
Abstract
Many of the chronic inflammatory and degenerative disorders that present to clinical rheumatologists have a complex genetic aetiology. Over the past decade a dramatic improvement in technology and methodology has accelerated the pace of gene discovery in complex disorders in an exponential fashion. In this review, we focus on rheumatoid arthritis, systemic lupus erythematosus and ankylosing spondylitis and describe some of the recently described genes that underlie these conditions and the extent to which they overlap. The next decade will witness a full account of the main disease susceptibility genes in these diseases and progress in establishing the molecular basis by which genetic variation contributes to pathogenesis.
Similar content being viewed by others

Genetics of rheumatic disease
The spectrum of rheumatic disease is wide and includes conditions with diverse pathology, although most have in common a heritable risk with a complex genetic basis. There has therefore been intense effort to understand the contribution of genotype to the expression of disease in terms of both basic pathogenesis and clinical characteristics. Recent technical advances in genotyping and statistical analysis and international collaborations assembling large cohorts of patients have led to a wealth of new data. In this review we describe insights gained into the pathogenesis of autoimmune rheumatic disease by the techniques of modern genetics, in particular evidence from genome-wide association (GWA) studies, which provide support for the existence of a common genetic risk basis to several diseases. To reflect the new data from GWA studies, our discussion will be confined to rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), and ankylosing spondylitis (AS), which in some cases share a common autoimmune pathogenesis. Osteoarthritis and osteoporosis are also complex genetic traits but limitations of space are such that these two conditions will not be considered in this review.
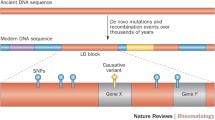
The concept of a systematic, GWA study became practical with the cataloguing of libraries of common polymorphisms. Currently, over 20 million single nucleotide polymorphisms (SNPs) have been identified [1] and platforms are available to type up to 1 million of these in a single reaction. Although not all SNPs are currently genotyped, as the human genome is arranged into haplotype blocks in linkage disequilibrium, it is only necessary to type so-called tag SNPs, which identify these areas of limited variability [2], to achieve good representation of the total amount of genetic variation. Most typed SNPs are relatively common (minor allele frequency of > 5%) and if associated with disease are likely, therefore, to have only modest pathogenic effects (odds ratios (ORs) usually between 1.2 and 2), as otherwise they would become depleted in a population due to natural selection. It is necessary, therefore, to invoke the 'common-disease common-variant' (CD-CV) model [3], which assumes an accumulation of risk caused by the carriage of multiple deleterious alleles, to explain current experimental findings.
One of the revolutionary advantages of the GWA study is the freedom from a required gene-centric hypothesis, which provides an unprecedentedly effective technique for risk gene discovery. Many disease-associated genes identified by GWA studies were completely unsuspected to be relevant - for example, the autophagy system in Crohn's disease [4]. However, because in essence up to 1 million independent hypotheses are being tested in each genotyping reaction, sample sizes powered to detect even the stronger associations must be large, and criteria for significance stringent. The general consensus is that significance can be defined as a P-value smaller than 5 × 10-7, which in a cohort such as the Wellcome Trust Case Control Consortium (WTCCC) of 2,000 cases, for example, approximates to a power of 43% rising to 80% to detect alleles with ORs of 1.3 and 1.5, respectively [5]. However, the genome is subject to variation at more than the SNP level, and individuals also differ in the copy number of sections of DNA of greater than several kilobases in size, so called copy number variation (CNV), which in fact accounts for more total nucleotide difference between individuals than SNPs [6, 7]. CNV can affect gene expression levels [8] and has been linked to autoimmune disease [9, 10], including SLE [11]. Whilst the latest genotyping platforms include assessment of CNV, earlier products actively excluded SNPs within regions of the most variation as they were more likely to fail quality control steps. Association studies based on CNV are, therefore, in their relative infancy. Finally, the genome is subject to modification without a change in DNA sequence; epigenetic mechanisms can have profound effects on gene expression. These include DNA methylation and changes in chromatin structure [12].
It has become apparent that SLE, RA, and AS, which have divergent clinical features, may share a common genetic risk framework, and we aim in our review to illustrate this.
The MHC region and antigen processing
The major histocompatibility complex (MHC) region on chromosome 6 contributes to the risk of almost all autoimmune diseases, and its role in immunity in mice was recognized over 60 years ago. In humans, the MHC locus is also known as the HLA (human leukocyte antigen) region, reflecting the initial identification of MHC gene products on the surface of white blood cells. The classical MHC extends over around 4 megabases, and comprises three clusters: class I, II, and III. Class I and II regions include genes that encode the α- and β-chains of the MHC I and II complexes, and flank the class III region, which contains an assortment of immunologically relevant genes. Despite extensive study, the mechanisms that link the MHC to disease are largely unknown, although it is supposed that variation in the MHC peptide binding cleft facilitates presentation of self-antigen to autoreactive lymphocytes.
These difficulties in understanding the MHC are not without reason; it contains some of the most polymorphic loci described in the genome, and has a highly complicated genetic architecture, with some regions exhibiting extended linkage disequilibrium [13].
In RA, the MHC accounts for around a third of the genetic liability [14]. Alleles at HLA-DRB1 contribute much of this risk - for example, DRB1*0401 carries an OR of 3. GWA studies confirm the strong association with MHC variants; risk alleles confer an OR of around 2 to 3 in homozygotes [15], with very high statistical significance (P < 10-100). Additional loci contributing to the risk of RA identified by high-density genotyping include HLA-DP in patients with anti-cyclic citrullinated peptide antibodies [16]. SLE not only has strongly associated alleles in the class II region, HLA-DR2 (DRB1*1501) and DR3 (DRB1*0301) [14], with ORs of 2 [17], but also risk variants in the class III cluster, which encodes genes such as TNF and the complement components C2, C4A and C4B. C4 is crucial in the classical and mannose-binding lectin pathways of complement activation, and complete deficiency of C4 or indeed other components of the classical pathway are rare, but strong, risk factors for SLE [18]. The C4 gene is subject to CNV and is of two isotypes, C4A and C4B. It is an attractive hypothesis that CNV at C4 affects expression and contributes to SLE risk. However, it remains to be established whether haplotypes carrying partial C4 deficiency exert their risk via an influence on complement or through other genetic variants that are in linkage disequilibrium. Other loci in the class III region have been implicated in SLE, including the SKIV2L gene, SNPs in which carry an OR of 2 in a family-based analysis [19]. SKIV2L encodes superkiller viralicidic activity 2-like, the human homologue of which is a DEAD box protein that may have nucleic acid processing activity. The second MHC III signal for SLE we will consider was identified in the International Consortium on the Genetics of Systemic Lupus Erythematosus (SLEGEN) GWA study [17, 20]. The SNP rs3131379 in mutS homologue 5 (MSH5) has an OR of 1.82. There is evidence that MSH5 has a role in immunoglobulin class switch variation [21]. Again, further work is required to definitively implicate this gene rather than variants in linkage disequilibrium, which include HLA-DRB1*0301 and C4A deletions.
Clearly, HLA-B27 is the overwhelming association in AS, with an OR of 200 to 300. In the MHC, other genetic risk variants have been identified, including HLA-B60 (OR 3.6) [22] and various HLA-DR genes with relatively minor contributions [23]. The pathogenic mechanism for these risk alleles is unknown. Outside of the MHC, two significant genes have so far been identified in AS: ARTS1 and IL-23R [24], the latter of which will be discussed below and has been associated with several different autoimmune diseases. ARTS1 has two identified functions. Its first is in the processing of peptide for presentation via MHC I. It is localised in the endoplasmic reticulum, and is upregulated by IFNγ. It acts as an amino-terminal aminopeptidase and in mice is essential for the display of the normal peptide repertoire. In its absence, many unstable and highly immunogenic MHC-peptide complexes are presented [25]. A hypothetical connection with HLA-B27 can thus be drawn. Its other function is to downregulate signalling by IL-1, IL-6, and TNFα through surface receptor cleavage [26–28]. The most associated SNP rs30187 risk allele has an OR of 1.4, and is of unknown functional significance.
