
Abstract
This article will review how epidemiological studies have advanced our knowledge of both genetic and environmental risk factors for rheumatic diseases over the past decade. The major rheumatic diseases, including rheumatoid arthritis, juvenile idiopathic arthritis, psoriatic arthritis, ankylosing spondylitis, systemic lupus erythematosus, scleroderma, osteoarthritis, gout, and fibromyalgia, and chronic widespread pain, will be covered. Advances discussed will include how a number of large prospective studies have improved our knowledge of risk factors, including diet, obesity, hormones, and smoking. The change from small-scale association studies to genome-wide association studies using gene chips to reveal new genetic risk factors will also be reviewed.
Similar content being viewed by others


Introduction
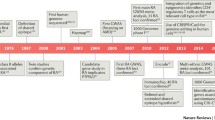
This article will review epidemiological studies that have advanced the knowledge of both genetic and environmental risk factors for the rheumatic diseases, outlining the major advances that have been achieved over the past decade (Table 1). It will focus on the following diseases: rheumatoid arthritis (RA), juvenile idiopathic arthritis (JIA), psoriatic arthritis (PsA), ankylosing spondylitis (AS), systemic lupus erythematosus (SLE), scleroderma (Scl), osteoarthritis (OA), gout, and fibromyalgia (FM) and chronic widespread pain (CWP).
A number of large prospective studies have improved our knowledge of risk factors: the Framingham Study [1] and the Chingford 1000 Women Study [2] for OA, the Nurses' Health Study cohort for RA [3] and SLE [4], the European Prospective Investigation of Cancer in Norfolk (EPIC-Norfolk) for inflammatory polyarthritis [5], and the Health Professionals Follow-up Study for gout [6]. These types of studies provide valuable and robust information. Unfortunately, epidemiological data often are obtained from retrospective studies and underpowered case-control studies, resulting in contradictory findings (for example, studies on the role of caffeine in RA). Although some of the studies have found significant associations with novel risk factors, these studies often suffer from poor design. Meta-analyses have also been performed in an attempt to form conclusions from the available epidemiological data and these are also discussed.
Over the past decade, genetic research has moved from the approach of small-scale association studies, to test for candidate genes in case-control studies, to whole-genome scans of linkage based on sibling pairs which proved to be limited in the small numbers of both pairs and markers (both in the hundreds). The more recent and exciting approach has been genome-wide association studies using gene chips which have allowed hundreds of thousands of single-nucleotide polymorphisms (SNPs) to be investigated as exemplified by the Wellcome Trust Case-Control Consortium (WTCCC) study of common diseases (including RA) [7]. The advantage to this approach is clearly the opportunity to identify novel genes for the diseases; however, the disadvantage is that it results in large numbers of hints that require verification in further studies to validate the results.
In general, the studies discussed in this review identify risk factors in whole populations of patients with the disease but it is more likely that each of the individual disease phenotypes results from a number of different combinations of genetic and environmental risk factors. Thus, some risk factors may have a strong effect but only in a small proportion of patients, whereas others will have weak effects and be present in a greater number of individuals but require the involvement of other risk factors. Thus, the size of any increased risk is not a reflection of the level of its attribution to disease causation. However, the sense of strength of risk in this review has been split arbitrarily into three groups based on the typically reported strength of association: 'small' (odds ratio [OR] or relative risk [RR] of less than 2), 'moderate' (OR or RR of between 2 and 5), or 'substantial' (OR or RR of greater than 5).
Rheumatoid arthritis
Environmental risk factors
Studies of environmental risk factors in RA have focused on diet, smoking, and hormones [8]. Several studies have investigated consumption of coffee/tea/caffeine as a risk factor but with mixed conclusions. Caffeine has been reported to moderately increase the risk of rheumatoid factor (RF)-positive RA, but no increased risk for RF-negative RA was found [9]. Decaffeinated coffee has been associated with a moderately increased risk of RA, whereas tea has been shown to have a protective effect [10]. The authors suggest that the decaffeination process (use of industrial solvents) and small traces of solvents may play a role in the disease whereas tea may have both anti-inflammatory and antioxidative properties [10]. However, other studies have found no association of caffeine/coffee consumption with RA [3]. Clearly, studies that are more robust are needed to verify these results.
The so-called 'Mediterranean diet' has been linked with health benefits for a number of diseases and this is also true for RA [11, 12]. High consumption of olive oil, oil-rich fish, fruit and vegetables [13], or vitamin D [14] has been shown to have a protective role in the development of RA. High consumption of red meat and meat products [5] has been associated with a moderately increased risk of inflammatory polyarthritis, but no risk was found in a more recent study [15].
Data on the link between smoking and RA are more compelling and include recent studies implicating a gene-environment interaction (see below). The duration and intensity of smoking have been linked to the development of RA in postmenopausal women [16]. Current smokers and those who had quit for 10 years or less were found to have a small increased risk of RA, whereas those who had quit for more than 10 years had no increased risk. Heavy cigarette smoking has been linked with a substantial increased risk of RA [17] (over 13-fold) and there was an increasing association between increasing pack-years of smoking and RA. Current smoking has been found to be a risk factor for RA, with the risk moderately increased in men and more so in men with seropositive RA [18]. Other studies have also shown a small increased risk due to smoking for seropositive RA in both women and men but have not shown an increased risk for seronegative RA [19]. This risk was evident in subjects who had long-term smoking habits (>20 years) and was evident even if daily smoking intensity was only moderate. Duration of smoking rather than intensity has also been found to be a risk factor in a study of female health professionals [20]. Smoking has also been linked with an increase in both the severity of RA and disease activity [21, 22], supporting a role for smoking in the development of RA. Other host factors that have been associated with RA include blood transfusion and obesity [23] and (high) birth weight [24], which have been linked with a moderate increased risk, and breast-feeding [25] and alcohol [26], which have been linked with a decreased risk/protective role. Stress has also been reported to have a role in the development of RA [27].
Genetic risk factors
Genetic factors implicated in RA have been widely studied using both candidate genes and whole-genome screens [28]. Whereas the strongest genetic risk factor for RA remains the HLA DRB1 shared epitope (SE), other candidate genes have been consistently implicated. In particular, an SNP (R620W) in the protein tyrosine phosphatase (PTPN22) gene, which has regulatory activities for both T and B cells, has been associated with RA [29]; furthermore, this has been replicated in well-powered studies in different populations [30–33]. This polymorphism has been associated with other autoimmune diseases, including JIA and SLE [28]. Studies on peptidyl arginine (PADI4) have shown a significant association [34] but so far this has been replicated in one other Japanese study [35] only and not in populations from the UK [36], France [37], or Spain [38]. A recent meta-analysis of three Asian and six European studies has shown that PADI4 polymorphisms were associated with Asian populations; in European populations, only PADI4_94 had a significant association [39]. Genes such as CTLA4, FCRL3, and major histocompatibility complex 2A (MHC2A) have also been the focus of recent research [28].
The search for novel genes has been advanced by the powerful approach of genome-wide association studies as typified by the UK WTCCC. This has identified three genes with independent associations for RA: two that have been reported to have strong associations (HLA-DRB1 and PTPN22) and a further one on chromosome 7 that had different genetic effects between genders with a strong and apparently additive effect on disease status in females [7]. Further susceptibility loci are likely to be discovered using this approach. Similarly, alleles from 14 genes from over 2,300 cases and 1,700 controls from the North American Rheumatoid Arthritis Consortium (NARAC) (the US version of the WTCCC) and the Swedish Epidemiological Investigation of Rheumatoid Arthritis (EIRA) collections have supported evidence for association of RA with PTPN22, CTLA4, and PADI4 (NARAC cohort only) [40]. There is also evidence that there is a genetic overlap with other autoimmune diseases (SLE, AS, multiple sclerosis, and inflammatory bowel disease) [41]. One of the newer and possibly more exciting areas of research focuses on evidence that certain polymorphisms can predict the response of a patient to treatment [42] and this is likely to be the focus of a number of future studies.
Gene-environment interactions
One of the most interesting studies has shown evidence of an important gene-environment interaction between the SE and smoking [43]. This Swedish population-based case-control study showed that the risk of developing RF-positive RA substantially increased in smokers carrying double copies of SE genes (RR = 15.7) compared with smokers with no copies of SE genes (RR = 2.4). Recent research has also shown additive and multiplicative interactions between PTPN22 and heavy cigarette smoking [44]. It has also been proposed that risk factors such as smoking, alcohol and coffee consumption, obesity, and oral contraceptive use may depend on the presence or absence of autoantibodies to cyclic citrullinated peptides [45, 46].
