
Abstract
Interstitial lung disease is a common manifestation of autoimmune myositis that confers significant morbidity and mortality. The vulnerability of the lung may offer insight into the etiology of this autoimmune disease. The frequency and patterns of lung injury vary based on the autoantibody. Antibodies against the aminoacyl-tRNA synthetases and melanoma differentiation-induced gene-5 are frequently associated with interstitial lung disease. Although the mechanisms underlying these associations have not been fully elucidated, emerging data highlight the importance of autoantigen expression and conformation in the target tissue (lung and muscle, in this case), as well as identifying relevant amplifying pathways (such as regeneration).
Similar content being viewed by others

Pathology of the lung in myositis
Epidemiology of lung involvement in autoimmune myositis
The interstitial lung diseases (ILDs) encompass a large number of distinct disorders that are characterized by inflammation and/or fibrosis involving the lung parenchyma. These disorders result from a myriad of potential etiologies including medications, environmental exposures and autoimmune diseases such as myositis. Regardless of etiology, these lung injuries result in a limited number of patterns that are classified based on pathologic appearance into several major subgroups (Table 1). In general, the pathology impacts prognosis regardless of the etiology of the ILD. The target of injury includes the critical alveolar-capillary interface responsible for gas exchange. As a group, therefore, ILDs result in com-promised oxygen absorption as well as decreased lung compliance and increased work of breathing. The clinical manifestation of ILD is typically dyspnea on exertion, cough and fatigue.
Lung involvement in autoimmune myositis was first systematically reviewed in 1974 [1], although it had been described in individuals and case series since the mid-1960s. Since that time, there has been an increasing appreciation of the prevalence and clinical significance of ILD. Recent studies suggest that ILD complications occur in 20 to 65% of individuals with myositis (reviewed in [2]). The wide variation in prevalence reflects in part the lack of a standard approach to screening for ILD in newly diagnosed patients with myositis. A recent study of 90 histidyl-tRNA synthetase (Jo-1) patients found ILD present in 86% [3]. Alanyl-tRNA synthetase (PL-12) is similarly associated with ILD in 90% or more of patients based on two recent case series [4, 5]. The apparently increasing rates of ILD in patients with myositis represent a combination of improved diagnostic techniques and increasing awareness of ILD as a presenting symptom of autoimmune myositis. The mortality associated with ILD has been anecdotally described since the earliest reports. Prospective long-term studies suggest a 40% increase in mortality attributable to ILD [6]. ILD is thus both a common and a highly lethal manifestation of auto-immune myositis.
Cellular and cytokine makeup of the normal and injured lung
The normal alveolus has a simple cellular makeup with resident alveolar type I cells arranged along a basement membrane lining the alveolar airspaces. Type II cells occupy the corners of the alveoli and are the source of type I cells in the event of cellular damage. Surrounding the alveoli are fine capillary networks lined by endothelial cells. The alveolar-capillary interface is a dynamic barrier. Inflammatory cells including neutrophils, lymphocytes and macrophages travel through the capillary vessels and transit into the alveolar air space. Mature fibroblasts and circulating fibrocytes can also transit through the alveolar basement membrane. Resident antigen-presenting cells, including dendritic cells, serve as key sensors for danger signaling and are principally responsible for inflammatory cell influx.
Because the lung is in constant contact with the exterior, much like the skin, it is in a permanent state of cellular injury and regeneration. Under normal circumstances, type I cells are generated by type II cells and injury does not result in fibrosis. In the setting of more severe injury, or in certain vulnerable hosts, injured type I cells are instead replaced by fibroblasts, possibly from marrow-derived circulating fibrocytes. The constant cycle of injury and repair in the lung creates a unique opportunity for the development of neoantigens, which is compounded by the frequency of viral encounters in the lung. Indeed, there have been a number of studies that associate the presence of ILD with specific viral pathogens [7–9].
Patterns of lung injury in myositis
A number of pathologic patterns are associated with myositis ILD. The most common patterns documented by surgical lung biopsy in a recent study of Jo-1-associated ILD showed diffuse alveolar damage in 55%, usual interstitial pneumonia in 45% and nonspecific interstitial pneumonia in 14% [3]. Interestingly, the diagnoses based on chest CT patterns rather than tissue biopsy favored a preponderance of nonspecific interstitial pneumonia (50%) while only 30% of CTs were suggestive of usual interstitial pneumonia. This high prevalence of diffuse alveolar damage on surgical biopsy probably reflects a tendency toward performing biopsies in patients who are failing therapy or who present with acute interstitial pneumonia as a first manifestation of myositis. In a recent multicenter series of patients with PL-12, 45% of patients had either biopsy or CT consistent with usual interstitial pneumonia, 16% with nonspecific interstitial pneumonia and 16% with cryptogenic organizing pneumonia [4]. The pattern of lung injury in myositis ILD thus appears to span the spectrum, but with a higher frequency of usual interstitial pneumonia than is commonly seen in other autoimmune ILDs.
