitivity reactions triggered by rituximab in CSS [82]. In both situations, antihistamines but not steroids were administered at the time of rituximab infusion, and it may be that co-administration of intravenous steroids may reduce this risk.
IL-5 is an important survival factor for eosinophils, and mepolizumab (a monoclonal antibody directed against IL-5) is a promising novel therapeutic agent. Two pilot studies, using monthly mepolizumab infusions but involving only 17 patients in total, have demonstrated the efficacy and steroid sparing potential of mepolizumab in CSS [83, 84]. Relapses were common on discontinuation of the drug, however, and larger, longer-term studies of this approach are required.
Future therapies
With the demonstration of the efficacy of rituximab in AAV, other B-cell-targeted therapies hold potential. The humanised anti-CD20 and anti-CD22 antibodies ocrelizumab, ofatumumab and epratuzumab have differences in pharmacodynamics and frequency of antiglobulin responses compared with rituximab [85]. The B-lymphocyte-stimulating protein (BLyS; also known as BAFF), a B-cell survival factor, is a further target. BLyS exerts its modulatory effect on B cells via three receptors; the transmembrane activator and calcium-modulator and cyclophilin inhibitor; B-cell maturation protein; and the BAFF receptor. Patients with active untreated GPA have significantly higher serum BLyS levels than healthy controls [86]. Also, following B-cell depletion with rituximab in SLE, BLyS levels significantly increase and then normalise as B-cell populations recover [87]. B-cell maturation protein is found on the surface of plasma cells and BLyS seems to protect lymphoid resident B cells from depletion, so BLyS inhibition may reduce plasma cell numbers [88] and enhance the susceptibility of tissue-residing B cells to depletion [89]. Belimumab is a humanised monoclonal antibody that inhibits BLyS. Efficacy and tolerability when added to standard therapy, but without a steroid-sparing effect, have recently been demonstrated in two phase III clinical trials in patients with SLE, without significant renal involvement. Belimumab may yet prove to be very useful in AAV.
Co-stimulatory blockade, which aims to prevent autoreactive T-cell activation by B cells or other antigen-presenting cells, should work in AAV. Abatacept (a CTLA4-Ig fusion protein) is under evaluation in GPA. Further biological agents targeting downstream cytokines are in trials in other autoimmune diseases, such as tocilizimab (an anti-IL-6 agent) in rheumatoid arthritis and IL-17 blocking agents in Crohn's disease. Historically, many treatments for AAV have been adopted from other autoimmune diseases, and at least some of these newer biological agents used presently for other indications will probably be tried in AAV.
Other therapeutic mechanisms of relevance to vasculitis include inhibitors of downstream inflammatory signalling pathways. After phosphorylation, spleen tyrosine kinase initiates several signalling cascades involving phosphatidylinositol-3-kinase that lead to aberrant neutrophil responses - including release of superoxide, which in turn leads to tissue damage. Inhibition of spleen tyrosine kinase in both mouse models and in human rheumatoid arthritis has exerted a positive clinical effect [90, 91]. Inhibition of phosphatidylinositol-3-kinase suppresses inflammation in mouse models of rheumatoid arthritis and SLE [92]. Although the pathology of AAV contains few immune deposits, complement depletion has abrogated experimental myeloperoxidase-ANCA renal vasculitis and is a potential target in human disease.
Discussion
The introduction of combined glucocorticoid and immunosuppressive therapy transformed the survival of patients with systemic vasculitis. Although there has been little change in the actual agents used to treat AAV, there has been considerable progress in optimising treatment regimens to minimise toxicity [93]. Despite these efforts, however, cyclophosphamide and high-dose glucocorticoids remain toxic and 10% of patients are intolerant of these therapies. In addition, relapsing disease is common, with over 50% of patients experiencing a relapse within 5 years despite continued immunosuppression. Furthermore, refractory disease affects 10 to 20% of patients and poses a significant management challenge. There is therefore a clear need for more efficacious and safer therapies.
Rituximab is the best studied biological agent in AAV. The recently published randomised controlled trials RITUXVAS and RAVE have shown that rituximab is similar to cyclophosphamide, in terms of both efficacy and safety, for induction of remission in AAV in the short term. Subgroup analysis in the RAVE trial found the efficacy of rituximab to be superior to at 6 months for patients with relapsing disease.
Potential adverse effects of any new therapy must be carefully evaluated prior to recommendation of use. Infection, particularly in the context of hypogamma-globulinaemia, is a concern following administration of rituximab. In a retrospective cohort of 105 patients with primary systemic vasculitis or SLE, 39% developed a mild infection, most commonly of the urinary tract, chest or upper respiratory tract. A total 28.5% of patients developed a severe infection, which is higher than in studies of rituximab in rheumatoid arthritis but lower than in vasculitis trials using alemtuzumab or deoxyspergualin. Patients with low IgG and IgM levels have been shown to be at significantly increased risk of infection, and immunoglobulin levels, as well as white cell counts, should be routinely monitored following rituximab administration [94]. In a retrospective series, late-onset neutropaenia occurred in 3/13 patients with GPA, all of whom developed infection needing intravenous antibiotics [95]. Further study is required into longer-term outcomes after rituximab treatment, including cardiovascular disease, infertility and malignancy.