Innate-adaptive interface
Interferon signalling: IRF5
It is clear that type 1 interferons (IFNα and IFNβ) are of great importance in the pathogenesis of SLE. Patients with active disease have high levels of IFNα, which has multiple immunomodulatory actions [29], including the induction of dendritic cell differentiation, the upregulation of innate immune receptors such as toll-like receptors (TLRs), the polarization of T cells towards a TH1 phenotype, and the activation of B cells. Type I interferons are produced by all cells in response to viral infection, but particularly by plasmacytoid dendritic cells in response to unmethylated CpG oligonucleotides binding to TLR-9, or RNA to TLR-7. Using a candidate gene approach targeting the IFN signalling pathway, the SNP rs2004640 in IRF5 (interferon regulatory factor 5) was found to be significantly associated with SLE (OR 1.6) [30], a risk gene confirmed in several other studies [17, 31–35]. The functional consequences for IRF5 of the identified mutations are variable, but include the creation of a 5' donor splice site in an alternative exon 1, allowing the expression of several isoforms [35], a 30 base-pair in-frame insertion/deletion variant of exon 6, a change in the 3' untranslated region, and a CGGGG insertion-deletion (indel) polymorphism, the latter two affecting mRNA stability [32, 36]. Interestingly, these mutations may occur together in a haplotype, with varying degrees of associated risk. The exact role of IRF5 in IFN signalling has not been fully elucidated, but it is also critical for the gene induction programme activated by TLRs [37], providing further biological plausibility for its importance in the pathogenesis of SLE. Haplotypes of IRF5 are also implicated in RA, and may confer either protection (OR 0.76) or predisposition (OR 1.8) [38]. The same CGGGG indel allele described above also carries risk for multiple sclerosis and inflammatory bowel disease [36].
TNF-associated signalling pathway: TNFAIP3 and TRAF1-C5
TNF-associated signalling pathway genes play a prominent role in the risk for both SLE and RA, and associations with variants in TNFAIP3, and the TRAF1-C5 locus have been identified [39, 40]. TNFα-induced protein-3 (TNFAIP3; also known as A20) is a ubiquitin editing enzyme that acts as a negative regulator of NFκB. A20 can disassemble Lys63-linked polyubiquitin chains from targets such as TRAF6 and RIP1. A second region of A20 catalyses Lys48-linked ubiquitination that targets the molecule for degradation by the proteasome [41]. A20 modifies key mediators in the downstream signalling of TLRs that use MyD88, TNF receptors, the IL-1 receptor family, and nucleotide-oligomerization domain protein 2 (NOD2) [42]. Tnfaip3 knockout mice develop severe multi-organ inflammatory disease, and the phenotype is lethal [43]. The SNP rs10499194 in TNFAIP3 carries an OR of 1.33 for RA, and rs5029939 an OR of 2.29 for SLE [44], the latter also conferring an increased risk of haematologic or renal complications [45].
On chromosome 9, the region containing TRAF1 (TNF receptor associated factor 1) and C5 (complement component 5) genes is associated with significant risk for RA (risk SNP OR of approximately 1.3) in most [15, 40, 46–48], but not all [5], studies. Due to linkage disequilibrium, the functional variant remains elusive. TRAF1 is principally expressed in lymphocytes, and inhibits NFκB signalling by TNF. This pathway is blocked in TRAF1 overexpression [49] whilst, conversely, Traf1-/- mice are sensitized to TNF and have exaggerated TNF-induced skin necrosis [50].
The complement system has long been known to be involved in the pathogenesis of RA. In the collagen-induced arthritis model of RA, C5 deficiency prevents disease de novo and ameliorates existing symptoms and signs [51, 52]. Interestingly, GG homozygotes at the TRAF1-C5 SNP rs3761847 with RA have a significantly increased risk of death (hazard ratio 3.96, 95% confidence interval 1.24 to 12.6, P = 0.02) from malignancy or sepsis, potentially allowing identification of patients for appropriate screening [53].
Immunomodulatory adhesion molecule: ITGAM
Integrin-α-M (ITGAM), variants of which are strongly associated with SLE, forms a heterodimer with integrin-β-2 to produce αMβ2-integrin (also known as CD11b, Mac-1, or complement receptor-3), which mediates the adhesion of myeloid cells to the endothelium via ICAM-1 (Intercellular adhesion molecule-1) and recognizes the complement component iC3b. It not only has a role in cell trafficking and phagocytosis [54], but also has other immunomodulatory functions. Antigen-presenting cells produce tolerogenic IL-10 and transforming growth factor-β on iC3b binding to CD11b [55], and mice deficient in this receptor upregulate expression of IL-6, favouring a pro-inflammatory TH17 response [56]. Despite its implication in defective immune complex clearance in SLE, experimental evidence for a role was lacking. GWA studies, however, demonstrate a strong and significant association [17, 33, 44], with an OR of 1.83 (P = 7 × 10-50) in meta-analysis [57]. The implicated SNP rs1143679 is non-synonymous, causing the substitution of histidine for arginine at amino acid 77, although this change does not affect the iC3b binding site [58]. Furthermore, although this SNP is disease associated in European and Hispanic patients, it is monomorphic in Japanese and Korean populations [59]; an explanation of its effect is therefore outstanding. It has been mentioned that CNV is important in C4 expression; the same is true for the Fcγ receptor IIIb (FCGR3B) [60], which relies on CD11b for function. Fcγ receptor IIIb is principally present on neutrophils and is important in the binding and clearance of immune complexes, therefore marking itself as a potential SLE risk gene. There is a significant association between low FCGR3B copy number and SLE. Patients with two or fewer copies of FCGR3B have an OR of 2.43 for SLE with nephritis, and 2.21 for SLE without nephritis [61].
Lymphocyte differentiation
T cell receptor signalling: PTPN22
Outside the HLA region, the first reproducible genetic association for RA came with the implication of PTPN22 from a candidate gene approach [62] based on linkage analysis identification of a susceptibility locus at 1p13 [63]. It has remained the strongest and most consistent association mapped by GWA studies in RA. A role in SLE has also been identified [17]. The OR for the risk allele is around 1.75 in RA, and 1.5 in SLE. However, it should be noted that this allele (encoding the R620W mutation) is monomorphic or not disease associated in Korean or Japanese patients [64, 65]. PTPN22 encodes lymphoid tyrosine phosphatase (LYP), a protein tyrosine phosphatase that inhibits T cell receptor signalling, decreasing IL-2 production. The disease associated SNP is responsible for a change from arginine to tryptophan at position 620, which inhibits binding to the SH3 domain of carboxy-terminal Src kinase. This in turn appears to enhance dephosphorylation of tyrosine residues in the Src family kinases Lck, FynT, and ZAP-70 [66, 67]. The overall effect of the mutation is a reduction in T cell receptor signalling. The pathogenic effect of this is unclear, but may relate to impaired negative selection in the thymus, or lead to a reduction in regulatory T cells [68]. Conversely, the R623Q variant of PTPN22, which is a loss-of-function mutation affecting the phosphatase activity of LYP, is protective against SLE [69]. PTPN22 does not appear to be a risk gene for AS [70].