Unique clinical patterns associated with interstitial lung disease
While myositis is traditionally defined by the presence of muscle or skin inflammation, it is increasingly apparent that a subset of patients with myositis-associated antibodies presents either initially or uniquely with ILD. This pattern has been highlighted by patients with clinically amyopathic dermatomyositis (CADM), with rapidly progressive and frequently fatal ILD (reviewed in [10]). These patients might previously have been considered to have idiopathic acute interstitial pneumonia. However, increasing awareness of CADM and the discovery of additional autoantibodies associated with this clinical presentation (see below) will allow more accurate ascertainment of this presentation. The occurrence of such a dramatic pulmonary-centric presentation raises the question of whether the primary site of injury might in fact be the lung rather than the muscle.
Prominent autoantibody specificities found in patients with myositis and lung disease
Across the spectrum of rheumatic autoimmune diseases, distinct patterns of antibody reactivity are strongly associated with specific clinical subgroups - a feature that makes autoantibodies of diagnostic and prognostic use. For example, autoantibodies against topoisomerase I are found in patients with diffuse scleroderma and lung disease, whereas antibodies against centromere protein B are found in patients with limited scleroderma and are predictive of digit loss. In patients with idiopathic inflammatory myopathies, such associations are also found. A well-recognized example of this is the strong association of myositis-specific anti-aminoacyl-tRNA synthetase (anti-ARS) antibodies with ILD. Interestingly, emerging data from another recently described dermatomyositis (DM)-specific antibody - melanoma differentiation-induced gene-5 (MDA-5) - indicates that it is also associated with ILD. Details of these two distinct myositis-specific autoantibody types are discussed below.
Aminoacyl-tRNA synthetase autoantibodies
ARSs are ubiquitously expressed, highly conserved cytoplasmic enzymes that perform an essential cellular function: they catalyze the esterification of specific amino acids to their cognate tRNA, thus forming an aminoacyl-tRNA complex. There is a distinct ARS for each of the 20 amino acids. The complex transfers the correct amino acid to an elongating polypeptide chain as the ribosome transcribes the coding sequence of mRNA into the appropriate protein. To date, autoantibodies to eight of the ARS enzymes have been identified; these include antibodies against histidyl-tRNA synthetase (Jo-1) [11], alanyl-tRNA synthetase (PL-12) [12], threonyl-tRNA synthetase (PL-7) [13], glycyl-tRNA synthetase (EJ) [14], isoleucyl-tRNA synthetase (OJ) [14], asparaginyl-tRNA synthetase (KS) [15], phenylalanyl-tRNA synthetase (Zo) [16] and tyrosyl-tRNA synthetase (YRS) [17]. Of these, anti-Jo-1 is by far the most frequently found specificity, occurring in 15 to 20% of myositis patients, whereas the other anti-ARS autoantibodies are found in 1 to 4% of myositis patients [18]. Although anti-ARS antibodies may be found together with other antibodies in some patients (for example, frequently with Ro52), it is noteworthy and intriguing that individuals with anti-ARS antibodies rarely target more than a single synthetase.
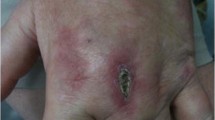
Anti-ARS antibodies are associated with a distinct clinical syndrome referred to as the antisynthetase syndrome. Characteristic features (shown in Figure 1) include myositis, ILD, nonerosive arthritis, Raynaud's phenomenon, fever and mechanic's hands (hyperkeratotic lesions along the radial and palmar aspects of the fingers) [19]. While clinical similarities between patients with different ant-ARSs are striking, noteworthy distinctions between these patient groups have been described.

Characteristic features of antisynthetase syndrome.