Concern has been expressed regarding the development of progressive multifocal leucoencephalopathy, an opportunistic infection caused by the JC virus. At least 57 cases have been reported following rituximab therapy, four in patients with autoimmune disease (two SLE patients, one rheumatoid arthritis patient and one immune thrombocytopaenia patient) [96]. This association is confounded by the use of immunosuppression prior to the administration of rituximab, and the observation that progressive multifocal leucoencephalopathy is seen in patients with autoimmune disease not treated with rituximab. Unfortunately, these reports highlight the possibility of rare, severe, adverse effects that may not be detected in clinical trials. It is important that patients who receive new biological agents enter registry follow-up, to document late outcomes and side effects. Relapses are common after rituximab, and maintenance strategies are required that keep patients well yet avoid complications of prolonged B-cell depletion.
Improved patient subgrouping and tailoring of therapy will reduce unnecessary exposure to toxic therapies. Older people pose particular challenges. The peak incidence of AAV occurs in the age group of 65 to 70 years. One-year mortality is 23% in those aged over 60 years, compared with just 5% in the under-60 age group. Much of the excess mortality is due to infection. More attention is being focused on older people. The CORTAGE study, coordinated by the French Vasculitis Study Group, is comparing low-dose glucocorticoids with standard-dose glucocorticoids, with standard immunosuppression in patients over 65 years old. Newer biomarkers are required to predict treatment response, relapse risk and drug selection. Urinary monocyte chemoattractant protein-1 correlates with the degree of active vasculitis on renal biopsy and may become useful in renal vasculitis [97]. With advances in understanding of the genetic basis of disease and early results of transcriptome analysis, there is a real possibility that other clinically useful tests will become available.
Advances in classification and evidence-based therapy of vasculitis have resulted from collaborative networks both within and between countries. To date, there has been little pharmaceutical investment in these disorders. With increasing physician collaboration and industry interest in rare diseases, progress is likely to accelerate. Consensus recommendation statements have been published that will lead to harmonisation of therapy. With the establishment of registries and quality indicators there is hope that the variable quality of care that patients have received in the past will be improved.
Conclusion
Considerable progress has been made over recent years in refining the use of traditional immunosuppressive agents, namely glucocorticoids and cyclophosphamide, in systemic vasculitis, with the objective of minimising exposure and hence toxicity. Progress in classifying disease severity has provided the rationale to tailor the potency of immunosuppressive regimes to the extent of disease. This has facilitated the stratification of patients into a number of randomised controlled trials looking at newer immunosuppressive agents (Table 2).
However, the pace of change is accelerating with greater understanding of the pathogenesis of vasculitis, and a number of targeted biological agents are entering clinical use. There is an ongoing need for improved therapies for patients who are intolerant of current immunosuppression, and for those who pursue a relapsing or refractory course. A range of therapeutic agents is likely to be required for a condition as heterogeneous as systemic vasculitis. The efficacy, safety and tolerability of each agent must be thoroughly assessed, however, and the international collaborative clinical networks established in the vasculitis community will make it possible to develop this evidence base.
As the number of therapeutic options grows, the treatment of vasculitis will probably become more complex for clinicians. With the introduction of biomarkers to more accurately assess disease activity and relapse risk, however, we should aim to simplify treatment for patients by individually tailoring their immunosuppressive regimen. As disease control improves, we should focus more on longer term causes of morbidity such as cardiovascular disease and on important patient-centred outcomes, including quality of life.
Note
Autoimmune Basis of Rheumatic Diseases
This article is part of a review series on Vasculitis, edited by Cees Kallenberg, which can be found online at http://arthritis-research.com/series/vasculitis
This series forms part of a special collection of reviews covering major autoimmune rheumatic diseases, available at: http://arthritis-research.com/series/abrd
Abbreviations
- AAV:
-
ANCA-associated vasculitis
- ANCA:
-
autoantibodies to neutrophil cytoplasmic antigens
- BLyS:
-
B-lymphocyte-stimulating protein
- CSS:
-
Churg-Strauss syndrome
- GPA:
-
granulomatosis with polyangiitis
- IL:
-
interleukin
- i.v.:
-
intravenous
- IVIg:
-
intravenous immunoglobulin
- MMF:
-
mycophenolate mofetil
- MPA:
-
microscopic polyangiitis
- SLE:
-
systemic lupus erythematosus
- TNF:
-
tumour necrosis factor.
References
Jennette JC, Falk RJ, Andrassy K, Bacon PA, Churg J, Gross WL, Hagen EC, Hoffman GS, Hunder GG, Kallenberg CG, McCluskey RT, Alberto Sinico R, Rees AJ, van Es LA, Waldherr R, Wiik A: Nomenclature of systemic vasculitides. Proposal of an international consensus conference. Arthritis Rheum. 1994, 37: 187-192.