Juvenile idiopathic arthritis
Epidemiological studies of JIA have been hampered by a lack of standardised criteria and case ascertainment, resulting in wide-ranging results: reported prevalence ranges from 0.07 to 4.01 per 1,000 children, and annual incidence varies from 0.008 to 0.226 per 1,000 children [47]. Hopefully, the development of new diagnostic criteria will aid future studies in having results that are more consistent. Ethnicity has been studied and European descent has been associated with a moderately increased risk of JIA; additionally, JIA subtypes differed significantly between ethnic groups [48]. There have been few developments in terms of environmental risk factors, although infection remains the most favoured hypothesis.
Genetic risk factors
Major advances in epidemiological studies of JIA have focused mainly on genetic aspects. A genome-wide scan in 121 families (247 affected children) confirmed linkage of juvenile RA to the HLA region [49]. In addition, early-onset polyarticular disease has been linked to chromosome 7q11 and pauciarticular disease has been linked to chromosome 19p13, suggesting that multiple genes are involved in the susceptibility to juvenile RA. Other candidate genes, including polymorphisms in the migration inhibitory factor (MIF) gene, have been associated with JIA. A study of UK JIA patients showed that patients with an MIF-173*C allele had a small increased risk of JIA [50], and serum MIF levels were also higher in patients with this allele. An SNP in the PTPN22 gene (a gene associated with both RA and SLE) has also been shown to have a novel association with JIA [30]. A recent meta-analysis has confirmed that the T allele and the T/T genotype of PTPN22 C1858T are associated with JIA [51]. Polymorphisms in the NRAMP1 gene may also play a role in the pathogenesis of JIA [52]. There is some evidence that a potentially protective CC genotype of the interleukin-6 (IL-6) gene is reduced in young patients [53].
Psoriatic arthritis
Epidemiologically, PsA is a complex disease to study as it is not simple to disentangle whether the risk factors revealed are for the complete disease phenotype of PsA or for one of its two components. Studies that compare PsA with healthy controls are not able to address this.
Environmental risk factors
Studies of environmental risk factors for PsA have focused on infection-related triggers and hormones. In a recent case-control study, exposure to rubella vaccination substantially increased the risk of PsA whereas injury requiring medical consultation, recurrent oral ulcers, and moving house all moderately increased the risk of PsA [54]. The strongest associations were with trauma, adding support to the hypothesis of a 'deep Koebner phenomenon' in PsA. These data suggest that infection-related triggers may be relevant and further studies are required to verify these results. In a nested case-control study, corticosteroid use (moderate increased risk) and pregnancy (decreased risk) were both associated with PsA, suggesting that changes to the immune system may play a role in this disease [55].
Genetic risk factors
Developments in the pathogenesis of PsA again have been mainly in the genetic field. There is evidence that caspase recruitment domain 15 (CARD15), a susceptibility gene for Crohn's disease, has a role in PsA, and this is supported by the fact that patients with Crohn's disease have an increased incidence of psoriasis. Initial reports suggested that over 38% of probands with PsA had at least one variant of the CARD15 gene compared with 12% of controls [56]. This pleiotropic autoimmune gene was proposed as the first non-MHC gene to be associated with PsA. Unfortunately, this has not been replicated in German [57] and Italian [58] cohorts; in these cohorts, no such association was found. A novel model that suggests that PsA susceptibility is determined by the balance of activating and inhibitory composite killer Ig-like receptor-HLA genotypes has been proposed [59]. Class I MHC chain-related gene A (MICA) may confer additional susceptibility to PsA. The MICA-A9 triplet repeat polymorphisms were present at a substantially higher frequency in PsA patients [60]. A linkage scan reported evidence that suggests that a locus on chromosome 16q is implicated in PsA; furthermore, the logarithm of the odds (LOD) score is much higher for paternal transmission than maternal transmission (4.19 and 1.03) [61]. Functional cytokine gene polymorphisms have also been associated with PsA [62], with tumour necrosis factor-alpha (TNF-α) -308 and TNF-β +252 polymorphisms being significantly associated with age at psoriasis onset, presence of joint erosions in PsA, and progression of joint erosions in early PsA. A genome-wide association study recently replicated associations of PsA with IL-23 receptor and IL-12B polymorphisms and also identified a novel locus on chromosome 4q27 [63]. A case-control study found evidence that HLA-Cw*06 and HLA-DRB1*07 are associated with the occurrence of type I psoriasis in patients with PsA, suggesting that the primary association is with age of onset of psoriasis [64].
Ankylosing spondylitis
Most of the epidemiological advances in AS have come from the ascertainment of novel genetic associations. Few environmental risk factors have been studied.
Genetic risk factors
Epidemiological studies have focused on the genetics behind AS. Twin studies have estimated the influence of genetics on the aetiopathogenesis of AS, indicating that additive genetic effects account for 94% of the variance in the causation of AS [65]. Genome-wide scans have confirmed the strong linkage of the MHC with AS, which is not surprising given the overwhelming relationship between HLA B27 and AS. However, this study suggested that only 31% of the susceptibility to AS is from genes in the MHC [66]. Thus, the search for non-MHC genes has gained much interest [67]. One of the most exciting developments has been the identification of two new loci for AS from a major genetic association scan: ARTS1 and IL-23R [68]. It was calculated from these studies that these genes are responsible for 26% (ARTS1) and 9% (IL-23R) of the population-attributable risk of AS. Another strong non-MHC linkage lies on chromosome 16q (overall LOD score of 4.7) [69]. Other scans have identified regions on chromosomes 6q and 11q [70]. Combined analysis of three whole-genome scans by the International Genetics of Ankylosing Spondylitis Consortium showed that regions on chromosomes 10q and 16q had evidence suggestive of linkage. Other regions showing nominal linkage (in two or more scans) were 1q, 3q, 5q, 6q, 9q, 17q, and 19q. Evidence was also confirmed for regions previously associated with AS on chromosomes 2q (the IL-1 gene cluster) and 22q (cytochrome P450 2D6 [CYP2D6]) [71].
A linkage study of chromosome 22 in families with AS-affected sibling pairs found that homozygosity for poor-metaboliser alleles in the CYP2D6 (debrisoquine hydroxylase) gene was associated with AS. The authors of that study postulated that altered metabolism of a natural toxin or antigen by this gene may increase the susceptibility to AS [72]. AS has also been linked to the IL-1RN*2 allele [73] as have other inflammatory diseases such as ulcerative colitis and Crohn's disease.
Systemic lupus erythematosus
Environmental risk factors
The majority of research into environmental risk factors for SLE has focused on the role of hormones due to the higher prevalence of this disease in women. In a recent population case-control study, breast-feeding was found to be associated with a reduced risk of SLE, with a trend for the number of babies fed and total weeks of breast-feeding [74]. Women who developed SLE had an earlier natural menopause whereas there was little association with current use or duration of use of hormonal replacement therapy or oral contraceptive pill and no association with the use of fertility drugs. The authors of that study proposed that early natural menopause may be a marker for susceptibility to SLE. However, another study has shown that risk of SLE or discoid lupus was moderately increased among current users of estrogens who had exposure of at least 2 years [75]. A prospective cohort study of women found no relationship between oral contraceptive use, either with duration or time since first use [4].
There has been a long-standing interest in the role of chemical exposures causing SLE. An interesting association has been found with lipstick use and SLE [76]. Researchers found that using lipstick 3 days per week was significantly associated with a small increased risk of SLE and this may be worth replicating in future studies on environmental risk factors. The authors suggest that chemicals (these include eosin, 2-octynoic acid [a xenobiotic], and phthalate isomers) present in lipsticks may be absorbed across the buccal mucosa and have a biological effect on disease development. Other risk factors associated with an increased risk of SLE include history of hypertension, drug allergy, type I/II sun-reactive skin type, and blood transfusions (all moderately increasing the risk) and family history substantially increasing the risk of SLE [77]. Consumption of alcohol has been inversely associated with the risk of SLE [77]. A small increased risk was found with smoking, but exposure to estrogen or hair-colouring dyes, both of which previously have been proposed as risk factors, was not associated.