For example, Kalluri and colleagues recently reported a detailed analysis of the clinical features of 31 subjects with anti-PL-12 antibodies [4]. They found a higher frequency of ILD in these patients compared with those with Jo-1 antibodies (90% versus 50 to 75%), whereas the association with myositis/myalgia was less strong (52% versus 90%). These findings are consistent with those reported by other workers, including a large study of Japanese myositis patients with anti-ARS antibodies performed by Yoshifuji and colleagues [20]. In addition to the clinical experience linking anti-ARS antibodies with lung pathology, a mouse model of anti-Jo-1 immune response demonstrates predominant lung disease [21]. This study as well as others correlating anti-Jo1 antibody levels with disease activity [22] raise the possibility that anti-ARS antibodies are pathogenic. However, this remains to be resolved. For years it has been noted that a significant proportion of myositis patients have ILD without autoantibodies [2], raising the possibility of additional specificities that are as yet unidentified. Recent findings show that this may indeed be the case: new myositis-specific antibodies continue to be described [23], including MDA-5 (see below), which was also recently found to associate with ILD. Of interest, these antibodies are detected in patients that do not have anti-ARS antibodies.
Melanoma differentiation-induced gene-5 antibodies
MDA-5 antibodies were first described in 2005 by Sato and colleagues in a study of 103 adult Japanese myositis patients (seen at Keio University in Tokyo) [24]. At the time, these antibodies were named anti-CADM-140 because they recognized a 140 kDa protein in patients and were associated with CADM; the antibodies were found in 8/42 (19%) DM patients and in 0/61 (0%) poly-myositis patients, with all eight of the antibody-positive patients having CADM. In this study, the authors reported that the presence of these antibodies was associated with rapidly progressive ILD, with 50% of the antibody-positive patients having ILD compared with 6% in the antibody-negative group [24]. These authors subsequently identified the 140 kDa autoantigen as a cytoplasmic RNA helicase encoded by MDA-5 [25]. This protein is a known RNA virus receptor, thus potentially providing a mechanistic linkage between the development of myositis ILD and viral infection [26].
These findings have been validated by those of Nakashima and colleagues, who independently and almost simultaneously identified the CADM-140 autoantigen as MDA-5 [27]. They studied an adult Japanese patient cohort seen at Kyoto University Hospital. Eighty-four myositis patients were studied, and anti-MDA-5 antibodies were found in 13/37 (35%) and 0/47 (0%) of DM patients and polymyositis patients, respectively. Of those patients with the antibody, 2/13 had typical DM and 11/13 had CADM. Interestingly, 54% of the anti-MDA-5-positive group had rapidly progressive ILD compared with 4% of the anti-MDA-5-negative sera, consistent with the findings of Sato and colleagues [24].
Gono and colleagues evaluated the clinical features of patients with MDA-5 antibodies and compared them with those of patients with anti-ARS antibodies in a retrospective study (at Tokyo Women's Medical University) [28]. They identified 65 patients with DM/ CADM, 46 (71%) of whom also had ILD. Amongst this latter group, 24/46 (52%) had either anti-MDA-5 or anti-ARS antibodies. Interesting differences were noted when clinical features of the 14 anti-MDA-5-positive patients were compared with the 10 anti-ARS-positive patients. In the former group, the frequency of acute/subacute interstitial pneumonia was significantly higher (71% compared with 20%), the cumulative 60-month survival rate was lower (63% versus 100%), serum ferritin levels were higher (680 ng/ml versus 171 ng/ml) and the median creatinine kinase values were lower (198 IU compared with 1,513 IU). While the numbers in the groups are small, making caution necessary in drawing conclusions, these findings indicate that serum ferritin levels and MDA-5 antibodies may be useful indicators for the early diagnosis of acute/subacute interstitial pneumonia with DM. It is also noteworthy that all three of the above studies showed that MDA-5 antibodies are found exclusively in patients with DM.
New data continue to expand the clinical phenotype associated with MDA-5 autoantibodies. For example, findings from a recent study of DM patients seen at the Stanford University Department of Dermatology out-patient clinic [29] show that the presence of anti-MDA-5 antibodies is associated with very distinctive cutaneous ulcerations and palmar papules. In this cohort, MDA-5 antibodies also identified a population of DM patients at increased risk for rapid lung disease (22.2% in the anti-MDA-5-positive DM group versus 4.5% in the anti-MDA-5-negative group). Identifying antibodies against MDA-5 in DM patients is therefore of clinical importance, as this specificity associates with high risk of ILD - including rapidly progressing ILD, which can lead to patient mortality, as well as cutaneous ulcerations and palmar papules. To date, there are no commercial assays that can be used to identify these antibodies in the clinical setting. Additionally, as mentioned above, it is noteworthy that other as yet unidentified autoantibodies probably do exist in patients with idiopathic inflammatory myopathies and ILD that are at present classified as antibody-negative. Identification of these antibodies may well yield further fine specificity of use for predicting and monitoring distinct clinical subgroups within the spectrum of DM patients with lung disease. Additional studies are also needed to understand why antigens as apparently unrelated as ARS and MDA-5 are such specific targets of the autoimmune response in this disease spectrum. Interestingly, several recent reports link anti-polymyositis Scl antibody with a similar phenotype to that of the antisynthetase syndrome [30, 31]. The mechanism underlying such similarity is unclear, but might include a shared HLA type or other shared genetic factors.