Polarization towards TH1 and TH17 phenotypes: STAT4 and IL23R
STAT4 encodes signal transducer and activation of transcription factor-4, responsible for signalling by IL-12, IL-23, and type 1 IFNs [71]. STAT4 polarizes T cells towards TH1 and TH17 phenotypes, which has the potential to promote autoimmunity [72]. In RA the OR for the risk allele of SNP rs7574865 is 1.32 in one case-control study [73], with a less strong disease association at rs11893432 in a meta-analysis of GWA studies (OR 1.14) [15]. There is convincing evidence that STAT4 is a risk locus for SLE in multiple racial groups [33, 74], and it may be theorized that interference in type I IFN signalling may be the underlying pathogenic mechanism in this case. Distinctive disease pathways could, therefore, emerge from mutations in a single gene. The WTCCC AS study identified IL23R as a risk gene in AS [24]. IL-23 is instrumental in the development of T cells with the pro-inflammatory TH17 phenotype [75], and IL23R has been linked to psoriasis, ulcerative colitis, and Crohn's disease in GWA studies [5, 76, 77]. An interesting connection between these conditions, all of which may share common clinical features, is thus made. In AS the risk SNP rs11209032 confers an OR of 1.3.
B cell activation
B cells are a population long suspected to be important in autoimmune rheumatic disease, and the benefits of their depletion in RA and SLE has resurrected interest in their pathogenic role. The risk genes identified so far are involved in signalling from the B cell receptor (BCR). BLK encodes a Src family tyrosine kinase restricted to the B cell lineage and is poorly understood. Risk alleles in the region upstream of the transcription initiation site are associated with SLE (OR 1.39, P = 1 × 10-10) and reduce levels of BLK mRNA [33]. BANK1 (B cell scaffold protein with ankyrin repeats-1) undergoes tyrosine phosphorylation upon B cell activation by the BCR, leading to an increase in intracellular calcium through the inositol trisphosphate mechanism [78]. The non-synonymous SNP rs10516487 in BANK1, which substitutes histidine for arginine at amino acid 61, also has disease association (OR 1.38) [79]. The functional consequence of this may be higher affinity for the inositol trisphosphate receptor, as the substitution is located in the binding site.
Lyn, another Src tyrosine kinase, is important in determining signalling thresholds for myeloid and B cells. On BCR ligation, it phosphorylates tyrosine residues of Syk, an activating tyrosine kinase, CD19, and the immunoreceptor tyrosine-based activation motifs (ITAMs) of the Igα/Igβ subunits of the BCR. However, it also has a critical regulatory role, mediated by phosphorylation of the inhibitory motifs of CD22 and Fcγ RIIB, which in turn activate SH2-domain containing phosphatases, leading to dephosphorylation and deactivation of a number of signalling intermediaries [80]. Lyn-/- mice develop severe autoimmunity associated with glomerulonephritis [81]. An association between SNPs in LYN and SLE, identified initially in the SLEGEN GWA study [17], has been recently confirmed in a case-control study [82]. The most associated SNP, rs6983130, is near the primary transcription initiation site.
OX40L, a member of the TNF super-family encoded by TNFSF4 (TNF superfamily 4), is associated with SLE. The cross-talk between B lymphocytes and dendritic cells expressing OX40L, and T cells that express its receptor, OX40, serves to enhance the adaptive immune response [83]. An upstream TNFSF4 haplotype, associated with SLE, enhances gene expression in vitro [84, 85], although the mechanism responsible for the deleterious effects observed remains to be established.
Despite the importance of B cells in the pathogenesis of RA, none of the gene effects described above have been identified in the current generation of GWA studies. However, variants at CD40 in European patients do carry risk [15]. CD40 expressed on B cells, via interaction with its ligand CD154 on CD4+ T cells, promotes immunoglobulin class switching, and germinal centre formation. B cells, however, also have a regulatory role, likely to be mediated by IL-10, and disruption of this function may be another route to autoimmune disease [86].
Post-translational modification: PADI4
Peptidyl arginine deiminase-4 (PADI4) is a member of the enzyme family responsible for the post-translational citrullination of arginine residues in RA synovium, subsequently recognized by anti-cyclic citrullinated protein antibodies. In Japanese [87] and Korean patients [88], case-control association studies have identified functional haplotypes of PADI4 conferring risk of RA. However, in Caucasian populations this association is inconsistent [89–91].
Conclusion
Even with the proliferation of new genetic associations discovered in the past few years by GWA studies, only around 10 to 15% of the inherited risk for SLE and RA can be currently explained. This may be accounted for, in part, by a number of factors, some related to limitations of recent study design. As mentioned above, even the largest current GWA cohorts have limited power to detect associations with ORs < 1.3, potentially losing multiple risk genes. By definition, most genotyped SNPs are common, and so rare but causal variants have a tendency to be missed. These rarer SNPs may be either those with a low minor allele frequency (< 5%), or occur de novo, of which 200 to 500 non-synonymous SNPs are expected per individual [92]. In many cases, it is far from certain if the associated SNP is functional, or in linkage disequilibrium with the true cause. Finally, the great majority of GWA studies have been conducted on European populations, thereby excluding carriers of many potential risk variants from analysis. However, it is unfortunately the case that current genotyping platforms often have poor coverage of tagging SNPs within populations that exhibit low levels of genomic linkage disequilibrium, such as those of African ancestry [93]. For example, the latest high-density genotyping chips from Affymetrix (6.0) and Illumina (1 M) may capture fewer than half the SNPs identified through re-sequencing in Yoruban Nigerians [94]. Given that clear differences exist in the risk of autoimmune disease according to ethnicity, and that not all disease risk alleles are in common, it is imperative that full account of this variation is made. Structural genetic differences have only recently begun to be assessed by modern genotyping platforms, and the contribution of, for example, CNV to inherited disease risk is largely unquantified. Even more difficult to appreciate is the influence of heritable epigenetic factors, and the exact relationship between genotype and phenotype. Nevertheless, although it will probably not be possible to explain all the observed genetic risk in the near future, we are rapidly moving towards the ability to quickly and cheaply fully sequence individual genomes [95], with all the advantages that brings [96]. In the meantime, understanding the functional basis of the disease risk variants so far identified presents an outstanding challenge. Integration of genotypic with RNA and protein expression data in a systems biologic approach represents one potentially valuable methodology [97]. Exploring and therapeutically utilizing the genetic differences between individuals is axiomatic to personalized medicine, and will undoubtedly lead to better outcomes in the management of autoimmune disease.
Note
The Scientific Basis of Rheumatology: A Decade of Progress
This article is part of a special collection of reviews, The Scientific Basis of Rheumatology: A Decade of Progress, published to mark Arthritis Research & Therapy's 10th anniversary.
Other articles in this series can be found at: http://arthritis-research.com/sbr
Abbreviations
- AS:
-
ankylosing spondylitis
- BCR:
-
B cell receptor
- CNV:
-
copy number variation
- GWA:
-
genome-wide association
- HLA:
-
human leukocyte antigen
- IFN:
-
interferon
- IL:
-
interleukin
- IRF:
-
interferon regulatory factor
- MHC:
-
major histocompatibility complex
- NF:
-
nuclear factor
- OR:
-
odds ratio
- RA:
-
rheumatoid arthritis
- SLE:
-
systemic lupus erythematosus
- SNP:
-
single nucleotide polymorphism
- STAT:
-
signal transducer and activation of transcription factor
- TLR:
-
toll-like receptor
- TNF:
-
tumour necrosis factor
- TNFAIP:
-
TNFα-induced protein-3
- TRAF:
-
TNF receptor associated factor
- WTCCC:
-
Wellcome Trust Case Control Consortium.
References
Entrez SNP. [http://www.ncbi.nlm.nih.gov/sites/entrez?db=snp]
International HapMap Consortium, Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, Gibbs RA, Belmont JW, Boudreau A, Hardenbol P, Leal SM, Pasternak S, Wheeler DA, Willis TD, Yu F, Yang H, Zeng C, Gao Y, Hu H, Hu W, Li C, Lin W, Liu S, Pan H, Tang X, Wang J, Wang W, Yu J, Zhang B, Zhang Q, et al: A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007, 449: 851-861. 10.1038/nature06258.
Risch N, Merikangas K: The future of genetic studies of complex human diseases. Science. 1996, 273: 1516-1517. 10.1126/science.273.5281.1516.
Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, Green T, Kuballa P, Barmada MM, Datta LW, Shugart YY, Griffiths AM, Targan SR, Ippoliti AF, Bernard EJ, Mei L, Nicolae DL, Regueiro M, Schumm LP, Steinhart AH, Rotter JI, Duerr RH, Cho JH, Daly MJ, Brant SR: Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. 2007, 39: 596-604. 10.1038/ng2032.
Wellcome Trust Case Control Consortium: Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007, 447: 661-678. 10.1038/nature05911.
Sebat J, Lakshmi B, Troge J, Alexander J, Young J, Lundin P, Månér S, Massa H, Walker M, Chi M, Navin N, Lucito R, Healy J, Hicks J, Ye K, Reiner A, Gilliam TC, Trask B, Patterson N, Zetterberg A, Wigler M: Large-scale copy number polymorphism in the human genome. Science. 2004, 305: 525-528. 10.1126/science.1098918.
Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, Chen W, Cho EK, Dallaire S, Freeman JL, González JR, Gratacòs M, Huang J, Kalaitzopoulos D, Komura D, MacDonald JR, Marshall CR, Mei R, Montgomery L, Nishimura K, Okamura K, Shen F, Somerville MJ, Tchinda J, Valsesia A, Woodwark C, Yang F, et al: Global variation in copy number in the human genome. Nature. 2006, 444: 444-454. 10.1038/nature05329.
Stranger BE, Forrest MS, Dunning M, Ingle CE, Beazley C, Thorne N, Redon R, Bird CP, de Grassi A, Lee C, Tyler-Smith C, Carter N, Scherer SW, Tavaré S, Deloukas P, Hurles ME, Dermitzakis ET: Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science. 2007, 315: 848-853. 10.1126/science.1136678.
Aitman TJ, Dong R, Vyse TJ, Norsworthy PJ, Johnson MD, Smith J, Mangion J, Roberton-Lowe C, Marshall AJ, Petretto E, Hodges MD, Bhangal G, Patel SG, Sheehan-Rooney K, Duda M, Cook PR, Evans DJ, Domin J, Flint J, Boyle JJ, Pusey CD, Cook HT: Copy number polymorphism in Fcgr3 predisposes to glomerulonephritis in rats and humans. Nature. 2006, 439: 851-855. 10.1038/nature04489.
Hollox EJ, Huffmeier U, Zeeuwen PL, Palla R, Lascorz J, Rodijk-Olthuis D, Kerkhof van de PC, Traupe H, de Jongh G, den Heijer M, Reis A, Armour JA, Schalkwijk J: Psoriasis is associated with increased beta-defensin genomic copy number. Nat Genet. 2008, 40: 23-25. 10.1038/ng.2007.48.
Yang Y, Chung EK, Wu YL, Savelli SL, Nagaraja HN, Zhou B, Hebert M, Jones KN, Shu Y, Kitzmiller K, Blanchong CA, McBride KL, Higgins GC, Rennebohm RM, Rice RR, Hackshaw KV, Roubey RA, Grossman JM, Tsao BP, Birmingham DJ, Rovin BH, Hebert LA, Yu CY: Gene copy-number variation and associated polymorphisms of complement component C4 in human systemic lupus erythematosus (SLE): low copy number is a risk factor for and high copy number is a protective factor against SLE susceptibility in European Americans. Am J Hum Genet. 2007, 80: 1037-1054. 10.1086/518257.
Felsenfeld G, Groudine M: Controlling the double helix. Nature. 2003, 421: 448-453. 10.1038/nature01411.
Horton R, Wilming L, Rand V, Lovering RC, Bruford EA, Khodiyar VK, Lush MJ, Povey S, Talbot CC, Wright MW, Wain HM, Trowsdale J, Ziegler A, Beck S: Gene map of the extended human MHC. Nat Rev Genet. 2004, 5: 889-899. 10.1038/nrg1489.
Fernando MM, Stevens CR, Walsh EC, De Jager PL, Goyette P, Plenge RM, Vyse TJ, Rioux JD: Defining the role of the MHC in autoimmunity: a review and pooled analysis. PLoS Genet. 2008, 4: e1000024-10.1371/journal.pgen.1000024.
Raychaudhuri S, Remmers EF, Lee AT, Hackett R, Guiducci C, Burtt NP, Gianniny L, Korman BD, Padyukov L, Kurreeman FA, Chang M, Catanese JJ, Ding B, Wong S, Helmvan Mil van der AH, Neale BM, Coblyn J, Cui J, Tak PP, Wolbink GJ, Crusius JB, Horst-Bruinsma van der IE, Criswell LA, Amos CI, Seldin MF, Kastner DL, Ardlie KG, Alfredsson L, Costenbader KH, Altshuler D, et al: Common variants at CD40 and other loci confer risk of rheumatoid arthritis. Nat Genet. 2008, 40: 1216-1223. 10.1038/ng.233.
Ding B, Padyukov L, Lundström E, Seielstad M, Plenge RM, Oksenberg JR, Gregersen PK, Alfredsson L, Klareskog L: Different patterns of associations with anti-citrullinated protein antibody-positive and anti-citrullinated protein antibody-negative rheumatoid arthritis in the extended major histocompatibility complex region. Arthritis Rheum. 2009, 60: 30-38. 10.1002/art.24135.
International Consortium for Systemic Lupus Erythematosus Genetics (SLEGEN), Harley JB, Alarcón-Riquelme ME, Criswell LA, Jacob CO, Kimberly RP, Moser KL, Tsao BP, Vyse TJ, Lange-feld CD, Nath SK, Guthridge JM, Cobb BL, Mirel DB, Marion MC, Williams AH, Divers J, Wang W, Frank SG, Namjou B, Gabriel SB, Lee AT, Gregersen PK, Behrens TW, Taylor KE, Fernando M, Zidovetzki R, Gaffney PM, Edberg JC, Rioux JD, et al: Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet. 2008, 40: 204-210. 10.1038/ng.81.
Lewis MJ, Botto M: Complement deficiencies in humans and animals: links to autoimmunity. Autoimmunity. 2006, 39: 367-378. 10.1080/08916930600739233.
Fernando MM, Stevens CR, Sabeti PC, Walsh EC, McWhinnie AJ, Shah A, Green T, Rioux JD, Vyse TJ: Identification of two independent risk factors for lupus within the MHC in United Kingdom families. PLoS Genet. 2007, 3: e192-10.1371/journal.pgen.0030192.
Harley IT, Kaufman KM, Langefeld CD, Harley JB, Kelly JA: Genetic susceptibility to SLE: new insights from fine mapping and genome-wide association studies. Nat Rev Genet. 2009, 10: 285-290. 10.1038/nrg2571.
Sekine H, Ferreira RC, Pan-Hammarström Q, Graham RR, Ziemba B, de Vries SS, Liu J, Hippen K, Koeuth T, Ortmann W, Iwahori A, Elliott MK, Offer S, Skon C, Du L, Novitzke J, Lee AT, Zhao N, Tompkins JD, Altshuler D, Gregersen PK, Cunningham-Rundles C, Harris RS, Her C, Nelson DL, Hammarström L, Gilkeson GS, Behrens TW: Role for Msh5 in the regulation of Ig class switch recombination. Proc Natl Acad Sci USA. 2007, 104: 7193-7198. 10.1073/pnas.0700815104.
Brown MA, Pile KD, Kennedy LG, Calin A, Darke C, Bell J, Wordsworth BP, Cornélis F: HLA class I associations of ankylosing spondylitis in the white population in the United Kingdom. Ann Rheum Dis. 1996, 55: 268-270. 10.1136/ard.55.4.268.
Brown MA, Kennedy LG, Darke C, Gibson K, Pile KD, Shatford JL, Taylor A, Calin A, Wordsworth BP: The effect of HLA-DR genes on susceptibility to and severity of ankylosing spondylitis. Arthritis Rheum. 1998, 41: 460-465. 10.1002/1529-0131(199803)41:3<460::AID-ART12>3.0.CO;2-X.