Genetic risk factors
There has been a major increase in the understanding of the genetics behind SLE, particularly over the last year, and this topic is concisely summarised in a recent review [78]. Two high-density case-control genome-wide association analyses have been published [79, 80]. From these studies, overwhelming evidence for the association of various genes with SLE (MHC, ITGAM, IRF5, BLK, and STAT4 [79, 80]) and strong evidence for a role for PTPN22 and FCGR2A [51, 79, 81] have emerged. Other genes for which there is evidence of an association, including the TNF superfamily gene [82], in which the upstream region of TNFSF4 contains a single risk haplotype for SLE, have also emerged. Gene copy number variation may lead to variation in disease susceptibility as highlighted in studies on the complement component C4 in which patients with SLE had a lower gene copy number of total C4 and C4A [83]. Zero copies or one copy of the C4A gene increased the risk of disease susceptibility, whereas three or more copies appeared to have a protective role. The risk of SLE was substantially greater in subjects with only two copies of total C4, but those with five or more copies of C4 had a reduced risk of disease. Another area of research focus has been on the role of sex chromosomes in the development of SLE, especially given the high incidence in females. An interesting observation was the increased incidence of Klinefelter's syndrome (47, XXY) in male patients with SLE, in whom the frequency was substantially increased (14-fold) compared with men without SLE, suggesting that the susceptibility to SLE could be due to an X-chromosome gene-dose effect [84].
Scleroderma
Environmental risk factors
Epidemiological studies of Scl have focused on the role for toxic environmental exposures. Specifically, studies have carefully investigated silica and organic solvents as both are thought to stimulate the immune system and cause inflammation and increase antibody production. Recent reports show that occupational silica exposure moderately increases the risk of Scl, with medium exposure increasing the risk twofold and high exposure increasing the risk fourfold [85]. There is still interest in the relationship of silicone breast implants and Scl. However, a recent meta-analysis of nine cohorts, nine case-control studies, and two cross-sectional studies found no association with Scl or other connective tissue diseases [86]. Exposure to organic solvents remains a moderate risk factor and the presence of anti-Scl-70 autoantibodies may be an effect modifier as the association was stronger in patients with these antibodies [87]. However, such studies are difficult to undertake as exposure to other chemicals cannot be controlled.
Genetic risk factors
There is increasing evidence for a genetic role in Scl development [88]. The familial risk of Scl has been investigated in three large US cohorts with a significant increase in risk observed: 2.6% in families with Scl compared with 0.026% in the general public [89]. Studies of HLA alleles suggest that the DQA1*0501 allele is significantly increased in men with Scl compared with healthy men. This allele was found to be moderately associated with diffuse Scl in men but not with limited Scl [90]. HLA associations have also been studied in mutually exclusive autoantibody subgroups, lending support to the theory that Scl in subgroups are actually separate diseases [91]. Transforming growth factor-beta (TGF-β) and connective tissue growth factor may have roles in Scl but further studies are required [92, 93]. Increased expression of TGF receptors may account for the increased production of collagen type I by Scl fibroblasts [94]. Fibrillin-1 SNPs haplotypes have been strongly associated with Scl in Choctaw and Japanese populations [95]. Long-term foetal microchimerism is also still being investigated as a potential risk factor [96, 97].
Osteoarthritis
Environmental risk factors
Studies on environmental risk factors for OA have focused on obesity, physical activity, and prior joint injury, all of which may increase stress on the joints. There have been several major cohort studies of OA, including the Framingham Study [1], the Chingford 1000 Women Study [2], Bristol OA 500 [98], and the North Staffordshire Osteoarthritis Project (NorSTOP) [99]. From these and other studies, a number of risk factors, including high body mass index (BMI), previous injury, and regular sports participation, have been found [100, 101]. The main preventable risk factor, and hence the subject of many reports, is obesity, which has been shown to substantially increase the risk of knee OA [100, 102]. A moderate influence of obesity has also been found with hip OA [103]. Data from adult twins (St. Thomas' Hospital Adult Twin Registry) have shown a moderate association between high BMI and knee OA (OR = 3.9) [104]. Manek and colleagues, who gathered those data, also concluded that this association was not influenced by shared genetic factors. Other influences have been the effect of physical activity on OA [105]. One study found a moderate association between heavy physical workload and hip OA [106]. High levels of physical activity were found to be a moderate risk factor for OA of the knee/hip joints in men younger than 50 years [107].
Men with maximal grip strength have been found to have a moderately increased risk of OA in the proximal interphalangeal, metacarpophalangeal (MCP), and thumb base joints, and women with maximal grip strength have been found to have a moderately increased risk of OA in the MCP joints [108]. There is some evidence that occupation can increase the risk of hand OA. A recent case-control study showed that occupations involving repetitive thumb use and jobs in which there were perceived to be insufficient breaks were associated with OA of the carpometacarpal (CMC) joints [109]. However, not all studies agree and a cross-sectional study found no association with occupation, physical activity, or sports participation but found a moderate increase in risk for hand OA for self-reported digital fracture [110].
Genetic risk factors
Genetic studies in female twins have estimated that the genetic contribution to radiographic hip OA is 58% for OA overall and 64% for joint space narrowing [111]. Studies have revealed that disease risk differs for males and females at different sites and thus there may be specific genes rather than a single OA phenotype [112]. The IL-1 gene cluster is a key regulator in a number of chronic disease processes, and within this cluster, haplotypes such as IL1A-IL1B-IL1RN, which confers a moderate increase in the risk of OA, and IL1B-IL1RN, which confers a fivefold reduced risk, have been identified [113]. This cluster has also been proposed to confer susceptibility for knee OA but not hip OA [114]. Functional polymorphisms in the frizzled motif associated with bone development (FRZB) genes have been found to confer susceptibility to hip OA in females [115]. Radiographic OA is also associated with genotypes of the insulin-like growth factor I gene [116].
Data from the Rotterdam study showed that polymorphisms in the estrogen receptor-alpha (ESR1) gene are associated with radiographic knee OA in elderly men and women [117]. In a case-control study, several candidate genes were investigated: the strongest associations with clinical knee OA were found with a haplotype in ADAM12 (a disintegrin and metalloproteinase domain 12) and ESR1 in women [118] and again with ADAM12 in men along with the CILP (cartilage intermediate layer protein) haplotype. There is also evidence that the cyclooxygenase-2 enzyme encoded by PTGS2 has a role in the pathogenesis of knee OA [119]. The iodothyronine-deiodinase enzyme type 2 (DIO2) gene has been identified as a new susceptibility locus for OA, using a genome-wide linkage scan [120]. A meta-analysis of more than 11,000 individuals provided evidence for an SNP in GDF5 having a positive association with knee OA in both European and Asian cohorts [121]. Other genes so far implicated include the IL-1 gene cluster, matrilin-3 gene, IL-4 receptor, frizzled-related protein-3 (FRZB) gene, metalloproteinase gene ADAM12, and the asporin (ASPN) gene [122]. An ambitious study that will screen over 8,000 people with hip or knee OA and 6,000 healthy controls – arcOGEN (Arthritis Research Campaign Osteoarthritis GENetics) [123] – has been recently been announced and is likely to lead to the identification of further genes associated with OA.
The Dutch GARP (Genetics, Arthrosis, and Progression) study has shown that there is a moderate increased risk for familial aggregation of both hand and hip OA whereas there was no increased risk for knee OA [124]. That there should be greater genetic effects on OA of the hand compared with other sites is not surprising given the relatively weaker role for environmental (including mechanical) factors. The familial risk of hand OA has shown a moderate increase in risk in sisters of women affected with hand OA and this risk was substantially increased with the severity of the disease, with sisters of those with severe first CMC OA having an RR of 6.9 [125]. Whole-genome linkage scans on female twins have shown significant linkage of distal interphalangeal (DIP) OA on chromosome 2 and Tot-KL (Kellgren-Lawrence score for both hands) on chromosome 19 [126]. Polymorphisms in the vitamin D receptor (VDR) gene have also been associated with symmetrical hand OA, with a novel finding of a joint effect of low calcium intake and VDR polymorphisms (aT haplotype) having a moderate increased risk of symmetrical hand OA [127]. Data from the Framingham Study have shown that several chromosomes (DIP joint on chromosome 7, first CMC joint on chromosome 15, and two sites in the female DIP joint on chromosome 1 and first CMC joint on chromosome 20) contain susceptibility genes for hand OA and that a joint-specific approach rather than a global approach to hand OA may be more useful in further investigations of these regions [128]. Genome-wide scans have also revealed linkage peaks on chromosomes 4q, 3p, and the short arm of chromosome 2 for idiopathic hand OA [129]. Genome-wide significance was reached for a locus on chromosome 2 for first CMC and DIP joints coinciding with the MATN3 gene, which encodes the extracellular matrix protein, matrilin-3.