Mechanisms regulating expression of ARS and MDA-5
While the mechanisms underlying the association of immune responses against specific, ubiquitously expressed autoantigens with distinct clinical disease phenotypes remain undefined, emerging data are focusing attention on unique expression patterns of the myositis-specific autoantigens themselves. Thus, although most autoantigens are ubiquitously expressed, recent in vivo findings show that significant differences in expression levels exist in unique microenvironments. These concepts are well illustrated by the examples of ARS and MDA-5, as described below. Such findings of local elevated expression levels of specific antigens are of great potential importance in understanding why certain antigens are targeted in specific autoimmune diseases, and why the feed-forward loop of autoimmunity is propagated so effectively.
Elevated levels of ARS are expressed in normal lung and regenerating muscle cells
Important new insights into the in vivo expression of autoantigens in unique microenvironments within relevant target tissues themselves were made several years ago [32]. While these studies indicate a probable role for damaged muscle in initiating/propagating myositis, they do not address the role of lung; however, the emerging principles may well be applicable to the lung, and are therefore reviewed briefly below. The authors performed immunoblots on lysates made from control and myositis patient muscle biopsies. Unexpectedly, the data showed very low expressed levels of myositis autoantigens such as the ARSs in control muscle. In contrast, these autoantigens were robustly expressed in lysates made from muscle biopsies obtained from myositis patients. Immunohistochemical studies confirmed that the enhanced autoantigen expression was detected in muscle cells themselves, as well as in infiltrating inflammatory cells. Importantly, further characterization of the muscle cell staining confirmed that the enhanced autoantigen expression was found in the regenerating muscle cells, and not in mature myotubes. These findings demonstrate that mature, healthy muscle is probably not the target of autoimmunity. Instead, injured muscle in which re-generating muscle cells (expressing high levels of mysoitis-specific autoantigens such as the ARS molecules) are replacing injured cells may be the primary target. The direction of the immune response against regenerating muscle cells, critical participants in the healing of muscle injury, probably enables a feed-forward cycle where tissue damage drives antigen expression, immune response and additional tissue damage.
Little is currently known about autoantigen expression in lung. The limited data examining levels of myositis-specific autoantigens in the lung available to date provide tantalizing evidence that levels of Jo-1 are increased in normal lung relative to the two other normal tissues that were studied (muscle and breast), and that Jo-1 expression in normal lung is robust whereas expression of other myositis autoantigens is minimal (Mi-2 and the catalytic subunit of DNA-dependent protein kinase were the other two autoantigens examined) [32]. Future studies will be important to evaluate whether and which cells in the lung express altered myositis autoantigen levels. The regeneration of alveolar type I cells from type II cells mirrors the regeneration of myocytes from muscle satellite cells in some aspects, thus providing a potentially analogous model for antigen upregulation in the lung.
In addition to autoantigen levels, the autoantigen structure plays a critical role in shaping the immune response. Changes in autoantigen structure in response to immune effector pathways are particularly relevant to autoimmune tissue injury. Noteworthy in this regard are the structural changes (including generation of novel cleavage fragments) induced when target cells are killed by the cytotoxic lymphocyte granule exocytosis pathway. Granzyme B is a protease that is abundantly expressed in cytotoxic lymphocyte granules. This essential host defense mechanism is mediated by cytotoxic lympho-cytes and natural killer cells, and has been documented to exist in myositis tissues [33]. Many myositis auto-antigens (including Jo-1) are cleaved by granzyme B [34]. Interestingly, a novel form of Jo-1 that is more susceptible to cleavage by granzyme B in the human lung has been demonstrated [35], raising the possibility that novel Jo-1 cleavage fragments might be generated uniquely in the lung. This would result in distinct fragments and consequently cryptic epitope generation during antigen processing.