Wellcome Trust Case Control Consortium; Australo-Anglo-American Spondylitis Consortium (TASC), Burton PR, Clayton DG, Cardon LR, Craddock N, Deloukas P, Duncanson A, Kwiatkowski DP, McCarthy MI, Ouwehand WH, Samani NJ, Todd JA, Donnelly P, Barrett JC, Davison D, Easton D, Evans DM, Leung HT, Marchini JL, Morris AP, Spencer CC, Tobin MD, Attwood AP, Boorman JP, Cant B, Everson U, Hussey JM, Jolley JD, Knight AS, et al: Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat Genet. 2007, 39: 1329-1337. 10.1038/ng.2007.17.
Hammer GE, Gonzalez F, James E, Nolla H, Shastri N: In the absence of aminopeptidase ERAAP, MHC class I molecules present many unstable and highly immunogenic peptides. Nat Immunol. 2007, 8: 101-108. 10.1038/ni1409.
Cui X, Rouhani FN, Hawari F, Levine SJ: Shedding of the type II IL-1 decoy receptor requires a multifunctional aminopeptidase, aminopeptidase regulator of TNF receptor type 1 shedding. J Immunol. 2003, 171: 6814-6819.
Cui X, Rouhani FN, Hawari F, Levine SJ: An aminopeptidase, ARTS-1, is required for interleukin-6 receptor shedding. J Biol Chem. 2003, 278: 28677-28685. 10.1074/jbc.M300456200.
Cui X, Hawari F, Alsaaty S, Lawrence M, Combs CA, Geng W, Rouhani FN, Miskinis D, Levine SJ: Identification of ARTS-1 as a novel TNFR1-binding protein that promotes TNFR1 ectodomain shedding. J Clin Invest. 2002, 110: 515-526.
Ronnblom L, Eloranta ML, Alm GV: The type I interferon system in systemic lupus erythematosus. Arthritis Rheum. 2006, 54: 408-420. 10.1002/art.21571.
Sigurdsson S, Nordmark G, Göring HH, Lindroos K, Wiman AC, Sturfelt G, Jönsen A, Rantapää-Dahlqvist S, Möller B, Kere J, Koskenmies S, Widén E, Eloranta ML, Julkunen H, Kristjansdottir H, Steinsson K, Alm G, Rönnblom L, Syvänen AC: Polymorphisms in the tyrosine kinase 2 and interferon regulatory factor 5 genes are associated with systemic lupus erythematosus. Am J Hum Genet. 2005, 76: 528-537. 10.1086/428480.
Sigurdsson S, Padyukov L, Kurreeman FA, Liljedahl U, Wiman AC, Alfredsson L, Toes R, Rönnelid J, Klareskog L, Huizinga TW, Alm G, Syvänen AC, Rönnblom L: Association of a haplotype in the promoter region of the interferon regulatory factor 5 gene with rheumatoid arthritis. Arthritis Rheum. 2007, 56: 2202-2210. 10.1002/art.22704.
Graham RR, Kyogoku C, Sigurdsson S, Vlasova IA, Davies LR, Baechler EC, Plenge RM, Koeuth T, Ortmann WA, Hom G, Bauer JW, Gillett C, Burtt N, Cunninghame Graham DS, Onofrio R, Petri M, Gunnarsson I, Svenungsson E, Rönnblom L, Nordmark G, Gregersen PK, Moser K, Gaffney PM, Criswell LA, Vyse TJ, Syvänen AC, Bohjanen PR, Daly MJ, Behrens TW, Altshuler D: Three functional variants of IFN regulatory factor 5 (IRF5) define risk and protective haplotypes for human lupus. Proc Natl Acad Sci USA. 2007, 104: 6758-6763. 10.1073/pnas.0701266104.
Hom G, Graham RR, Modrek B, Taylor KE, Ortmann W, Garnier S, Lee AT, Chung SA, Ferreira RC, Pant PV, Ballinger DG, Kosoy R, Demirci FY, Kamboh MI, Kao AH, Tian C, Gunnarsson I, Bengtsson AA, Rantapää-Dahlqvist S, Petri M, Manzi S, Seldin MF, Rönnblom L, Syvänen AC, Criswell LA, Gregersen PK, Behrens TW: Association of systemic lupus erythematosus with C8orf13-BLK and ITGAM-ITGAX. N Engl J Med. 2008, 358: 900-909. 10.1056/NEJMoa0707865.
Cunninghame Graham DS, Manku H, Wagner S, Reid J, Timms K, Gutin A, Lanchbury JS, Vyse TJ: Association of IRF5 in UK SLE families identifies a variant involved in polyadenylation. Hum Mol Genet. 2007, 16: 579-591. 10.1093/hmg/ddl469.
Graham RR, Kozyrev SV, Baechler EC, Reddy MV, Plenge RM, Bauer JW, Ortmann WA, Koeuth T, González Escribano MF, Argentine and Spanish Collaborative Groups, Pons-Estel B, Petri M, Daly M, Gregersen PK, Martín J, Altshuler D, Behrens TW, Alarcón-Riquelme ME: A common haplotype of interferon regulatory factor 5 (IRF5) regulates splicing and expression and is associated with increased risk of systemic lupus erythematosus. Nat Genet. 2006, 38: 550-555. 10.1038/ng1782.
Sigurdsson S, Göring HH, Kristjansdottir G, Milani L, Nordmark G, Sandling JK, Eloranta ML, Feng D, Sangster-Guity N, Gunnarsson I, Svenungsson E, Sturfelt G, Jönsen A, Truedsson L, Barnes BJ, Alm G, Rönnblom L, Syvänen AC: Comprehensive evaluation of the genetic variants of interferon regulatory factor 5 (IRF5) reveals a novel 5 bp length polymorphism as strong risk factor for systemic lupus erythematosus. Hum Mol Genet. 2008, 17: 872-881. 10.1093/hmg/ddm359.
Takaoka A, Yanai H, Kondo S, Duncan G, Negishi H, Mizutani T, Kano S, Honda K, Ohba Y, Mak TW, Taniguchi T: Integral role of IRF-5 in the gene induction programme activated by Toll-like receptors. Nature. 2005, 434: 243-249. 10.1038/nature03308.
Dieguez-Gonzalez R, Calaza M, Perez-Pampin E, de la Serna AR, Fernandez-Gutierrez B, Castañeda S, Largo R, Joven B, Narvaez J, Navarro F, Marenco JL, Vicario JL, Blanco FJ, Fernandez-Lopez JC, Caliz R, Collado-Escobar MD, Carreño L, Lopez-Longo J, Cañete JD, Gomez-Reino JJ, Gonzalez A: Association of interferon regulatory factor 5 haplotypes, similar to that found in systemic lupus erythematosus, in a large subgroup of patients with rheumatoid arthritis. Arthritis Rheum. 2008, 58: 1264-1274. 10.1002/art.23426.
Plenge RM, Cotsapas C, Davies L, Price AL, de Bakker PI, Maller J, Pe'er I, Burtt NP, Blumenstiel B, DeFelice M, Parkin M, Barry R, Winslow W, Healy C, Graham RR, Neale BM, Izmailova E, Roubenoff R, Parker AN, Glass R, Karlson EW, Maher N, Hafler DA, Lee DM, Seldin MF, Remmers EF, Lee AT, Padyukov L, Alfredsson L, Coblyn J, et al: Two independent alleles at 6q23 associated with risk of rheumatoid arthritis. Nat Genet. 2007, 39: 1477-1482. 10.1038/ng.2007.27.
Plenge RM, Seielstad M, Padyukov L, Lee AT, Remmers EF, Ding B, Liew A, Khalili H, Chandrasekaran A, Davies LR, Li W, Tan AK, Bonnard C, Ong RT, Thalamuthu A, Pettersson S, Liu C, Tian C, Chen WV, Carulli JP, Beckman EM, Altshuler D, Alfredsson L, Criswell LA, Amos CI, Seldin MF, Kastner DL, Klareskog L, Gregersen PK: TRAF1-C5 as a risk locus for rheumatoid arthritis - a genomewide study. N Engl J Med. 2007, 357: 1199-1209. 10.1056/NEJMoa073491.