Watts R, Lane S, Hanslik T, Hauser T, Hellmich B, Koldingsnes W, Mahr A, Segelmark M, Cohen-Tervaert JW, Scott D: Development and validation of a consensus methodology for the classification of the ANCA-associated vasculitides and polyarteritis nodosa for epidemiological studies. Ann Rheum Dis. 2007, 66: 222-227.
Xiao H, Heeringa P, Hu P, Liu Z, Zhao M, Aratani Y, Maeda N, Falk RJ, Jennette JC: Antineutrophil cytoplasmic autoantibodies specific for myeloperoxidase cause glomerulonephritis and vasculitis in mice. J Clin Invest. 2002, 110: 955-963.
Feehally J, Wheeler DC, Walls J, Jones S, Lockwood CM, Savage CO: A case of microscopic polyarteritis associated with antineutrophil cytoplasmic antibodies. Clin Nephrol. 1987, 27: 214-215.
Lane SE, Watts R, Scott DG: Epidemiology of systemic vasculitis. Curr Rheumatol Rep. 2005, 7: 270-275.
Mukhtyar C, Flossmann O, Hellmich B, Bacon P, Cid M, Cohen-Tervaert JW, Gross WL, Guillevin L, Jayne D, Mahr A, Merkel PA, Raspe H, Scott D, Witter J, Yazici H, Luqmani RA, European Vasculitis Study Group (EUVAS): Outcomes from studies of antineutrophil cytoplasm antibody associated vasculitis: a systematic review by the European League Against Rheumatism systemic vasculitis task force. Ann Rheum Dis. 2008, 67: 1004-1010.
Frohnert PP, Sheps SG: Long-term follow-up of periarteritis nodosa. Am J Med. 1967, 43: 8-11.
Talar-Williams C, Hijazi YM, Walther MM, Linehan WM, Hallahan CW, Lubensky I, Kerr GS, Hoffman GS, Fauci AS, Sneller MC: Cyclophosphamide-induced cystitis and bladder cancer in patients with Wegener granulomatosis. Ann Intern Med. 1996, 124: 477-484.
Hoffman GS, Kerr GS, Leavitt RY, Hallahan CW, Lebovics RS, Travis WD, Rottem M, Fauci AS: Wegener granulomatosis: an analysis of 158 patients. Ann Intern Med. 1992, 116: 488-498.
de Leeuw K, Kallenberg C, Bijl M: Acclerated atherosclerosis in patients with Wegner's granulomatosis. Ann N Y Acad Sci. 2005, 1051: 362-371.
Little MA, Nightingale P, Verburgh CA, Hauser T, De Groot K, Savage C, Jayne D, Harper L, European Vasculitis Study (EUVAS) Group: Early mortality in systemic vasculitis: relative contribution of adverse events and active vasculitis. Ann Rheum Dis. 2010, 69: 1036-1043.
Booth AD, Almond MK, Burns A, Ellis P, Gaskin G, Neild GH, Plaisance M, Pusey CD, Jayne DR: Outcome of ANCA-associated renal vasculitis: a 5-year retrospective study. Am J Kidney Dis. 2003, 41: 776-784.
Little MA, Al-Ani B, Ren S, Al-Nuaimi H, Leite M, Alpers CE, Savage CO, Duffield JS: Anti-proteinase 3 anti-neutrophil cytoplasm autoantibodies recapitulate systemic vasculitis in mice with a humanized immune system. PloS One. 2012, 7: e28626-
Pendergraft WF 3rd, Preston GA, Shah RR, Tropsha A, Carter CW, Jennette JC, Falk RJ: Autoimmunity is triggered by cPR-3(105-201), a protein complementary to human autoantigen proteinase-3. Nat Med. 2004, 10: 72-79.
Kain R, Exner M, Brandes R, Ziebermayr R, Cunningham D, Alderson CA, Davidovits A, Raab I, Jahn R, Ashour O, Spitzauer S, Sunder-Plassmann G, Fukuda M, Klemm P, Rees AJ, Kerjaschki D: Molecular mimicry in pauciimmune focal necrotizing glomerulonephritis. Nat Med. 2008, 14: 1088-1096.
Matsubara K, Fukaya T, Miwa K, Shibayama N, Nigami H, Harigaya H, Nozaki H, Hirata T, Baba K, Suzuki T, Ishiguro A: Development of serum IgM antibodies against superantigens of Staphylococcus aureus and Streptococcus pyogenes in Kawasaki disease. Clin Exp Immunol. 2006, 143: 427-434.
Popa ER, Stegeman CA, Kallenberg CG, Tervaert JW: Staphylococcus aureus and Wegener's granulomatosis. Arthritis Res. 2002, 4: 77-79.
Stegeman CA, Tervaert JW, Sluiter WJ, Manson WL, de Jong PE, Kallenberg CG: Association of chronic nasal carriage of Staphylococcus aureus and higher relapse rates in Wegener granulomatosis. Ann Intern Med. 1994, 120: 12-17.