Gout
Environmental risk factors
Studies on environmental risk factors for gout have focused mainly on the long-established risk factors of high purine diet and diuretic use. The incidence of gout is increasing [130] and high alcohol consumption is no longer the only risk factor for the disease [131]. Other risk factors that have been proposed include longevity, metabolic syndromes [132], and use of certain pharmacologic agents [133]. The high incidence in some ethnic groups has no obvious host factor, and genetic factors may be implicated in these groups.
Dietary factors have a strong association with gout. Much of the research in this area has been conducted by Choi and colleagues [6, 134–137]. As part of a large prospective study in men (the Health Professionals Follow-up Study), a number of factors were associated with an increased risk of gout. Higher adiposity, hypertension, and diuretic use were all moderate risk factors, whereas weight loss had a protective role [136]. High intake of sugar-sweetened drinks and high fructose intake from fruit juice and fruit have been associated with a small increased risk of gout [137]. High meat intake and seafood intake (purine intake) have also been positively associated with gout with a small increase in risk [6]. In the same study, long-term coffee consumption was inversely associated with gout [138]. Consumption of low-fat dairy products has been shown to decrease the risk of gout [6]; milk proteins (casein and lactalbumin) can reduce serum uric acid levels in healthy individuals.
Genetic risk factors
Advances in the genetic factors behind gout have included a variation in the SLC2A gene, which appears to make it more difficult for uric acid to be removed from the blood [139]. A polymorphism in the TNF-α promoter gene has been shown to be significantly associated with gout [140]. Genetic studies have included families with purine metabolism defects and case-control studies of isolated aboriginal cohorts with primary gout [133].
Fibromyalgia and chronic widespread pain
These poorly defined conditions are nonetheless the target of many investigations seeking to unravel risk factors for their causation or severity.
Environmental risk factors
Studies on environmental risk factors for FM and CWP have focused on physical trauma and psychosocial factors. Physical trauma in the months prior to disease onset has been significantly associated with FM [141]. FM was found to be 13 times more likely in patients who had a prior injury to the cervical spine compared with those with injuries to the lower extremities [142]. In a population-based prospective study, three psychosocial factors independently predicted a moderate increased risk of the development of CWP: somatisation, health-seeking behaviour, and poor sleep [143].
Subjects with all three factors had a substantial increased risk of developing CWP.
There may be biologically based risk factors. Thus, abnormalities in the hypothalamic-pituitary-adrenal (HPA) stress-response system may predict the onset of CWP. In a recent study, high levels of cortisol after dexamethasone and high levels in evening saliva moderately increased the risk of CWP [144]. Low levels in morning saliva were also associated with a small increase in risk. These factors were both independent and additive predictors of CWP, with over 90% of new-onset cases of CWP being identified by one or more of these HPA factors.
Genetic risk factors
Perhaps surprisingly, there have been some interesting suggestions of a genetic basis to FM. FM has been shown to aggregate strongly in families: the odds of FM in a relative of a proband with FM versus the odds of FM in a relative of a proband with RA was 8.5 [145]. Genotypes in the promoter region of the serotonin transporter gene (5-HTT) were analysed in FM patients. A higher frequency of the S/S genotype was found in patients compared with controls [146], supporting the hypothesis of altered serotonin metabolism in FM patients. Family studies have also shown significant genetic linkage of the HLA region to FM [147]. Polymorphisms in the gene encoding the COMT (catechol-O-methyltransferase) enzyme may also have a role in FM as certain genotypes combined are higher in patients than controls and a third genotype was significantly lower in control groups [148].
Conclusion
Over the last 10 years, there have been some major epidemiological advances, particularly in the field of genetic risk factors, in which new candidate genes have been identified and useful gene-environment interactions have been studied. Studying lone environmental factors has been less fruitful. The problem epidemiologically is that these factors often explain only a small number of cases, and on their own, they are not sufficient to cause the disease; both of these issues present considerable epidemiological challenges. The hope is that, as we begin to understand more about the genetics behind the diseases and genetic studies become more technically practical, it will enable stratification by genetic subgroups to identify environmental triggers (such as smoking). However, in other disease areas, progress has been very slow and we still understand very little.
Note
The Scientific Basis of Rheumatology: A Decade of Progress
This article is part of a special collection of reviews, The Scientific Basis of Rheumatology: A Decade of Progress, published to mark Arthritis Research & Therapy's 10th anniversary.
Other articles in this series can be found at: http://arthritis-research.com/sbr
Abbreviations
- ADAM12 :
-
a disintegrin and metalloproteinase domain 12
- AS:
-
ankylosing spondylitis
- BMI:
-
body mass index
- CARD15:
-
caspase recruitment domain 15
- CMC:
-
carpometacarpal
- CWP:
-
chronic widespread pain
- CYP2D6:
-
cytochrome P450 2D6
- DIP:
-
distal interphalangeal
- FM:
-
fibromyalgia
- FRZB:
-
frizzled-related protein-3
- HPA:
-
hypothalamic-pituitary-adrenal
- IL:
-
interleukin
- JIA:
-
juvenile idiopathic arthritis
- LOD:
-
logarithm of the odds
- MCP:
-
metacarpophalangeal
- MHC:
-
major histocompatibility complex
- MICA:
-
class I major histocompatibility complex chain-related gene A
- MIF:
-
migration inhibitory factor
- NARAC:
-
North American Rheumatoid Arthritis Consortium
- OA:
-
osteoarthritis
- OR:
-
odds ratio
- PADI4:
-
peptidyl arginine
- PsA:
-
psoriatic arthritis
- PTPN22:
-
protein tyrosine phosphatase
- RA:
-
rheumatoid arthritis
- RF:
-
rheumatoid factor
- RR:
-
relative risk
- Scl:
-
scleroderma
- SE:
-
shared epitope
- SLE:
-
systemic lupus erythematosus
- SNP:
-
single-nucleotide polymorphism
- TGF:
-
transforming growth factor
- TNF:
-
tumour necrosis factor
- VDR:
-
vitamin D receptor
- WTCCC:
-
Wellcome Trust Case-Control Consortium.
References
Felson DT, Zhang Y, Hannan MT, Naimark A, Weissman B, Aliabadi P, Levy D: Risk factors for incident radiographic knee osteoarthritis in the elderly: the Framingham Study. Arthritis Rheum. 1997, 40: 728-733. 10.1002/art.1780400420.
Hart DJ, Doyle DV, Spector TD: Incidence and risk factors for radiographic knee osteoarthritis in middle-aged women: the Chingford Study. Arthritis Rheum. 1999, 42: 17-24. 10.1002/1529-0131(199901)42:1<17::AID-ANR2>3.0.CO;2-E.
Karlson EW, Mandl LA, Aweh GN, Grodstein F: Coffee consumption and risk of rheumatoid arthritis. Arthritis Rheum. 2003, 48: 3055-3060. 10.1002/art.11306.
Sanchez Guerrero J, Karlson EW, Liang MH, Hunter DJ, Speizer FE, Colditz GA: Past use of oral contraceptives and the risk of developing systemic lupus erythematosus. Arthritis Rheum. 1997, 40: 804-808. 10.1002/art.1780400505.
Pattison DJ, Symmons DP, Lunt M, Welch A, Luben R, Bingham SA, Khaw KT, Day NE, Silman AJ: Dietary risk factors for the development of inflammatory polyarthritis: evidence for a role of high level of red meat consumption. Arthritis Rheum. 2004, 50: 3804-3812. 10.1002/art.20731.
Choi HK, Atkinson K, Karlson EW, Willett W, Curhan G: Purine-rich foods, dairy and protein intake, and the risk of gout in men. N Engl J Med. 2004, 350: 1093-1103. 10.1056/NEJMoa035700.
The Wellcome Trust Case-Control Consortium: Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007, 447: 661-678. 10.1038/nature05911.
Oliver JE, Silman AJ: Risk factors for the development of rheumatoid arthritis. Scand J Rheumatol. 2006, 35: 169-174. 10.1080/03009740600718080.
Heliovaara M, Aho K, Knekt P, Impivaara O, Reunanen A, Aromaa A: Coffee consumption, rheumatoid factor, and the risk of rheumatoid arthritis. Ann Rheum Dis. 2000, 59: 631-635. 10.1136/ard.59.8.631.
Mikuls TR, Cerhan JR, Criswell LA, Merlino L, Mudano AS, Burma M, Folsom AR, Saag KG: Coffee, tea, and caffeine consumption and risk of rheumatoid arthritis: results from the Iowa women's health study. Arthritis Rheum. 2002, 46: 83-91. 10.1002/1529-0131(200201)46:1<83::AID-ART10042>3.0.CO;2-D.