Of note, the cleavable Jo-1 conformation was not detected in muscle, thus focusing attention on the lung as the possible initiation site for anti-ARS autoimmunity associated with ILD. In this scenario, muscle might become targeted at a later stage, potentially in the setting of later muscle damage, where muscle regeneration induces high levels of Jo-1 - and other myositis-specific autoantigen - expression and autoimmune injury. It is of some interest that Jo-1 has been shown to trigger chemokine receptors on T cells and immature dendritic cells [36], raising a possible mechanistic association between increased antigen expression and inflammatory cell recruitment. Whether cleavage of Jo-1 enhances its effect on chemokine receptors is not currently known. Further studies examining the expression and cleavability in both the lung and muscle of other ARS molecules and MDA-5 are needed to provide additional context and interpretation.
Substantial evidence has accumulated implicating type I interferons in the pathogenesis of DM [37]. It is noteworthy that both MDA-5 and Ro52 - frequent targets in DM - are type I interferon inducible. Evaluating whether MDA-5 expression is increased in target tissues in DM (lung, muscle and skin) with the ARS/ILD disease spectrum will be informative. Such observations may suggest a potential role for anti-interferon therapies in MDA-5-associated lung disease.
Whether autoantibodies have a direct role in generating and augmenting tissue injury in myositis associated ILD is very unclear, but there are no available data to support this role. Rather, it is more likely that autoantibody specificities reflect the antigen driving a T-cell response, which more directly results in tissue injury. Data about antigen specific T cells in human ILD in DM/poly-myositis are not available, but there are several intriguing studies suggesting that the fulminant onset of ILD associated with myositis is linked to the increased CD8 T cells in the lung that express cytotoxic properties [38, 39]. There are some interesting, although anecdotal, data that suggest treatment of such patients with the T-cell-directed agents cyclosporine or FK506 may have a beneficial effect [40, 41]. Defining antigen-specific CD4 and CD8 T cells in the lung of patients with myositis and ILD may provide very important insights into the mechanism and possibly therapy in these diseases.
Models for targeting the lung in myositis
Two distinct models currently exist, which differ in the initiation site, that may explain the targeting of the lung in patients with myositis.
One scenario is that the lung is the primary target of the autoimmune response. Here, the presence of supra-threshold levels of Jo-1 or MDA-5 potentially during infection or malignancy may produce novel autoantigen forms. In the setting of an appropriate immune response, autoreactivity to these novel antigens may be produced. In the setting of subsequent muscle injury (for example, muscle overuse or viral infection), the preformed immune response might be directed against regenerating muscle cells that express high levels of myositis auto-antigens, thereby focusing the subsequent amplification loop onto muscle.
An alternate model would invoke muscle as the primary target, with the lung being involved as a bystander. In this circumstance, enhanced expression of myositis autoantigens could be caused by muscle damage with increased numbers of regenerating cells, or as a consequence of interferon exposure. The subsequent immune response may become focused on the lung, where relevant myositis autoantigens are robustly expressed.
Conclusion
The striking targeting of the lung in autoimmune myopathies that are associated with specific autoantibody responses (noteworthy examples include MDA-5 and the ARS family of antigens) provides important pathogenic insights. Understanding antigen expression, conformations and modifications in the lung is key to defining relevant mechanisms. Whether involvement of the lung in this disease spectrum occurs because it is the primary target or the secondary target of the autoimmune response is currently unknown, but identifying amplifying pathways (for example, interferon or regeneration) may have therapeutic implications.
Note
Autoimmune Basis of Rheumatic Diseases
This article is part of a series on Myositis, edited by Ingrid Lundberg, which can be found online at http://arthritis-research.com/series/myositis
This series forms part of a special collection of reviews covering major autoimmune rheumatic diseases, available at: http://arthritis-research.com/series/abrd
Abbreviations
- ARS:
-
aminoacyl-tRNA synthetase
- CADM:
-
clinically amyopathic dermatomyositis
- CT:
-
computed tomography
- DM:
-
dermatomyositis
- ILD:
-
interstitial lung disease
- Jo-1:
-
histidyl-tRNA synthetase
- MDA-5:
-
melanoma differentiation-induced gene-5
- PL-12:
-
alanyl-tRNA synthetase.
References
Frazier AR, Miller RD: Interstitial pneumonitis in association with polymyositis and dermatomyositis. Chest. 1974, 65: 403-407. 10.1378/chest.65.4.403.