Komander D, Barford D: Structure of the A20 OTU domain and mechanistic insights into deubiquitination. Biochem J. 2008, 409: 77-85. 10.1042/BJ20071399.
Sun SC: Deubiquitylation and regulation of the immune response. Nat Rev Immunol. 2008, 8: 501-511. 10.1038/nri2337.
Lee EG, Boone DL, Chai S, Libby SL, Chien M, Lodolce JP, Ma A: Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science. 2000, 289: 2350-2354. 10.1126/science.289.5488.2350.
Graham RR, Cotsapas C, Davies L, Hackett R, Lessard CJ, Leon JM, Burtt NP, Guiducci C, Parkin M, Gates C, Plenge RM, Behrens TW, Wither JE, Rioux JD, Fortin PR, Graham DC, Wong AK, Vyse TJ, Daly MJ, Altshuler D, Moser KL, Gaffney PM: Genetic variants near TNFAIP3 on 6q23 are associated with systemic lupus erythematosus. Nat Genet. 2008, 40: 1059-1061. 10.1038/ng.200.
Bates JS, Lessard CJ, Leon JM, Nguyen T, Battiest LJ, Rodgers J, Kaufman KM, James JA, Gilkeson GS, Kelly JA, Humphrey MB, Harley JB, Gray-McGuire C, Moser KL, Gaffney PM: Meta-analysis and imputation identifies a 109 kb risk haplotype spanning TNFAIP3 associated with lupus nephritis and hematologic manifestations. Genes Immun. 2009, 10: 470-477. 10.1038/gene.2009.31.
Kurreeman FA, Padyukov L, Marques RB, Schrodi SJ, Seddighzadeh M, Stoeken-Rijsbergen G, Helmvan Mil van der AH, Allaart CF, Verduyn W, Houwing-Duistermaat J, Alfredsson L, Begovich AB, Klareskog L, Huizinga TW, Toes RE: A candidate gene approach identifies the TRAF1/C5 region as a risk factor for rheumatoid arthritis. PLoS Med. 2007, 4: e278-10.1371/journal.pmed.0040278.
Kurreeman FA, Rocha D, Houwing-Duistermaat J, Vrijmoet S, Teixeira VH, Migliorini P, Balsa A, Westhovens R, Barrera P, Alves H, Vaz C, Fernandes M, Pascual-Salcedo D, Michou L, Bombardieri S, Radstake T, van Riel P, Putte van de L, Lopes-Vaz A, Prum B, Bardin T, Gut I, Cornelis F, Huizinga TW, Petit-Teixeira E, Toes RE, European Consortium on Rheumatoid Arthritis Families: Replication of the tumor necrosis factor receptor-associated factor 1/complement component 5 region as a susceptibility locus for rheumatoid arthritis in a European family-based study. Arthritis Rheum. 2008, 58: 2670-2674. 10.1002/art.23793.
Barton A, Thomson W, Ke X, Eyre S, Hinks A, Bowes J, Gibbons L, Plant D, Wellcome Trust Case Control Consortium, Wilson AG, Marinou I, Morgan A, Emery P, YEAR consortium, Steer S, Hocking L, Reid DM, Wordsworth P, Harrison P, Worthington J: Re-evaluation of putative rheumatoid arthritis susceptibility genes in the post-genome wide association study era and hypothesis of a key pathway underlying susceptibility. Hum Mol Genet. 2008, 17: 2274-2279. 10.1093/hmg/ddn128.
Carpentier I, Beyaert R: TRAF1 is a TNF inducible regulator of NF-kappaB activation. FEBS Lett. 1999, 460: 246-250. 10.1016/S0014-5793(99)01356-3.
Tsitsikov EN, Laouini D, Dunn IF, Sannikova TY, Davidson L, Alt FW, Geha RS: TRAF1 is a negative regulator of TNF signaling. enhanced TNF signaling in TRAF1-deficient mice. Immunity. 2001, 15: 647-657. 10.1016/S1074-7613(01)00207-2.
Wang Y, Rollins SA, Madri JA, Matis LA: Anti-C5 monoclonal antibody therapy prevents collagen-induced arthritis and ameliorates established disease. Proc Natl Acad Sci USA. 1995, 92: 8955-8959. 10.1073/pnas.92.19.8955.
Wang Y, Kristan J, Hao L, Lenkoski CS, Shen Y, Matis LA: A role for complement in antibody-mediated inflammation: C5-deficient DBA/1 mice are resistant to collagen-induced arthritis. J Immunol. 2000, 164: 4340-4347.
Panoulas VF, Smith JP, Nightingale P, Kitas GD: Association of the TRAF1/C5 locus with increased mortality, particularly from malignancy or sepsis, in patients with rheumatoid arthritis. Arthritis Rheum. 2009, 60: 39-46. 10.1002/art.24176.
Larson RS, Springer TA: Structure and function of leukocyte integrins. Immunol Rev. 1990, 114: 181-217. 10.1111/j.1600-065X.1990.tb00565.x.
Sohn JH, Bora PS, Suk HJ, Molina H, Kaplan HJ, Bora NS: Tolerance is dependent on complement C3 fragment iC3b binding to antigen-presenting cells. Nat Med. 2003, 9: 206-212. 10.1038/nm814.
Ehirchiou D, Xiong Y, Xu G, Chen W, Shi Y, Zhang L: CD11b facilitates the development of peripheral tolerance by suppressing Th17 differentiation. J Exp Med. 2007, 204: 1519-1524. 10.1084/jem.20062292.
Han S, Kim-Howard X, Deshmukh H, Kamatani Y, Viswanathan P, Guthridge JM, Thomas K, Kaufman KM, Ojwang J, Rojas-Villarraga A, Baca V, Orozco L, Rhodes B, Choi CB, Gregersen PK, Merrill JT, James JA, Gaffney PM, Moser KL, Jacob CO, Kimberly RP, Harley JB, Bae SC, Anaya JM, Alarcón-Riquelme ME, Matsuda K, Vyse TJ, Nath SK: Evaluation of imputation-based association in and around the integrin-alpha-M (ITGAM) gene and replication of robust association between a non-synonymous functional variant within ITGAM and systemic lupus erythematosus (SLE). Hum Mol Genet. 2009, 18: 1171-1180. 10.1093/hmg/ddp007.
Nath SK, Han S, Kim-Howard X, Kelly JA, Viswanathan P, Gilkeson GS, Chen W, Zhu C, McEver RP, Kimberly RP, Alarcón-Riquelme ME, Vyse TJ, Li QZ, Wakeland EK, Merrill JT, James JA, Kaufman KM, Guthridge JM, Harley JB: A nonsynonymous functional variant in integrin-alpha(M) (encoded by ITGAM) is associated with systemic lupus erythematosus. Nat Genet. 2008, 40: 152-154. 10.1038/ng.71.
Han S, Kim-Howard X, Deshmukh H, Kamatani Y, Viswanathan P, Guthridge JM, Thomas K, Kaufman KM, Ojwang J, Rojas-Villarraga A, Baca V, Orozco L, Rhodes B, Choi CB, Gregersen PK, Merrill JT, James JA, Gaffney PM, Moser KL, Jacob CO, Kimberly RP, Harley JB, Bae SC, Anaya JM, Alarcón-Riquelme ME, Matsuda K, Vyse TJ, Nath SK: Evaluation of imputation-based association in and around the integrin-alpha-M (ITGAM) gene and replication of robust association between a non-synonymous functional variant within ITGAM and systemic lupus erythematosus (SLE). Hum Mol Genet. 2009, 18: 1171-180. 10.1093/hmg/ddp007.