Skoldstam L, Hagfors L, Johansson G: An experimental study of a Mediterranean diet intervention for patients with rheumatoid arthritis. Ann Rheum Dis. 2003, 62: 208-214. 10.1136/ard.62.3.208.
Linos A, Kaklamani VG, Kaklamani E, Koumantaki Y, Giziaki E, Papazoglou S, Mantzoros CS: Dietary factors in relation to rheumatoid arthritis: a role for olive oil and cooked vegetables?. Am J Clin Nutr. 1999, 70: 1077-1082.
Pattison DJ, Harrison RA, Symmons DP: The role of diet in susceptibility to rheumatoid arthritis: a systematic review. J Rheumatol. 2004, 31: 1310-1319.
Merlino LA, Curtis J, Mikuls TR, Cerhan JR, Criswell LA, Saag KG: Vitamin D intake is inversely associated with rheumatoid arthritis: results from the Iowa Women's Health Study. Arthritis Rheum. 2004, 50: 72-77. 10.1002/art.11434.
ito-Garcia E, Feskanich D, Hu FB, Mandl LA, Karlson EW: Protein, iron, and meat consumption and risk for rheumatoid arthritis: a prospective cohort study. Arthritis Res Ther. 2007, 9: R16-10.1186/ar2123.
Criswell LA, Merlino LA, Cerhan JR, Mikuls TR, Mudano AS, Burma M, Folsom AR, Saag KG: Cigarette smoking and the risk of rheumatoid arthritis among postmenopausal women: results from the Iowa women's health study. Am J Med. 2002, 112: 465-471. 10.1016/S0002-9343(02)01051-3.
Hutchinson D, Shepstone L, Moots R, Lear JT, Lynch MP: Heavy cigarette smoking is strongly associated with rheumatoid arthritis (RA), particularly in patients without a family history of RA. Ann Rheum Dis. 2001, 60: 223-227. 10.1136/ard.60.3.223.
Uhlig T, Hagen KB, Kvien TK: Current tobacco smoking, formal education, and the risk of rheumatoid arthritis. J Rheumatol. 1999, 26: 47-54.
Stolt P, Bengtsson C, Nordmark B, Lindblad S, Lundberg I, Klareskog L, Alfredsson L, EIRA study group: Quantification of the influence of cigarette smoking on rheumatoid arthritis: results from a population based case-control study, using incident cases. Ann Rheum Dis. 2003, 62: 835-841. 10.1136/ard.62.9.835.
Karlson EW, Lee IM, Cook NR, Manson JE, Buring JE, Hennekens CH: A retrospective cohort study of cigarette smoking and risk of rheumatoid arthritis in female health professionals. Arthritis Rheum. 1999, 42: 910-917. 10.1002/1529-0131(199905)42:5<910::AID-ANR9>3.0.CO;2-D.
Harrison BJ, Silman AJ, Wiles NJ, Scott DG, Symmons DP: The association of cigarette smoking with disease outcome in patients with early inflammatory polyarthritis. Arthritis Rheum. 2001, 44: 323-330. 10.1002/1529-0131(200102)44:2<323::AID-ANR49>3.0.CO;2-C.
Papadopoulos NG, Alamanos Y, Voulgari PV, Epagelis EK, Tsifetaki N, Drosos AA: Does cigarette smoking influence disease expression, activity and severity in early rheumatoid arthritis patients?. Clin Exp Rheumatol. 2005, 23: 861-866.
Symmons DP, Bankhead CR, Harrison BJ, Brennan P, Barrett EM, Scott DG, Silman AJ: Blood transfusion, smoking, and obesity as risk factors for the development of rheumatoid arthritis: results from a primary care-based incident case-control study in Norfolk, England. Arthritis Rheum. 1997, 40: 1955-1961. 10.1002/art.1780401106.
Mandl LA, Costenbader KH, Simard J, Karlson EW: Is birth-weight associated with risk of rheumatoid arthritis? Data from a large prospective Cohort Study. Ann Rheum Dis. 2008
Pikwer M, Bergström U, Nilsson JA, Jacobsson L, Berglund G, Turesson C: Breast-feeding, but not oral contraceptives, is associated with a reduced risk of rheumatoid arthritis. Ann Rheum Dis. 2008
Källberg H, Jacobsen S, Bengtsson C, Pedersen M, Padyukov L, Garred P, Frisch M, Karlson EW, Klareskog L, Alfredsson L: Alcohol consumption is associated with decreased risk of rheumatoid arthritis: results from two Scandinavian case-control studies. Ann Rheum Dis. 2009, 68: 222-227. 10.1136/ard.2007.086314.
Cutolo M, Straub RH: Stress as a risk factor in the pathogenesis of rheumatoid arthritis. Neuroimmunomodulation. 2006, 13: 277-282. 10.1159/000104855.
Oliver JE, Worthington J, Silman AJ: Genetic epidemiology of rheumatoid arthritis. Curr Opin Rheumatol. 2006, 18: 141-146. 10.1097/01.bor.0000209425.84775.23.
Begovich AB, Carlton VE, Honigberg LA, Schrodi SJ, Chokkalingam AP, Alexander HC, Ardlie KG, Huang Q, Smith AM, Spoerke JM, Conn MT, Chang M, Chang SY, Saiki RK, Catanese JJ, Leong DU, Garcia VE, McAllister LB, Jeffery DA, Lee AT, Batliwalla F, Remmers E, Criswell LA, Seldin MF, Kastner DL, Amos CI, Sninsky JJ, Gregersen PK: A missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associated with rheumatoid arthritis. Am J Hum Genet. 2004, 75: 330-337. 10.1086/422827.
Hinks A, Barton A, John S, Bruce I, Hawkins C, Griffiths CE, Donn R, Thomson W, Silman A, Worthington J: Association between the PTPN22 gene and rheumatoid arthritis and juvenile idiopathic arthritis in a UK population: further support that PTPN22 is an autoimmunity gene. Arthritis Rheum. 2005, 52: 1694-1699. 10.1002/art.21049.
Lee AT, Li W, Liew A, Bombardier C, Weisman M, Massarotti EM, Kent J, Wolfe F, Begovich AB, Gregersen PK: The PTPN22 R620W polymorphism associates with RF positive rheumatoid arthritis in a dose-dependent manner but not with HLA-SE status. Genes Immun. 2005, 6: 129-133. 10.1038/sj.gene.6364159.
Seldin MF, Shigeta R, Laiho K, Li H, Saila H, Savolainen A, Leirisalo-Repo M, Aho K, Tuomilehto-Wolf E, Kaarela K, Kauppi M, Alexander HC, Begovich AB, Tuomilehto J: Finnish case-control and family studies support PTPN22 R620W polymorphism as a risk factor in rheumatoid arthritis, but suggest only minimal or no effect in juvenile idiopathic arthritis. Genes Immun. 2005, 6: 720-722.
Orozco G, Sánchez E, González-Gay MA, López-Nevot MA, Torres B, Cáliz R, Ortego-Centeno N, Jiménez-Alonso J, Pascual-Salcedo D, Balsa A, de Pablo R, Nuñez-Roldan A, González-Escribano MF, Martín J: Association of a functional single-nucleotide polymorphism of PTPN22, encoding lymphoid protein phosphatase, with rheumatoid arthritis and systemic lupus erythematosus. Arthritis Rheum. 2005, 52: 219-224. 10.1002/art.20771.
Suzuki A, Yamada R, Chang X, Tokuhiro S, Sawada T, Suzuki M, Nagasaki M, Nakayama-Hamada M, Kawaida R, Ono M, Ohtsuki M, Furukawa H, Yoshino S, Yukioka M, Tohma S, Matsubara T, Wakitani S, Teshima R, Nishioka Y, Sekine A, Iida A, Takahashi A, Tsunoda T, Nakamura Y, Yamamoto K: Functional haplotypes of PADI4, encoding citrullinating enzyme peptidylarginine deiminase 4, are associated with rheumatoid arthritis. Nat Genet. 2003, 34: 395-402. 10.1038/ng1206.
Ikari K, Kuwahara M, Nakamura T, Momohara S, Hara M, Yamanaka H, Tomatsu T, Kamatani N: Association between PADI4 and rheumatoid arthritis: a replication study. Arthritis Rheum. 2005, 52: 3054-3057. 10.1002/art.21309.
Barton A, Bowes J, Eyre S, Spreckley K, Hinks A, John S, Worthington J: A functional haplotype of the PADI4 gene associated with rheumatoid arthritis in a Japanese population is not associated in a United Kingdom population. Arthritis Rheum. 2004, 50: 1117-1121. 10.1002/art.20169.