Labirua A, Lundberg IE: Interstitial lung disease and idiopathic inflammatory myopathies: progress and pitfalls. Curr Opin Rheumatol. 2010, 22: 633-638. 10.1097/BOR.0b013e32833f1970.
Richards TJ, Eggebeen A, Gibson K, Yousem S, Fuhrman C, Gochuico BR, Fertig N, Oddis CV, Kaminski N, Rosas IO, Ascherman DP: Characterization and peripheral blood biomarker assessment of anti-Jo-1 antibody-positive interstitial lung disease. Arthritis Rheum. 2009, 60: 2183-2192. 10.1002/art.24631.
Kalluri M, Sahn SA, Oddis CV, Gharib SL, Christopher-Stine L, Danoff SK, Casciola-Rosen L, Hong G, Dellaripa PF, Highland KB: Clinical profile of anti-PL-12 autoantibody. Cohort study and review of the literature. Chest. 2009, 135: 1550-1556. 10.1378/chest.08-2233.
Hervier B, Wallaert B, Hachulla E, Adoue D, Lauque D, Audrain M, Camara B, Fournie B, Couret B, Hatron PY, Dubucquoi S, Hamidou M: Clinical manifestations of anti-synthetase syndrome positive for anti-alanyl-tRNA synthetase (anti-PL12) antibodies: a retrospective study of 17 cases. Rheumatology (Oxford). 2010, 49: 972-976. 10.1093/rheumatology/kep455.
Cottin V, Thivolet-Bejui F, Reynaud-Gaubert M, Cadranel J, Delaval P, Ternamian PJ, Cordier JF, Groupe d'Etudes et de Recherche sur les Maladies 'Orphelines' Pulmonaires: Interstitial lung disease in amyopathic dermatomyositis, dermatomyositis and polymyositis. Eur Respir J. 2003, 22: 245-250. 10.1183/09031936.03.00026703.
Crowson AN, Magro CM, Dawood MR: A causal role for parvovirus B19 infection in adult dermatomyositis and other autoimmune syndromes. J Cutan Pathol. 2000, 27: 505-515. 10.1034/j.1600-0560.2000.027010505.x.
Kasifoglu T, Korkmaz C, Ozkan R: Cytomegalovirus-induced interstitial pneumonitis in a patient with dermatomyositis. Clin Rheumatol. 2006, 25: 731-733. 10.1007/s10067-005-0062-8.
Weidensaul D, Imam T, Holyst MM, King PD, McMurray RW: Polymyositis, pulmonary fibrosis, and hepatitis C. Arthritis Rheum. 1995, 38: 437-439. 10.1002/art.1780380324.
Sato S, Kuwana M: Clinically amyopathic dermatomyositis. Curr Opin Rheumatol. 2010, 22: 639-643. 10.1097/BOR.0b013e32833f1987.
Nishikai M, Reichlin M: Heterogeneity of precipitating antibodies in polymyositis and dermatomyositis. Characterization of the Jo-1 antibody system. Arthritis Rheum. 1980, 23: 881-888. 10.1002/art.1780230802.
Bunn CC, Bernstein RM, Mathews MB: Autoantibodies against alanyl-tRNA synthetase and tRNAAla coexist and are associated with myositis. J Exp Med. 1986, 163: 1281-1291. 10.1084/jem.163.5.1281.
Mathews MB, Reichlin M, Hughes GR, Bernstein RM: Anti-threonyl-tRNA synthetase, a second myositis-related autoantibody. J Exp Med. 1984, 160: 420-434. 10.1084/jem.160.2.420.
Targoff IN: Autoantibodies to aminoacyl-transfer RNA synthetases for isoleucine and glycine. Two additional synthetases are antigenic in myositis. J Immunol. 1990, 144: 1737-1743.
Hirakata M, Suwa A, Nagai S, Kron MA, Trieu EP, Mimori T, Akizuki M, Targoff IN: Anti-KS: identification of autoantibodies to asparaginyl-transfer RNA synthetase associated with interstitial lung disease. J Immunol. 1999, 162: 2315-2320.
Betteridge Z, Gunawardena H, North J, Slinn J, McHugh N: Anti-synthetase syndrome: a new autoantibody to phenylalanyl transfer RNA synthetase (anti-Zo) associated with polymyositis and interstitial pneumonia. Rheumatology (Oxford). 2007, 46: 1005-1008. 10.1093/rheumatology/kem045.