Willcocks LC, Lyons PA, Clatworthy MR, Robinson JI, Yang W, Newland SA, Plagnol V, McGovern NN, Condliffe AM, Chilvers ER, Adu D, Jolly EC, Watts R, Lau YL, Morgan AW, Nash G, Smith KG: Copy number of FCGR3B, which is associated with systemic lupus erythematosus, correlates with protein expression and immune complex uptake. J Exp Med. 2008, 205: 1573-1582. 10.1084/jem.20072413.
Fanciulli M, Norsworthy PJ, Petretto E, Dong R, Harper L, Kamesh L, Heward JM, Gough SC, de Smith A, Blakemore AI, Froguel P, Owen CJ, Pearce SH, Teixeira L, Guillevin L, Graham DS, Pusey CD, Cook HT, Vyse TJ, Aitman TJ: FCGR3B copy number variation is associated with susceptibility to systemic, but not organ-specific, autoimmunity. Nat Genet. 2007, 39: 721-723. 10.1038/ng2046.
Begovich AB, Carlton VE, Honigberg LA, Schrodi SJ, Chokkalingam AP, Alexander HC, Ardlie KG, Huang Q, Smith AM, Spoerke JM, Conn MT, Chang M, Chang SY, Saiki RK, Catanese JJ, Leong DU, Garcia VE, McAllister LB, Jeffery DA, Lee AT, Batliwalla F, Remmers E, Criswell LA, Seldin MF, Kastner DL, Amos CI, Sninsky JJ, Gregersen PK: A missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associated with rheumatoid arthritis. Am J Hum Genet. 2004, 75: 330-337. 10.1086/422827.
Jawaheer D, Seldin MF, Amos CI, Chen WV, Shigeta R, Etzel C, Damle A, Xiao X, Chen D, Lum RF, Monteiro J, Kern M, Criswell LA, Albani S, Nelson JL, Clegg DO, Pope R, Schroeder HW, Bridges SL, Pisetsky DS, Ward R, Kastner DL, Wilder RL, Pincus T, Callahan LF, Flemming D, Wener MH, Gregersen PK, North American Rheumatoid Arthritis Consortium: Screening the genome for rheumatoid arthritis susceptibility genes: a replication study and combined analysis of 512 multicase families. Arthritis Rheum. 2003, 48: 906-916. 10.1002/art.10989.
Lee HS, Korman BD, Le JM, Kastner DL, Remmers EF, Gregersen PK, Bae SC: Genetic risk factors for rheumatoid arthritis differ in Caucasian and Korean populations. Arthritis Rheum. 2009, 60: 364-371. 10.1002/art.24245.
Ikari K, Momohara S, Inoue E, Tomatsu T, Hara M, Yamanaka H, Kamatani N: Haplotype analysis revealed no association between the PTPN22 gene and RA in a Japanese population. Rheumatology (Oxford). 2006, 45: 1345-1348. 10.1093/rheumatology/kel169.
Cloutier JF, Veillette A: Cooperative inhibition of T-cell antigen receptor signaling by a complex between a kinase and a phosphatase. J Exp Med. 1999, 189: 111-121. 10.1084/jem.189.1.111.
Gjörloff-Wingren A, Saxena M, Williams S, Hammi D, Mustelin T: Characterization of TCR-induced receptor-proximal signaling events negatively regulated by the protein tyrosine phosphatase PEP. Eur J Immunol. 1999, 29: 3845-3854. 10.1002/(SICI)1521-4141(199912)29:12<3845::AID-IMMU3845>3.0.CO;2-U.
Vang T, Miletic AV, Arimura Y, Tautz L, Rickert RC, Mustelin T: Protein tyrosine phosphatases in autoimmunity. Annu Rev Immunol. 2008, 26: 29-55. 10.1146/annurev.immunol.26.021607.090418.
Orrú V, Tsai SJ, Rueda B, Fiorillo E, Stanford SM, Dasgupta J, Hartiala J, Zhao L, Ortego-Centeno N, D'Alfonso S, Italian Collaborative Group, Arnett FC, Wu H, Gonzalez-Gay MA, Tsao BP, Pons-Estel B, Alarcon-Riquelme ME, He Y, Zhang ZY, Allayee H, Chen XS, Martin J, Bottini N: A loss-of-function variant of PTPN22 is associated with reduced risk of systemic lupus erythematosus. Hum Mol Genet. 2009, 18: 569-579. 10.1093/hmg/ddn363.
Orozco G, García-Porrúa C, López-Nevot MA, Raya E, González-Gay MA, Martín J: Lack of association between ankylosing spondylitis and a functional polymorphism of PTPN22 proposed as a general susceptibility marker for autoimmunity. Ann Rheum Dis. 2006, 65: 687-688. 10.1136/ard.2005.046094.
Watford WT, Hissong BD, Bream JH, Kanno Y, Muul L, O'Shea JJ: Signaling by IL-12 and IL-23 and the immunoregulatory roles of STAT4. Immunol Rev. 2004, 202: 139-156. 10.1111/j.0105-2896.2004.00211.x.
Mathur AN, Chang HC, Zisoulis DG, Stritesky GL, Yu Q, O'Malley JT, Kapur R, Levy DE, Kansas GS, Kaplan MH: Stat3 and Stat4 direct development of IL-17-secreting Th cells. J Immunol. 2007, 178: 4901-4907.
Remmers EF, Plenge RM, Lee AT, Graham RR, Hom G, Behrens TW, de Bakker PI, Le JM, Lee HS, Batliwalla F, Li W, Masters SL, Booty MG, Carulli JP, Padyukov L, Alfredsson L, Klareskog L, Chen WV, Amos CI, Criswell LA, Seldin MF, Kastner DL, Gregersen PK: STAT4 and the risk of rheumatoid arthritis and systemic lupus erythematosus. N Engl J Med. 2007, 357: 977-986. 10.1056/NEJMoa073003.
Namjou B, Sestak AL, Armstrong DL, Zidovetzki R, Kelly JA, Jacob N, Ciobanu V, Kaufman KM, Ojwang JO, Ziegler J, Quismorio FP, Reiff A, Myones BL, Guthridge JM, Nath SK, Bruner GR, Mehrian-Shai R, Silverman E, Klein-Gitelman M, McCurdy D, Wagner-Weiner L, Nocton JJ, Putterman C, Bae SC, Kim YJ, Petri M, Reveille JD, Vyse TJ, Gilkeson GS, Kamen DL, et al: High-density genotyping of STAT4 reveals multiple haplotypic associations with systemic lupus erythematosus in different racial groups. Arthritis Rheum. 2009, 60: 1085-1095. 10.1002/art.24387.
Kastelein RA, Hunter CA, Cua DJ: Discovery and biology of IL-23 and IL-27: related but functionally distinct regulators of inflammation. Annu Rev Immunol. 2007, 25: 221-242. 10.1146/annurev.immunol.22.012703.104758.
Nair RP, Duffin KC, Helms C, Ding J, Stuart PE, Goldgar D, Gudjonsson JE, Li Y, Tejasvi T, Feng BJ, Ruether A, Schreiber S, Weichenthal M, Gladman D, Rahman P, Schrodi SJ, Prahalad S, Guthery SL, Fischer J, Liao W, Kwok PY, Menter A, Lathrop GM, Wise CA, Begovich AB, Voorhees JJ, Elder JT, Krueger GG, Bowcock AM, Abecasis GR, Collaborative Association Study of Psoriasis: Genome-wide scan reveals association of psoriasis with IL-23 and NF-kappaB pathways. Nat Genet. 2009, 41: 199-204. 10.1038/ng.311.
Silverberg MS, Cho JH, Rioux JD, McGovern DP, Wu J, Annese V, Achkar JP, Goyette P, Scott R, Xu W, Barmada MM, Klei L, Daly MJ, Abraham C, Bayless TM, Bossa F, Griffiths AM, Ippoliti AF, Lahaie RG, Latiano A, Paré P, Proctor DD, Regueiro MD, Steinhart AH, Targan SR, Schumm LP, Kistner EO, Lee AT, Gregersen PK, Rotter JI, et al: Ulcerative colitis-risk loci on chromosomes 1p36 and 12q15 found by genome-wide association study. Nat Genet. 2009, 41: 216-220. 10.1038/ng.275.