Popa ER, Franssen CF, Limburg PC, Huitema MG, Kallenberg CG, Tervaert JW: In vitro cytokine production and proliferation of T cells from patients with anti-proteinase 3- and antimyeloperoxidase-associated vasculitis, in response to proteinase 3 and myeloperoxidase. Arthritis Rheum. 2002, 46: 1894-1904.
Weyand CM, Goronzy JJ: Medium- and large-vessel vasculitis. N Engl J Med. 2003, 349: 160-169.
Lamprecht P: Off balance: T-cells in antineutrophil cytoplasmic antibody (ANCA)-associated vasculitides. Clin Exp Immunol. 2005, 141: 201-210.
McKinney EF, Lyons PA, Carr EJ, Hollis JL, Jayne DR, Willcocks LC, Koukoulaki M, Brazma A, Jovanovic V, Kemeny DM, Pollard AJ, Macary PA, Chaudhry AN, Smith KG: A CD8+ T cell transcription signature predicts prognosis in autoimmune disease. Nat Med. 2010, 16: 586-591.
Haubitz M, Koch KM, Brunkhorst R: Cyclosporin for the prevention of disease reactivation in relapsing ANCA-associated vasculitis. Nephrol Dial Transplant. 1998, 13: 2074-2076.
Walsh M, Chaudhry A, Jayne D: Long-term follow-up of relapsing/refractory anti-neutrophil cytoplasm antibody associated vasculitis treated with the lymphocyte depleting antibody alemtuzumab (CAMPATH-1H). Ann Rheum Dis. 2008, 67: 1322-1327.
Hellmich B, Flossmann O, Gross WL, Bacon P, Cohen-Tervaert JW, Guillevin L, Jayne D, Mahr A, Merkel PA, Raspe H, Scott DG, Witter J, Yazici H, Luqmani RA: EULAR recommendations for conducting clinical studies and/or clinical trials in systemic vasculitis: focus on anti-neutrophil cytoplasm antibody-associated vasculitis. Ann Rheum Dis. 2007, 66: 605-617.
Turnbull J, Harper L: Adverse effects of therapy for ANCA-associated vasculitis. Best Pract Res Clin Rheumatol. 2009, 23: 391-401.
Mukhtyar C, Guillevin L, Cid MC, Dasgupta B, de Groot K, Gross W, Hauser T, Hellmich B, Jayne D, Kallenberg CG, Merkel PA, Raspe H, Salvarani C, Scott DG, Stegeman C, Watts R, Westman K, Witter J, Yazici H, Luqmani R, European Vasculitis Study Group: EULAR recommendations for the management of primary small and medium vessel vasculitis. Ann Rheum Dis. 2009, 68: 310-317.
Jayne D, Rasmussen N, Andrassy K, Bacon P, Tervaert JW, Dadoniené J, Ekstrand A, Gaskin G, Gregorini G, de Groot K, Gross W, Hagen EC, Mirapeix E, Pettersson E, Siegert C, Sinico A, Tesar V, Westman K, Pusey C, European Vasculitis Study Group: A randomized trial of maintenance therapy for vasculitis associated with antineutrophil cytoplasmic autoantibodies. N Engl J Med. 2003, 349: 36-44.
de Groot K, Adu D, Savage CO: The value of pulse cyclophosphamide in ANCA-associated vasculitis: meta-analysis and critical review. Nephrol Dial Transplant. 2001, 16: 2018-2027.
de Groot K, Harper L, Jayne DR, Flores Suarez LF, Gregorini G, Gross WL, Luqmani R, Pusey CD, Rasmussen N, Sinico RA, Tesar V, Vanhille P, Westman K, Savage CO, EUVAS (European Vasculitis Study Group): Pulse versus daily oral cyclophosphamide for induction of remission in antineutrophil cytoplasmic antibody-associated vasculitis: a randomized trial. Ann Intern Med. 2009, 150: 670-680.
Harper L, Morgan MD, Walsh M, Hoglund P, Westman K, Flossmann O, Tesar V, Vanhille P, Groot KD, Luqmani R, Flores-Suarez LF, Watts R, Pusey C, Bruchfeld A, Rasmussen N, Blockmans D, Savage CO, Jayne D, on behalf of EUVAS investigators: Pulse versus daily oral cyclophosphamide for induction of remission in ANCA-associated vasculitis: long-term follow-up. Ann Rheum Dis. 2011, [Epub ahead of print]
De Groot K, Rasmussen N, Bacon PA, Tervaert JW, Feighery C, Gregorini G, Gross WL, Luqmani R, Jayne DR: Randomized trial of cyclophosphamide versus methotrexate for induction of remission in early systemic antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum. 2005, 52: 2461-2469.
de Lind van Wijngaarden RA, Hauer HA, Wolterbeek R, Jayne DR, Gaskin G, Rasmussen N, Noël LH, Ferrario F, Waldherr R, Bruijn JA, Bajema IM, Hagen EC, Pusey CD, EUVAS: Chances of renal recovery for dialysis-dependent ANCA-associated glomerulonephritis. J Am Soc Nephrol. 2007, 18: 2189-2197.