Hashish L, Trieu EP, Sadanandan P, Targoff IN: Identification of autoantibodies to tyrosyl-tRNA synthetase in dermatomyositis with features consistent with antisynthetase syndrome [abstract]. Arthritis Rheum. 2005, 52: S312-10.1002/art.20784.
Hengstman GJ, van Engelen BG, Vree Egberts WT, van Venrooij WJ: Myositis-specific autoantibodies: overview and recent developments. Curr Opin Rheumatol. 2001, 13: 476-482. 10.1097/00002281-200111000-00004.
Marguerie C, Bunn CC, Beynon HL, Bernstein RM, Hughes JM, So AK, Walport MJ: Polymyositis, pulmonary fibrosis and autoantibodies to aminoacyl-tRNA synthetase enzymes. Q J Med. 1990, 77: 1019-1038.
Yoshifuji H, Fujii T, Kobayashi S, Imura Y, Fujita Y, Kawabata D, Usui T, Tanaka M, Nagai S, Umehara H, Mimori T: Anti-aminoacyl-tRNA synthetase antibodies in clinical course prediction of interstitial lung disease complicated with idiopathic inflammatory myopathies. Autoimmunity. 2006, 39: 233-241. 10.1080/08916930600622884.
Katsumata Y, Ridgway WM, Oriss T, Gu X, Chin D, Wu Y, Fertig N, Oury T, Vandersteen D, Clemens P, Camacho CJ, Weinberg A, Ascherman DP: Species-specific immune responses generated by histidyl-tRNA synthetase immunization are associated with muscle and lung inflammation. J Autoimmun. 2007, 29: 174-186. 10.1016/j.jaut.2007.07.005.
Stone KB, Oddis CV, Fertig N, Katsumata Y, Lucas M, Vogt M, Domsic R, Ascherman DP: Anti-Jo-1 antibody levels correlate with disease activity in idiopathic inflammatory myopathy. Arthritis Rheum. 2007, 56: 3125-3131. 10.1002/art.22865.
Gunawardena H, Betteridge ZE, McHugh NJ: Newly identified autoantibodies: relationship to idiopathic inflammatory myopathy subsets and pathogenesis. Curr Opin Rheumatol. 2008, 20: 675-680. 10.1097/BOR.0b013e328313bff4.
Sato S, Hirakata M, Kuwana M, Suwa A, Inada S, Mimori T, Nishikawa T, Oddis CV, Ikeda Y: Autoantibodies to a 140-kd polypeptide, CADM-140, in Japanese patients with clinically amyopathic dermatomyositis. Arthritis Rheum. 2005, 52: 1571-1576. 10.1002/art.21023.
Sato S, Hoshino K, Satoh T, Fujita T, Kawakami Y, Fujita T, Kuwana M: RNA helicase encoded by melanoma differentiation-associated gene 5 is a major autoantigen in patients with clinically amyopathic dermatomyositis: association with rapidly progressive interstitial lung disease. Arthritis Rheum. 2009, 60: 2193-2200. 10.1002/art.24621.
Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh CS, Reis e Sousa C, Matsuura Y, Fujita T, Akira S: Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006, 441: 101-105. 10.1038/nature04734.
Nakashima R, Imura Y, Kobayashi S, Yukawa N, Yoshifuji H, Nojima T, Kawabata D, Ohmura K, Usui T, Fujii T, Okawa K, Mimori T: The RIG-I-like receptor IFIH1/ MDA5 is a dermatomyositis-specific autoantigen identified by the anti-CADM-140 antibody. Rheumatology (Oxford). 2010, 49: 433-440. 10.1093/rheumatology/kep375.
Gono T, Kawaguchi Y, Satoh T, Kuwana M, Katsumata Y, Takagi K, Masuda I, Tochimoto A, Baba S, Okamoto Y, Ota Y, Yamanaka H: Clinical manifestation and prognostic factor in anti-melanoma differentiation-associated gene 5 antibody-associated interstitial lung disease as a complication of dermatomyositis. Rheumatology (Oxford). 2010, 49: 1713-1719. 10.1093/rheumatology/keq149.
Fiorentino D, Chung L, Zwerner J, Rosen A, Casciola-Rosen L: The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM-140): a retrospective study. J Am Acad Dermatol. 2011, 65: 25-34. 10.1016/j.jaad.2010.09.016.
Marie I, Lahaxe L, Benveniste O, Delavigne K, Adoue D, Mouthon L, Hachulla E, Constans J, Tiev K, Diot E, Levesque H, Boyer O, Jouen F: Long-term outcome of patients with polymyositis/dermatomyositis and anti-PM-Scl antibody. Br J Dermatol. 2010, 162 (2): 337-344. 10.1111/j.1365-2133.2009.09484.x.