Caponi L, Petit-Teixeira E, Sebbag M, Bongiorni F, Moscato S, Pratesi F, Pierlot C, Osorio J, Chapuy-Regaud S, Guerrin M, Cornelis F, Serre G, Migliorini P, ECRAF: A family based study shows no association between rheumatoid arthritis and the PADI4 gene in a white French population. Ann Rheum Dis. 2005, 64: 587-593. 10.1136/ard.2004.026831.
Martinez A, Valdivia A, Pascual-Salcedo D, Lamas JR, Fernández-Arquero M, Balsa A, Fernández-Gutiérrez B, de la Concha EG, Urcelay E: PADI4 polymorphisms are not associated with rheumatoid arthritis in the Spanish population. Rheumatology (Oxford). 2005, 44: 1263-1266. 10.1093/rheumatology/kei008.
Lee YH, Rho YH, Choi SJ, Ji JD, Song GG: PADI4 polymorphisms and rheumatoid arthritis susceptibility: a meta-analysis. Rheumatol Int. 2007, 27: 827-833. 10.1007/s00296-007-0320-y.
Plenge RM, Padyukov L, Remmers EF, Purcell S, Lee AT, Karlson EW, Wolfe F, Kastner DL, Alfredsson L, Altshuler D, Gregersen PK, Klareskog L, Rioux JD: Replication of putative candidate-gene associations with rheumatoid arthritis in > 4,000 samples from North America and Sweden: association of susceptibility with PTPN22, CTLA4, and PADI4. Am J Hum Genet. 2005, 77: 1044-1060. 10.1086/498651.
Jawaheer D, Seldin MF, Amos CI, Chen WV, Shigeta R, Monteiro J, Kern M, Criswell LA, Albani S, Nelson JL, Clegg DO, Pope R, Schroeder HW, Bridges SL, Pisetsky DS, Ward R, Kastner DL, Wilder RL, Pincus T, Callahan LF, Flemming D, Wener MH, Gregersen PK: A genomewide screen in multiplex rheumatoid arthritis families suggests genetic overlap with other autoimmune diseases. Am J Hum Genet. 2001, 68: 927-936. 10.1086/319518.
Liu C, Batliwalla F, Li W, Lee A, Roubenoff R, Beckman E, Khalili H, Damle A, Kern M, Furie R, Dupuis J, Plenge RM, Coenen MJ, Behrens TW, Carulli JP, Gregersen PK: Genome-wide association scan identifies candidate polymorphisms associated with differential response to anti-TNF treatment in Rheumatoid Arthritis. Mol Med. 2008, 14: 575-581.
Padyukov L, Silva C, Stolt P, Alfredsson L, Klareskog L: A gene-environment interaction between smoking and shared epitope genes in HLA-DR provides a high risk of seropositive rheumatoid arthritis. Arthritis Rheum. 2004, 50: 3085-3092. 10.1002/art.20553.
Costenbader KH, Chang SC, De VI, Plenge R, Karlson EW: Genetic polymorphisms in PTPN22, PADI-4, and CTLA-4 and risk for rheumatoid arthritis in two longitudinal cohort studies: evidence of gene-environment interactions with heavy cigarette smoking. Arthritis Res Ther. 2008, 10: R52-10.1186/ar2421.
Pedersen M, Jacobsen S, Klarlund M, Pedersen BV, Wiik A, Wohlfahrt J, Frisch M: Environmental risk factors differ between rheumatoid arthritis with and without auto-antibodies against cyclic citrullinated peptides. Arthritis Res Ther. 2006, 8: R133-10.1186/ar2022.
Pedersen M, Jacobsen S, Garred P, Madsen HO, Klarlund M, Svejgaard A, Pedersen BV, Wohlfahrt J, Frisch M: Strong combined gene-environment effects in anti-cyclic citrullinated peptide-positive rheumatoid arthritis: a nationwide case-control study in Denmark. Arthritis Rheum. 2007, 56: 1446-1453. 10.1002/art.22597.
Manners PJ, Bower C: Worldwide prevalence of juvenile arthritis: why does it vary so much?. J Rheumatol. 2002, 29: 1520-1530.
Saurenmann RK, Rose JB, Tyrrell P, Feldman BM, Laxer RM, Schneider R, Silverman ED: Epidemiology of juvenile idiopathic arthritis in a multiethnic cohort: ethnicity as a risk factor. Arthritis Rheum. 2007, 56: 1974-1984. 10.1002/art.22709.
Thompson SD, Moroldo MB, Guyer L, Ryan M, Tombragel EM, Shear ES, Prahalad S, Sudman M, Keddache MA, Brown WM, Giannini EH, Langefeld CD, Rich SS, Nichols WC, Glass DN: A genome-wide scan for juvenile rheumatoid arthritis in affected sibpair families provides evidence of linkage. Arthritis Rheum. 2004, 50: 2920-2930. 10.1002/art.20425.
Donn R, Alourfi Z, De Benedetti F, Meazza C, Zeggini E, Lunt M, Stevens A, Shelley E, Lamb R, Ollier WE, Thomson W, Ray D, British Paediatric Rheumatology Study Group: Mutation screening of the macrophage migration inhibitory factor gene: positive association of a functional polymorphism of macrophage migration inhibitory factor with juvenile idiopathic arthritis. Arthritis Rheum. 2002, 46: 2402-2409. 10.1002/art.10492.
Lee YH, Rho YH, Choi SJ, Ji JD, Song GG, Nath SK, Harley JB: The PTPN22 C1858T functional polymorphism and autoimmune diseases-a meta-analysis. Rheumatology (Oxford). 2007, 46: 49-56. 10.1093/rheumatology/kel170.
Sanjeevi CB, Miller EN, Dabadghao P, Rumba I, Shtauvere A, Denisova A, Clayton D, Blackwell JM: Polymorphism at NRAMP1 and D2S1471 loci associated with juvenile rheumatoid arthritis. Arthritis Rheum. 2000, 43: 1397-1404. 10.1002/1529-0131(200006)43:6<1397::AID-ANR25>3.0.CO;2-6.
Fishman D, Faulds G, Jeffery R, Mohamed-Ali V, Yudkin JS, Humphries S, Woo P: The effect of novel polymorphisms in the interleukin-6 (IL-6) gene on IL-6 transcription and plasma IL-6 levels, and an association with systemic-onset juvenile chronic arthritis. J Clin Invest. 1998, 102: 1369-1376. 10.1172/JCI2629.
Pattison E, Harrison BJ, Griffiths CE, Silman AJ, Bruce IN: Environmental risk factors for the development of psoriatic arthritis: results from a case-control study. Ann Rheum Dis. 2008, 67: 672-676. 10.1136/ard.2007.073932.
Thumboo J, Uramoto K, Shbeeb MI, O'Fallon WM, Crowson CS, Gibson LE, Michet CJ, Gabriel SE: Risk factors for the development of psoriatic arthritis: a population based nested case control study. J Rheumatol. 2002, 29: 757-762.
Rahman P, Bartlett S, Siannis F, Pellett FJ, Farewell VT, Peddle L, Schentag CT, Alderdice CA, Hamilton S, Khraishi M, Tobin Y, Hefferton D, Gladman DD: CARD15: a pleiotropic autoimmune gene that confers susceptibility to psoriatic arthritis. Am J Hum Genet. 2003, 73: 677-681. 10.1086/378076.
Lascorz J, Burkhardt H, Hüffmeier U, Böhm B, Schürmeyer-Horst F, Lohmann J, Ständer M, Wendler J, Kelsch R, Baumann C, Küster W, Traupe H, Reis A: Lack of genetic association of the three more common polymorphisms of CARD15 with psoriatic arthritis and psoriasis in a German cohort. Ann Rheum Dis. 2005, 64: 951-954. 10.1136/ard.2004.029157.
Giardina E, Novelli G, Costanzo A, Nisticò S, Bulli C, Sinibaldi C, Sorgi ML, Chimenti S, Pallone F, Taccari E, Borgiani P: Psoriatic arthritis and CARD15 gene polymorphisms: no evidence for association in the Italian population. J Invest Dermatol. 2004, 122: 1106-1107. 10.1111/j.0022-202X.2004.22524.x.
Nelson GW, Martin MP, Gladman D, Wade J, Trowsdale J, Carrington M: Cutting edge: heterozygote advantage in autoimmune disease: hierarchy of protection/susceptibility conferred by HLA and killer Ig-like receptor combinations in psoriatic arthritis. J Immunol. 2004, 173: 4273-4276.