Jayne DR, Gaskin G, Rasmussen N, Abramowicz D, Ferrario F, Guillevin L, Mirapeix E, Savage CO, Sinico RA, Stegeman CA, Westman KW, van der Woude FJ, de Lind van Wijngaarden RA, Pusey CD, European Vasculitis Study Group: Randomized trial of plasma exchange or high-dosage methylprednisolone as adjunctive therapy for severe renal vasculitis. J Am Soc Nephrol. 2007, 18: 2180-2188.
Casian A: Plasma exchange for severe renal vasculitis: long-term follow-up of the MEPEX trial. Clin Exp Immunol. 2011, 164 (suppl 1): 50-61.
Szpirt WM, Heaf JG, Petersen J: Plasma exchange for induction and cyclosporine A for maintenance of remission in Wegener's granulomatosis - a clinical randomized controlled trial. Nephrol Dial Transplant. 2011, 26: 206-213.
Walsh M, Catapano F, Szpirt W, Thorlund K, Bruchfeld A, Guillevin L, Haubitz M, Merkel PA, Peh CA, Pusey C, Jayne D: Plasma exchange for renal vasculitis and idiopathic rapidly progressive glomerulonephritis: a meta-analysis. Am J Kidney Dis. 2011, 57: 566-574.
Gallagher H, Kwan JT, Jayne DR: Pulmonary renal syndrome: a 4-year, single-center experience. Am J Kidney Dis. 2002, 39: 42-47.
Klemmer PJ, Chalermskulrat W, Reif MS, Hogan SL, Henke DC, Falk RJ: Plasmapheresis therapy for diffuse alveolar hemorrhage in patients with small-vessel vasculitis. Am J Kidney Dis. 2003, 42: 1149-1153.
Smith RM, Jayne DR: B cell depletion in sytemic vasculitis. Curr Immunol Rev. 2011, 7: 415-422.
Jones RB, Ferraro AJ, Chaudhry AN, Brogan P, Salama AD, Smith KG, Savage CO, Jayne DR: A multicenter survey of rituximab therapy for refractory antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum. 2009, 60: 2156-2168.
Jones RB, Tervaert JW, Hauser T, Luqmani R, Morgan MD, Peh CA, Savage CO, Segelmark M, Tesar V, van Paassen P, Walsh D, Walsh M, Westman K, Jayne DR, European Vasculitis Study Group: Rituximab versus cyclophosphamide in ANCA-associated renal vasculitis. N Engl J Med. 2010, 363: 211-220.
Stone JH, Merkel PA, Spiera R, Seo P, Langford CA, Hoffman GS, Kallenberg CG, St Clair EW, Turkiewicz A, Tchao NK, Webber L, Ding L, Sejismundo LP, Mieras K, Weitzenkamp D, Ikle D, Seyfert-Margolis V, Mueller M, Brunetta P, Allen NB, Fervenza FC, Geetha D, Keogh KA, Kissin EY, Monach PA, Peikert T, Stegeman C, Ytterberg SR, Specks U, RAVE-ITN Research Group: Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med. 2010, 363: 221-232.
Stassen PM, Tervaert JW, Stegeman CA: Induction of remission in active anti-neutrophil cytoplasmic antibody-associated vasculitis with mycophenolate mofetil in patients who cannot be treated with cyclophosphamide. Ann Rheum Dis. 2007, 66: 798-802.
Silva F, Specks U, Kalra S, Hogan MC, Leung N, Sethi S, Fervenza FC: Mycophenolate mofetil for induction and maintenance of remission in microscopic polyangiitis with mild to moderate renal involvement - a prospective, open-label pilot trial. Clin J Am Soc Nephrol. 2010, 5: 445-453.
Hu W, Liu C, Xie H, Chen H, Liu Z, Li L: Mycophenolate mofetil versus cyclophosphamide for inducing remission of ANCA vasculitis with moderate renal involvement. Nephrol Dial Transplant. 2008, 23: 1307-1312.
European Vasculitis Study Group. [http://www.vasculitis.org/protocols/MYCYC.pdf]
Slot MC, Tervaert JW, Boomsma MM, Stegeman CA: Positive classic antineutrophil cytoplasmic antibody (C-ANCA) titer at switch to azathioprine therapy associated with relapse in proteinase 3-related vasculitis. Arthritis Rheum. 2004, 51: 269-273.
Sanders JS, Huitma MG, Kallenberg CG, Stegeman CA: Prediction of relapses in PR3-ANCA-associated vasculitis by assessing responses of ANCA titres to treatment. Rheumatology (Oxford). 2006, 45: 724-729.
Boomsma MM, Stegeman CA, van der Leij MJ, Oost W, Hermans J, Kallenberg CG, Limburg PC, Tervaert JW: Prediction of relapses in Wegener's granulomatosis by measurement of antineutrophil cytoplasmic antibody levels: a prospective study. Arthritis Rheum. 2000, 43: 2025-2033.