Lega JC, Cottin V, Fabien N, Thivolet-Bejui F, Cordier JF: Interstitial lung disease associated with anti-PM/Scl or anti-aminoacyl-tRNA synthetase autoantibodies: a similar condition?. J Rheumatol. 2010, 37: 1000-1009. 10.3899/jrheum.090652.
Casciola-Rosen L, Nagaraju K, Plotz P, Wang K, Levine S, Gabrielson E, Corse A, Rosen A: Enhanced autoantigen expression in regenerating muscle cells in idiopathic inflammatory myopathy. J Exp Med. 2005, 201: 591-601. 10.1084/jem.20041367.
Goebels N, Michaelis D, Engelhardt M, Huber S, Bender A, Pongratz D, Johnson MA, Wekerle H, Tschopp J, Jenne D, Hohlfeld R: Differential expression of perforin in muscle-infiltrating T cells in polymyositis and dermatomyositis. J Clin Invest. 1996, 97: 2905-2910. 10.1172/JCI118749.
Casciola-Rosen L, Andrade F, Ulanet D, Wong WB, Rosen A: Cleavage by granzyme B is strongly predictive of autoantigen status: implications for initiation of autoimmunity. J Exp Med. 1999, 190: 815-826. 10.1084/jem.190.6.815.
Levine SM, Raben N, Xie D, Askin FB, Tuder R, Mullins M, Rosen A, Casciola-Rosen LA: Novel conformation of histidyl-transfer RNA synthetase in the lung: the target tissue in Jo-1 autoantibody-associated myositis. Arthritis Rheum. 2007, 56: 2729-2739. 10.1002/art.22790.
Howard OM, Dong HF, Yang D, Raben N, Nagaraju K, Rosen A, Casciola-Rosen L, Hartlein M, Kron M, Yang D, Yiadom K, Dwivedi S, Plotz PH, Oppenheim JJ: Histidyl-tRNA synthetase and asparaginyl-tRNA synthetase, autoantigens in myositis, activate chemokine receptors on T lymphocytes and immature dendritic cells. J Exp Med. 2002, 196: 781-791. 10.1084/jem.20020186.
Greenberg SA, Pinkus JL, Pinkus GS, Burleson T, Sanoudou D, Tawil R, Barohn RJ, Saperstein DS, Briemberg HR, Ericsson M, Park P, Amato AA: Interferon-alpha/beta-mediated innate immune mechanisms in dermatomyositis. Ann Neurol. 2005, 57: 664-678. 10.1002/ana.20464.
Sauty A, Rochat T, Schoch OD, Hamacher J, Kurt AM, Dayer JM, Nicod LP: Pulmonary fibrosis with predominant CD8 lymphocytic alveolitis and anti-Jo-1 antibodies. Eur Respir J. 1997, 10: 2907-2912. 10.1183/09031936.97.10122907.
Yokoyama T, Sakamoto T, Shida N, Kinoshita M, Kunou Y, Karukaya R, Takamatsu M, Aizawa H: Fatal rapidly progressive interstitial pneumonitis associated with amyopathic dermatomyositis and CD8 T lymphocytes. J Intensive Care Med. 2005, 20: 160-163. 10.1177/0885066605275391.
Guglielmi S, Merz TM, Gugger M, Suter C, Nicod LP: Acute respiratory distress syndrome secondary to antisynthetase syndrome is reversible with tacrolimus. Eur Respir J. 2008, 31: 213-217. 10.1183/09031936.00014707.
Takada K, Nagasaka K, Miyasaka N: Polymyositis/dermatomyositis and interstitial lung disease: a new therapeutic approach with T-cell-specific immunosuppressants. Autoimmunity. 2005, 38: 383-392. 10.1080/08916930500124023.
Acknowledgements
The present work was supported by National Institutes of Health grant AR44684 (to LC-R) and by the Lisa Sandler Spaeth and Robert M. Fisher Funds for Pulmonary Fibrosis (to SKD). Images in Figure 1 were used with permission from Dr Christopher-Stine.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Danoff, S.K., Casciola-Rosen, L. The lung as a possible target for the immune reaction in myositis. Arthritis Res Ther 13, 230 (2011). https://doi.org/10.1186/ar3347
Published:
DOI: https://doi.org/10.1186/ar3347