Gonzalez S, Martinez-Borra J, Torre-Alonso JC, Gonzalez-Roces S, Sanchez del Río J, Rodriguez Pérez A, Brautbar C, López-Larrea C: The MICA-A9 triplet repeat polymorphism in the transmembrane region confers additional susceptibility to the development of psoriatic arthritis and is independent of the association of Cw*0602 in psoriasis. Arthritis Rheum. 1999, 42: 1010-1016. 10.1002/1529-0131(199905)42:5<1010::AID-ANR21>3.0.CO;2-H.
Karason A, Gudjonsson JE, Upmanyu R, Antonsdottir AA, Hauksson VB, Runasdottir EH, Jonsson HH, Gudbjartsson DF, Frigge ML, Kong A, Stefansson K, Valdimarsson H, Gulcher JR: A susceptibility gene for psoriatic arthritis maps to chromosome 16q: evidence for imprinting. Am J Hum Genet. 2003, 72: 125-131. 10.1086/345646.
Balding J, Kane D, Livingstone W, Mynett-Johnson L, Bresnihan B, Smith O, FitzGerald O: Cytokine gene polymorphisms: association with psoriatic arthritis susceptibility and severity. Arthritis Rheum. 2003, 48: 1408-1413. 10.1002/art.10935.
Liu Y, Helms C, Liao W, Zaba LC, Duan S, Gardner J, Wise C, Miner A, Malloy MJ, Pullinger CR, Kane JP, Saccone S, Worthington J, Bruce I, Kwok PY, Menter A, Krueger J, Barton A, Saccone NL, Bowcock AM: A genome-wide association study of psoriasis and psoriatic arthritis identifies new disease loci. PLoS Genet. 2008, 4: e1000041-10.1371/journal.pgen.1000041.
Ho PY, Barton A, Worthington J, Plant D, Griffiths CE, Young HS, Bradburn P, Thomson W, Silman AJ, Bruce IN: Investigating the role of the HLA-Cw*06 and HLA-DRB1 genes in susceptibility to psoriatic arthritis: comparison with psoriasis and undifferentiated inflammatory arthritis. Ann Rheum Dis. 2008, 67: 677-682. 10.1136/ard.2007.071399.
Pedersen OB, Svendsen AJ, Ejstrup L, Skytthe A, Harris JR, Junker P: Ankylosing spondylitis in Danish and Norwegian twins: occurrence and the relative importance of genetic vs. environmental effectors in disease causation. Scand J Rheumatol. 2008, 37: 120-126. 10.1080/03009740701824613.
Brown MA, Pile KD, Kennedy LG, Campbell D, Andrew L, March R, Shatford JL, Weeks DE, Calin A, Wordsworth BP: A genome-wide screen for susceptibility loci in ankylosing spondylitis. Arthritis Rheum. 1998, 41: 588-595. 10.1002/1529-0131(199804)41:4<588::AID-ART5>3.0.CO;2-0.
Brionez TF, Reveille JD: The contribution of genes outside the major histocompatibility complex to susceptibility to ankylosing spondylitis. Curr Opin Rheumatol. 2008, 20: 384-391. 10.1097/BOR.0b013e32830460fe.
Wellcome Trust Case Control Consortium; Australo-Anglo-American Spondylitis Consortium (TASC), Burton PR, Clayton DG, Cardon LR, Craddock N, Deloukas P, Duncanson A, Kwiatkowski DP, McCarthy MI, Ouwehand WH, Samani NJ, Todd JA, Donnelly P, Barrett JC, Davison D, Easton D, Evans DM, Leung HT, Marchini JL, Morris AP, Spencer CC, Tobin MD, Attwood AP, Boorman JP, Cant B, Everson U, Hussey JM, Jolley JD, Knight AS, Koch K, et al: Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat Genet. 2007, 39: 1329-1337. 10.1038/ng.2007.17.
Laval SH, Timms A, Edwards S, Bradbury L, Brophy S, Milicic A, Rubin L, Siminovitch KA, Weeks DE, Calin A, Wordsworth BP, Brown MA: Whole-genome screening in ankylosing spondylitis: evidence of non-MHC genetic-susceptibility loci. Am J Hum Genet. 2001, 68: 918-926. 10.1086/319509.
Zhang G, Luo J, Bruckel J, Weisman MA, Schumacher HR, Khan MA, Inman RD, Mahowald M, Maksymowych WP, Martin TM, Yu DT, Stone M, Rosenbaum JT, Newman P, Lee J, McClain JA, West OC, Jin L, Reveille JD: Genetic studies in familial ankylosing spondylitis susceptibility. Arthritis Rheum. 2004, 50: 2246-2254. 10.1002/art.20308.
Carter KW, Pluzhnikov A, Timms AE, Miceli-Richard C, Bourgain C, Wordsworth BP, Jean-Pierre H, Cox NJ, Palmer LJ, Breban M, Reveille JD, Brown MA: Combined analysis of three whole genome linkage scans for ankylosing spondylitis. Rheumatology (Oxford). 2007, 46: 763-771. 10.1093/rheumatology/kel443.
Brown MA, Edwards S, Hoyle E, Campbell S, Laval S, Daly AK, Pile KD, Calin A, Ebringer A, Weeks DE, Wordsworth BP: Polymorphisms of the CYP2D6 gene increase susceptibility to ankylosing spondylitis. Hum Mol Genet. 2000, 9: 1563-1566. 10.1093/hmg/9.11.1563.
Paardt van der M, Crusius JB, García-González MA, Baudoin P, Kostense PJ, Alizadeh BZ, Dijkmans BA, Peña AS, Horst-Bruinsma van der IE: Interleukin-1beta and interleukin-1 receptor antagonist gene polymorphisms in ankylosing spondylitis. Rheumatology (Oxford). 2002, 41: 1419-1423. 10.1093/rheumatology/41.12.1419.
Cooper GS, Dooley MA, Treadwell EL, St Clair EW, Gilkeson GS: Hormonal and reproductive risk factors for development of systemic lupus erythematosus: results of a population-based, case-control study. Arthritis Rheum. 2002, 46: 1830-1839. 10.1002/art.10365.
Meier CR, Sturkenboom MCJM, Cohen AS, Jick H: Post-menopausal estrogen replacement therapy and the risk of developing systemic lupus erythematosus or discoid lupus. J Rheumatol. 1998, 25: 1515-1519.
Wang J, Kay AB, Fletcher J, Formica MK, McAlindon TE: Is lipstick associated with the development of systemic lupus erythematosus (SLE)?. Clin Rheumatol. 2008, 27: 1183-1187. 10.1007/s10067-008-0937-6.
Bengtsson AA, Rylander L, Hagmar L, Nived O, Sturfelt G: Risk factors for developing systemic lupus erythematosus: a case-control study in southern Sweden. Rheumatology (Oxford). 2002, 41: 563-571. 10.1093/rheumatology/41.5.563.
Rhodes B, Vyse TJ: The genetics of SLE: an update in the light of genome-wide association studies. Rheumatology (Oxford). 2008, 47: 1603-1611. 10.1093/rheumatology/ken247.
International Consortium for Systemic Lupus Erythematosus Genetics (SLEGEN), Harley JB, Alarcón-Riquelme ME, Criswell LA, Jacob CO, Kimberly RP, Moser KL, Tsao BP, Vyse TJ, Langefeld CD, Nath SK, Guthridge JM, Cobb BL, Mirel DB, Marion MC, Williams AH, Divers J, Wang W, Frank SG, Namjou B, Gabriel SB, Lee AT, Gregersen PK, Behrens TW, Taylor KE, Fernando M, Zidovetzki R, Gaffney PM, Edberg JC, Rioux JD, et al: Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet. 2008, 40: 204-210. 10.1038/ng.81.
Hom G, Graham RR, Modrek B, Taylor KE, Ortmann W, Garnier S, Lee AT, Chung SA, Ferreira RC, Pant PV, Ballinger DG, Kosoy R, Demirci FY, Kamboh MI, Kao AH, Tian C, Gunnarsson I, Bengtsson AA, Rantapää-Dahlqvist S, Petri M, Manzi S, Seldin MF, Rönnblom L, Syvänen AC, Criswell LA, Gregersen PK, Behrens TW: Association of systemic lupus erythematosus with C8orf13-BLK and ITGAM-ITGAX. N Engl J Med. 2008, 358: 900-909. 10.1056/NEJMoa0707865.
Karassa FB, Trikalinos TA, Ioannidis JP: Role of the Fcgamma receptor IIa polymorphism in susceptibility to systemic lupus erythematosus and lupus nephritis: a meta-analysis. Arthritis Rheum. 2002, 46: 1563-1571. 10.1002/art.10306.