Finkielman JD, Merkel PA, Schroeder D, Hoffman GS, Spiera R, St Clair EW, Davis JC Jr, McCune WJ, Lears AK, Ytterberg SR, Hummel AM, Viss MA, Peikert T, Stone JH, Specks U, WGET Research Group: Antiproteinase 3 antineutrophil cytoplasmic antibodies and disease activity in Wegener granulomatosis. Ann Intern Med. 2007, 147: 611-619.
Walsh M, Merkel PA, Mahr A, Jayne D: Effects of duration of glucocorticoid therapy on relapse rate in antineutrophil cytoplasmic antibody-associated vasculitis: a meta-analysis. Arthritis Care Res (Hoboken). 2010, 62: 1166-1173.
Lapraik C, Watts R, Bacon P, Carruthers D, Chakravarty K, D'Cruz D, Guillevin L, Harper L, Jayne D, Luqmani R, Mooney J, Scott D, BSR and BHPR Standards Guidelines and Audit Working Group: BSR and BHPR guidelines for the management of adults with ANCA associated vasculitis. Rheumatology (Oxford). 2007, 46: 1615-1616.
The European Vasculitis Society. [http://www.vasculitis.org]
Pagnoux C, Mahr A, Hamidou MA, Boffa JJ, Ruivard M, Ducroix JP, Kyndt X, Lifermann F, Papo T, Lambert M, Le Noach J, Khellaf M, Merrien D, Puéchal X, Vinzio S, Cohen P, Mouthon L, Cordier JF, Guillevin L, French Vasculitis Study Group: Azathioprine or methotrexate maintenance for ANCA-associated vasculitis. N Engl J Med. 2008, 359: 2790-2803.
Metzler C, Miehle N, Manger K, Iking-Konert C, de Groot K, Hellmich B, Gross WL, Reinhold-Keller E, German Network of Rheumatic Diseases: Elevated relapse rate under oral methotrexate versus leflunomide for maintenance of remission in Wegener's granulomatosis. Rheumatology (Oxford). 2007, 46: 1087-1091.
Stegeman CA, Tervaert JW, de Jong PE, Kallenberg CG: Trimethoprim-sulfamethoxazole (co-trimoxazole) for the prevention of relapses of Wegener's granulomatosis. Dutch Co-Trimoxazole Wegener Study Group. N Engl J Med. 1996, 335: 16-20.
Nowack R, Gobel U, Klooker P, Hergesell O, Andrassy K, van der Woude FJ: Mycophenolate mofetil for maintenance therapy of Wegener's granulomatosis and microscopic polyangiitis: a pilot study in 11 patients with renal involvement. J Am Soc Nephrol. 1999, 10: 1965-1971.
Hiemstra TF, Walsh M, Mahr A, Savage CO, de Groot K, Harper L, Hauser T, Neumann I, Tesar V, Wissing KM, Pagnoux C, Schmitt W, Jayne DR, European Vasculitis Study Group (EUVAS): Mycophenolate mofetil vs azathioprine for remission maintenance in antineutrophil cytoplasmic antibody-associated vasculitis: a randomized controlled trial. JAMA. 2010, 304: 2381-2388.
Holle JU, Dubrau C, Herlyn K, Heller M, Ambrosch P, Noelle B, Reinhold-Keller E, Gross WL: Rituximab for refractory granulomatosis with polyangiitis (Wegener's granulomatosis): comparison of efficacy in granulomatous versus vasculitic manifestations. Ann Rheum Dis. 2012, 71: 327-333.
Jayne DR, Esnault VL, Lockwood CM: ANCA anti-idiotype antibodies and the treatment of systemic vasculitis with intravenous immunoglobulin. J Autoimmun. 1993, 6: 207-219.
Richter C, Schnabel A, Csernok E, De Groot K, Reinhold-Keller E, Gross WL: Treatment of anti-neutrophil cytoplasmic antibody (ANCA)-associated systemic vasculitis with high-dose intravenous immunoglobulin. Clin Exp Immunol. 1995, 101: 2-7.
Rossi F, Bellon B, Vial MC, Druet P, Kazatchkine MD: Beneficial effect of human therapeutic intravenous immunoglobulins (IVIg) in mercuric-chloride-induced autoimmune disease of Brown-Norway rats. Clin Exp Immunol. 1991, 84: 129-133.
Brooks CJ, King WJ, Radford DJ, Adu D, McGrath M, Savage CO: IL-1 beta production by human polymorphonuclear leucocytes stimulated by anti-neutrophil cytoplasmic autoantibodies: relevance to systemic vasculitis. Clin Exp Immunol. 1996, 106: 273-279.
Jayne DR, Chapel H, Adu D, Misbah S, O'Donoghue D, Scott D, Lockwood CM: Intravenous immunoglobulin for ANCA-associated systemic vasculitis with persistent disease activity. QJM. 2000, 93: 433-439.
Martinez V, Cohen P, Pagnoux C, Vinzio S, Mahr A, Mouthon L, Sailler L, Delaunay C, Sadoun A, Guillevin L, French Vasculitis Study Group: Intravenous immunoglobulins for relapses of systemic vasculitides associated with antineutrophil cytoplasmic autoantibodies: results of a multicenter, prospective, open-label study of twenty-two patients. Arthritis Rheum. 2008, 58: 308-317.