Yokoyama K, Su Ih IH, Tezuka T, Yasuda T, Mikoshiba K, Tarakhovsky A, Yamamoto T: BANK regulates BCR-induced calcium mobilization by promoting tyrosine phosphorylation of IP(3) receptor. EMBO J. 2002, 21: 83-92. 10.1093/emboj/21.1.83.
Kozyrev SV, Abelson AK, Wojcik J, Zaghlool A, Linga Reddy MV, Sanchez E, Gunnarsson I, Svenungsson E, Sturfelt G, Jönsen A, Truedsson L, Pons-Estel BA, Witte T, D'Alfonso S, Barizzone N, Danieli MG, Gutierrez C, Suarez A, Junker P, Laustrup H, González-Escribano MF, Martin J, Abderrahim H, Alarcón-Riquelme ME: Functional variants in the B-cell gene BANK1 are associated with systemic lupus erythematosus. Nat Genet. 2008, 40: 211-216. 10.1038/ng.79.
Kurosaki T, Hikida M: Tyrosine kinases and their substrates in B lymphocytes. Immunol Rev. 2009, 228: 132-148. 10.1111/j.1600-065X.2008.00748.x.
Hibbs ML, Tarlinton DM, Armes J, Grail D, Hodgson G, Maglitto R, Stacker SA, Dunn AR: Multiple defects in the immune system of Lyn-deficient mice, culminating in autoimmune disease. Cell. 1995, 83: 301-311. 10.1016/0092-8674(95)90171-X.
Lu R, Vidal GS, Kelly JA, Delgado-Vega AM, Howard XK, Macwana SR, Dominguez N, Klein W, Burrell C, Harley IT, Kaufman KM, Bruner GR, Moser KL, Gaffney PM, Gilkeson GS, Wakeland EK, Li QZ, Langefeld CD, Marion MC, Divers J, Alarcón GS, Brown EE, Kimberly RP, Edberg JC, Ramsey-Goldman R, Reveille JD, McGwin G, Vilá LM, Petri MA, Bae SC, et al: Genetic associations of LYN with systemic lupus erythematosus. Genes Immun. 2009, 10: 397-403. 10.1038/gene.2009.19.
Croft M: The role of TNF superfamily members in T-cell function and diseases. Nat Rev Immunol. 2009, 9: 271-285. 10.1038/nri2526.
Cunninghame Graham DS, Graham RR, Manku H, Wong AK, Whittaker JC, Gaffney PM, Moser KL, Rioux JD, Altshuler D, Behrens TW, Vyse TJ: Polymorphism at the TNF superfamily gene TNFSF4 confers susceptibility to systemic lupus erythematosus. Nat Genet. 2008, 40: 83-89. 10.1038/ng.2007.47.
Manku H, Graham DS, Vyse TJ: Association of the co-stimulator OX40L with systemic lupus erythematosus. J Mol Med. 2009, 87: 229-234. 10.1007/s00109-008-0431-2.
Fillatreau S, Gray D, Anderton SM: Not always the bad guys: B cells as regulators of autoimmune pathology. Nat Rev Immunol. 2008, 8: 391-397. 10.1038/nri2315.
Suzuki A, Yamada R, Chang X, Tokuhiro S, Sawada T, Suzuki M, Nagasaki M, Nakayama-Hamada M, Kawaida R, Ono M, Ohtsuki M, Furukawa H, Yoshino S, Yukioka M, Tohma S, Matsubara T, Wakitani S, Teshima R, Nishioka Y, Sekine A, Iida A, Takahashi A, Tsunoda T, Nakamura Y, Yamamoto K: Functional haplotypes of PADI4, encoding citrullinating enzyme peptidylarginine deiminase 4, are associated with rheumatoid arthritis. Nat Genet. 2003, 34: 395-402. 10.1038/ng1206.
Kang N, Clarke AJ, Nicholson IA, Chard RB: Circulatory arrest for repair of postcoarctation site aneurysm. Ann Thorac Surg. 2004, 77: 2029-2033. 10.1016/j.athoracsur.2003.11.007.
Gandjbakhch F, Fajardy I, Ferré B, Dubucquoi S, Flipo RM, Roger N, Solau-Gervais E: A functional haplotype of PADI4 gene in rheumatoid arthritis: positive correlation in a French population. J Rheumatol. 2009, 36: 881-886. 10.3899/jrheum.080398.
Martinez A, Valdivia A, Pascual-Salcedo D, Lamas JR, Fernández-Arquero M, Balsa A, Fernández-Gutiérrez B, de la Concha EG, Urcelay E: PADI4 polymorphisms are not associated with rheumatoid arthritis in the Spanish population. Rheumatology (Oxford). 2005, 44: 1263-1266. 10.1093/rheumatology/kei008.
Caponi L, Petit-Teixeira E, Sebbag M, Bongiorni F, Moscato S, Pratesi F, Pierlot C, Osorio J, Chapuy-Regaud S, Guerrin M, Cornelis F, Serre G, Migliorini P, ECRAF: A family based study shows no association between rheumatoid arthritis and the PADI4 gene in a white French population. Ann Rheum Dis. 2005, 64: 587-593. 10.1136/ard.2004.026831.
Ng PC, Levy S, Huang J, Stockwell TB, Walenz BP, Li K, Axelrod N, Busam DA, Strausberg RL, Venter JC: Genetic variation in an individual human exome. PLoS Genet. 2008, 4: e1000160-10.1371/journal.pgen.1000160.
Jakobsson M, Scholz SW, Scheet P, Gibbs JR, VanLiere JM, Fung HC, Szpiech ZA, Degnan JH, Wang K, Guerreiro R, Bras JM, Schymick JC, Hernandez DG, Traynor BJ, Simon-Sanchez J, Matarin M, Britton A, Leemput van de J, Rafferty I, Bucan M, Cann HM, Hardy JA, Rosenberg NA, Singleton AB: Genotype, haplotype and copy-number variation in worldwide human populations. Nature. 2008, 451: 998-1003. 10.1038/nature06742.
Bhangale TR, Rieder MJ, Nickerson DA: Estimating coverage and power for genetic association studies using near-complete variation data. Nat Genet. 2008, 40: 841-843. 10.1038/ng.180.
Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HR, Boutell JM, Bryant J, Carter RJ, Keira Cheetham R, Cox AJ, Ellis DJ, Flat-bush MR, Gormley NA, Humphray SJ, Irving LJ, Karbelashvili MS, Kirk SM, Li H, Liu X, Maisinger KS, Murray LJ, Obradovic B, Ost T, Parkinson ML, Pratt MR, et al: Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008, 456: 53-59. 10.1038/nature07517.
Kryukov GV, Shpunt A, Stamatoyannopoulos JA, Sunyaev SR: Power of deep, all-exon resequencing for discovery of human trait genes. Proc Natl Acad Sci USA. 2009, 106: 3871-3876. 10.1073/pnas.0812824106.
Chen Y, Zhu J, Lum PY, Yang X, Pinto S, MacNeil DJ, Zhang C, Lamb J, Edwards S, Sieberts SK, Leonardson A, Castellini LW, Wang S, Champy MF, Zhang B, Emilsson V, Doss S, Ghazalpour A, Horvath S, Drake TA, Lusis AJ, Schadt EE: Variations in DNA elucidate molecular networks that cause disease. Nature. 2008, 452: 429-435. 10.1038/nature06757.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Rights and permissions
About this article
Cite this article
Clarke, A., Vyse, T.J. Genetics of rheumatic disease. Arthritis Res Ther 11, 248 (2009). https://doi.org/10.1186/ar2781
Published:
DOI: https://doi.org/10.1186/ar2781