Little MA, Bhangal G, Smyth CL, Nakada MT, Cook HT, Nourshargh S, Pusey CD: Therapeutic effect of anti-TNF-alpha antibodies in an experimental model of anti-neutrophil cytoplasm antibody-associated systemic vasculitis. J Am Soc Nephrol. 2006, 17: 160-169.
Wegener's Granulomatosis Etanercept Trial (WGET) Research Group: Etanercept plus standard therapy for Wegener's granulomatosis. N Engl J Med. 2005, 352: 351-361.
Stone JH, Holbrook JT, Marriott MA, Tibbs AK, Sejismundo LP, Min YI, Specks U, Merkel PA, Spiera R, Davis JC, St Clair EW, McCune WJ, Ytterberg SR, Allen NB, Hoffman GS, Wegener's Granulomatosis Etanercept Trial Research Group: Solid malignancies among patients in the Wegener's Granulomatosis Etanercept Trial. Arthritis Rheum. 2006, 54: 1608-1618.
Silva F, Seo P, Schroeder DR, Stone JH, Merkel PA, Hoffman GS, Spiera R, Sebastian JK, Davis JC Jr, St Clair EW, Allen NB, McCune WJ, Ytterberg SR, Specks U, Wegener's Granulomatosis Etanercept Trial Research Group: Solid malignancies among etanercept-treated patients with granulomatosis with polyangiitis (Wegener's): long-term followup of a multicenter longitudinal cohort. Arthritis Rheum. 2011, 63: 2495-2503.
Booth A, Harper L, Hammad T, Bacon P, Griffith M, Levy J, Savage C, Pusey C, Jayne D: Prospective study of TNFα blockade with infliximab in anti-neutrophil cytoplasmic antibody-associated systemic vasculitis. J Am Soc Nephrol. 2004, 15: 717-721.
Morgan MD, Drayson MT, Savage CO, Harper L: Addition of infliximab to standard therapy for ANCA-associated vasculitis. Nephron Clin Pract. 2010, 117: c89-c97.
Laurino S, Chaudhry A, Booth A, Conte G, Jayne D: Prospective study of TNFalpha blockade with adalimumab in ANCA-associated systemic vasculitis with renal involvement. Nephrol Dial Transplant. 2010, 25: 3307-3314.
Schmitt WH, Hagen EC, Neumann I, Nowack R, Flores-Suarez LF, van der Woude FJ: Treatment of refractory Wegener's granulomatosis with antithymocyte globulin (ATG): an open study in 15 patients. Kidney Int. 2004, 65: 1440-1448.
Flossmann O, Baslund B, Bruchfeld A, Tervaert JW, Hall C, Heinzel P, Hellmich B, Luqmani RA, Nemoto K, Tesar V, Jayne DR: Deoxyspergualin in relapsing and refractory Wegener's granulomatosis. Ann Rheum Dis. 2009, 68: 1125-1130.
Flossmann O, Jayne DR: Long-term treatment of relapsing Wegener's granulomatosis with 15-deoxyspergualin. Rheumatology (Oxford). 2010, 49: 556-562.
Schmitt WH, Birck R, Heinzel PA, Göbel U, Choi M, Warnatz K, Peter HH, van der Woude FJ: Prolonged treatment of refractory Wegener's granulomatosis with 15-deoxyspergualin: an open study in seven patients. Nephrol Dial Transplant. 2005, 20: 1083-1092.
Daikeler T, Kotter I, Bocelli Tyndall C, Apperley J, Attarbaschi A, Guardiola P, Gratwohl A, Jantunen E, Marmont A, Porretto F, Musso M, Maurer B, Rinaldi N, Saccardi R, Tyndall A, EBMT Autoimmune Diseases Working Party: Haematopoietic stem cell transplantation for vasculitis including Behcet's disease and polychondritis: a retrospective analysis of patients recorded in the European Bone Marrow Transplantation and European League Against Rheumatism databases and a review of the literature. Ann Rheum Dis. 2007, 66: 202-207.
Pagnoux C, Guilpain P, Guillevin L: Churg-Strauss syndrome. Curr Opin Rheumatol. 2007, 19: 25-32.
Ribi C, Cohen P, Pagnoux C, Mahr A, Arène JP, Lauque D, Puéchal X, Letellier P, Delaval P, Cordier JF, Guillevin L, French Vasculitis Study Group: Treatment of Churg-Strauss syndrome without poor-prognosis factors: a multicenter, prospective, randomized, open-label study of seventy-two patients. Arthritis Rheum. 2008, 58: 586-594.
Cohen P, Pagnoux C, Mahr A, Arène JP, Mouthon L, Le Guern V, André MH, Gayraud M, Jayne D, Blöckmans D, Cordier JF, Guillevin L, French Vasculitis Study Group: Churg-Strauss syndrome with poor-prognosis factors: a prospective multicenter trial comparing glucocorticoids and six or twelve cyclophosphamide pulses in forty-eight patients. Arthritis Rheum. 2007, 57: 686-693.
Guerry MJ, Brogan P, Bruce IN, D'Cruz DP, Harper L, Luqmani R, Pusey CD, Salama AD, Scott DG, Savage CO, Watts RA, Jayne DR: Recommendations for the use of rituximab in anti-neutrophil cytoplasm antibody-associated vasculitis. Rheumatology. 2011, 51: 634-643.
Kim S, Marigowda G, Oren E, Israel E, Wechsler ME: Mepolizumab as a steroid-sparing treatment option in patients with Churg-Strauss syndrome. J Allergy Clin Immunol. 2010, 125: 1336-1343.
Moosig F, Gross WL, Herrmann K, Bremer JP, Hellmich B: Targeting interleukin-5 in refractory and relapsing Churg-Strauss syndrome. Ann Internal Med. 2011, 155: 341-343.
Dorner T, Kaufmann J, Wegener WA, Teoh N, Goldenberg DM, Burmester GR: Initial clinical trial of epratuzumab (humanized anti-CD22 antibody) for immunotherapy of systemic lupus erythematosus. Arthritis Res Ther. 2006, 8: R74-
Krumbholz M, Specks U, Wick M, Kalled SL, Jenne D, Meinl E: BAFF is elevated in serum of patients with Wegener's granulomatosis. J Autoimmun. 2005, 25: 298-302.
Cambridge G, Stohl W, Leandro MJ, Migone TS, Hilbert DM, Edwards JC: Circulating levels of B lymphocyte stimulator in patients with rheumatoid arthritis following rituximab treatment: relationships with B cell depletion, circulating antibodies, and clinical relapse. Arthritis Rheum. 2006, 54: 723-732.
O'Connor BP, Raman VS, Erickson LD, Cook WJ, Weaver LK, Ahonen C, Lin LL, Mantchev GT, Bram RJ, Noelle RJ: BCMA is essential for the survival of long-lived bone marrow plasma cells. J Exp Med. 2004, 199: 91-98.
Gong Q, Ou Q, Ye S, Lee WP, Cornelius J, Diehl L, Lin WY, Hu Z, Lu Y, Chen Y, Wu Y, Meng YG, Gribling P, Lin Z, Nguyen K, Tran T, Zhang Y, Rosen H, Martin F, Chan AC: Importance of cellular microenvironment and circulatory dynamics in B cell immunotherapy. J Immunol. 2005, 174: 817-826.
Pine PR, Chang B, Schoettler N, Banquerigo ML, Wang S, Lau A, Zhao F, Grossbard EB, Payan DG, Brahn E: Inflammation and bone erosion are suppressed in models of rheumatoid arthritis following treatment with a novel Syk inhibitor. Clin Immunol. 2007, 124: 244-257.
Weinblatt ME, Kavanaugh A, Burgos-Vargas R, Dikranian AH, Medrano-Ramirez G, Morales-Torres JL, Murphy FT, Musser TK, Straniero N, Vicente-Gonzales AV, Grossbard E: Treatment of rheumatoid arthritis with a Syk kinase inhibitor: a twelve-week, randomized, placebo-controlled trial. Arthritis Rheum. 2008, 58: 3309-3318.
Hirsch E, Katanaev VL, Garlanda C, Azzolino O, Pirola L, Silengo L, Sozzani S, Mantovani A, Altruda F, Wymann MP: Central role for G protein-coupled phosphoinositide 3-kinase gamma in inflammation. Science. 2000, 287: 1049-1053.
Holle JU, Gross WL, Latza U, Nölle B, Ambrosch P, Heller M, Fertmann R, Reinhold-Keller E: Improved outcome in 445 patients with Wegener's granulomatosis in a German vasculitis center over four decades. Arthritis Rheum. 2011, 63: 257-266.
Catapano F, Jones R, Walsh M, Chaudhry A, Jayne D: Safety and tolerability of rituximab in vasculitis and systemic lupus erythematosus. APMIS. 2009, 117: 67-180.
Tesfa D, Ajeganova S, Hagglund H, Sander B, Fadeel B, Hafström I, Palmblad J: Late-onset neutropenia following rituximab therapy in rheumatic diseases: association with B lymphocyte depletion and infections. Arthritis Rheum. 2011, 63: 2209-2214.
Harris HE: Progressive multifocal leucoencephalopathy in a patient with systemic lupus erythematosus treated with rituximab. Rheumatology (Oxford). 2008, 47: 224-225.
Tam FW, Sanders JS, George A, Hammad T, Miller C, Dougan T, Cook HT, Kallenberg CG, Gaskin G, Levy JB, Pusey CD: Urinary monocyte chemoattractant protein-1 (MCP-1) is a marker of active renal vasculitis. Nephrol Dial Transplant. 2004, 19: 2761-2768.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Rights and permissions
About this article
Cite this article
Smith, R.M., Jones, R.B. & Jayne, D.R. Progress in treatment of ANCA-associated vasculitis . Arthritis Res Ther 14, 210 (2012). https://doi.org/10.1186/ar3797
Published:
DOI: https://doi.org/10.1186/ar3797