Graham DSC, Graham RR, Manku H, Wong AK, Whittaker JC, Gaffney PM, Moser KL, Rioux JD, Altshuler D, Behrens TW, Vyse TJ: Polymorphism at the TNF superfamily gene TNFSF4 confers susceptibility to systemic lupus erythematosus. Nat Genet. 2008, 40: 83-89. 10.1038/ng.2007.47.
Yang Y, Chung EK, Wu YL, Savelli SL, Nagaraja HN, Zhou B, Hebert M, Jones KN, Shu Y, Kitzmiller K, Blanchong CA, McBride KL, Higgins GC, Rennebohm RM, Rice RR, Hackshaw KV, Roubey RA, Grossman JM, Tsao BP, Birmingham DJ, Rovin BH, Hebert LA, Yu CY: Gene copy-number variation and associated polymorphisms of complement component C4 in human systemic lupus erythematosus (SLE): low copy number is a risk factor for and high copy number is a protective factor against SLE susceptibility in European Americans. Am J Hum Genet. 2007, 80: 1037-1054. 10.1086/518257.
Scofield RH, Bruner GR, Namjou B, Kimberly RP, Ramsey-Goldman R, Petri M, Reveille JD, Alarcón GS, Vilá LM, Reid J, Harris B, Li S, Kelly JA, Harley JB: Klinefelter's syndrome (47,XXY) in male systemic lupus erythematosus patients: support for the notion of a gene-dose effect from the X chromosome. Arthritis Rheum. 2008, 58: 2511-2517. 10.1002/art.23701.
Parks CG, Cooper GS, Nylander-French LA, Sanderson WT, Dement JM, Cohen PL, Dooley MA, Treadwell EL, St Clair EW, Gilkeson GS, Hoppin JA, Savitz DA: Occupational exposure to crystalline silica and risk of systemic lupus erythematosus: a population-based, case-control study in the southeastern United States. Arthritis Rheum. 2002, 46: 1840-1850. 10.1002/art.10368.
Janowsky EC, Kupper LL, Hulka BS: Meta-analyses of the relation between silicone breast implants and the risk of connective-tissue diseases. N Engl J Med. 2000, 342: 781-790. 10.1056/NEJM200003163421105.
Nietert PJ, Sutherland SE, Silver RM, Pandey JP, Knapp RG, Hoel DG, Dosemeci M: Is occupational organic solvent exposure a risk factor for scleroderma?. Arthritis Rheum. 1998, 41: 1111-1118. 10.1002/1529-0131(199806)41:6<1111::AID-ART19>3.0.CO;2-J.
Tan FK, Arnett FC: Genetic factors in the etiology of systemic sclerosis and Raynaud phenomenon. Curr Opin Rheumatol. 2000, 12: 511-519. 10.1097/00002281-200011000-00007.
Arnett FC, Cho M, Chatterjee S, Aguilar MB, Reveille JD, Mayes MD: Familial occurrence frequencies and relative risks for systemic sclerosis (scleroderma) in three United States cohorts. Arthritis Rheum. 2001, 44: 1359-1362. 10.1002/1529-0131(200106)44:6<1359::AID-ART228>3.0.CO;2-S.
Lambert NC, Distler O, Muller-Ladner U, Tylee TS, Furst DE, Nelson JL: HLA-DQA1*0501 is associated with diffuse systemic sclerosis in Caucasian men. Arthritis Rheum. 2000, 43: 2005-2010. 10.1002/1529-0131(200009)43:9<2005::AID-ANR11>3.0.CO;2-#.
Fanning GC, Welsh KI, Bunn C, Du Bois R, Black CM: HLA associations in three mutually exclusive autoantibody subgroups in UK systemic sclerosis patients. Br J Rheumatol. 1998, 37: 201-207. 10.1093/rheumatology/37.2.201.
Cotton SA, Herrick AL, Jayson MIV, Freemont AJ: TGF beta: a role in systemic sclerosis?. J Pathol. 1998, 184: 4-6. 10.1002/(SICI)1096-9896(199801)184:1<4::AID-PATH968>3.0.CO;2-0.
Denton CP, Abraham DJ: Transforming growth factor-beta and connective tissue growth factor: key cytokines in scleroderma pathogenesis. Curr Opin Rheumatol. 2001, 13: 505-511. 10.1097/00002281-200111000-00010.
Kawakami T, Ihn H, Xu W, Smith E, LeRoy C, Trojanowska M: Increased expression of TGF-beta receptors by scleroderma fibroblasts: evidence for contribution of autocrine TGF-beta signaling to scleroderma phenotype. J Invest Dermatol. 1998, 110: 47-51. 10.1046/j.1523-1747.1998.00073.x.
Tan FK, Wang N, Kuwana M, Chakraborty R, Bona CA, Milewicz DM, Arnett FC: Association of fibrillin 1 single-nucleotide polymorphism haplotypes with systemic sclerosis in Choctaw and Japanese populations. Arthritis Rheum. 2001, 44: 893-901. 10.1002/1529-0131(200104)44:4<893::AID-ANR146>3.0.CO;2-3.
Evans PC, Lambert N, Maloney S, Furst DE, Moore JM, Nelson JL: Long-term fetal microchimerism in peripheral blood mononuclear cell subsets in healthy women and women with scleroderma. Blood. 1999, 93: 2033-2037.
Artlett CM, Smith JB, Jimenez SA: Identification of fetal DNA and cells in skin lesions from women with systemic sclerosis. N Engl J Med. 1998, 338: 1186-1191. 10.1056/NEJM199804233381704.
Dieppe P, Cushnaghan J, Tucker M, Browning S, Shepstone L: The Bristol 'OA500 study': progression and impact of the disease after 8 years. Osteoarthritis Cartilage. 2000, 8: 63-68. 10.1053/joca.1999.0272.
Thomas E, Wilkie R, Peat G, Hill S, Dziedzic K, Croft P: The North Staffordshire Osteoarthritis Project-NorStOP: prospective, 3-year study of the epidemiology and management of clinical osteoarthritis in a general population of older adults. BMC Musculoskelet Disord. 2004, 5: 2-10.1186/1471-2474-5-2.
Cooper C, Snow S, McAlindon TE, Kellingray S, Stuart B, Coggon D, Dieppe PA: Risk factors for the incidence and progression of radiographic knee osteoarthritis. Arthritis Rheum. 2000, 43: 995-1000. 10.1002/1529-0131(200005)43:5<995::AID-ANR6>3.0.CO;2-1.
Cooper C, Inskip H, Croft P, Campbell L, Smith G, McLaren M, Coggon D: Individual risk factors for hip osteoarthritis: obesity, hip injury and physical activity. Am J Epidemiol. 1998, 147: 516-522.
Lohmander LS, Gerhardsson M, Rollof J, Nilsson PM, Engstrom G: Incidence of severe knee and hip osteoarthritis in relation to different measures of body mass. A population-based prospective cohort study. Ann Rheum Dis. 2008
Lievense AM, Bierma-Zeinstra SMA, Verhagen AP, van Baar ME, Verhaar JAN, Koes BW: Influence of obesity on the development of osteoarthritis of the hip: a systematic review. Rheumatology. 2002, 41: 1155-1162. 10.1093/rheumatology/41.10.1155.
Manek NJ, Hart D, Spector TD, MacGregor AJ: The association of body mass index and osteoarthritis of the knee joint: an examination of genetic and environmental influences. Arthritis Rheum. 2003, 48: 1024-1029. 10.1002/art.10884.
McAlindon TE, Wilson PW, Aliabadi P, Weissman B, Felson DT: Level of physical activity and the risk of radiographic and symptomatic knee osteoarthritis in the elderly: the Framingham study. Am J Med. 1999, 106: 151-157. 10.1016/S0002-9343(98)00413-6.
Lievense A, Bierma-Zeinstra S, Verhagen A, Verhaar J, Koes B: Influence of work on the development of osteoarthritis of the hip: a systematic review. J Rheumatol. 2001, 28: 2520-2528.
Cheng YL, Macera CA, Davis DR, Ainsworth BE, Troped PJ, Blair SN: Physical activity and self-reported, physician-diagnosed osteoarthritis: is physical activity a risk factor?. J Clin Epidemiol. 2000, 53: 315-322. 10.1016/S0895-4356(99)00168-7.
Chaisson CE, Zhang YQ, Sharma L, Kannel W, Felson DT: Grip strength and the risk of developing radiographic hand osteoarthritis: results from the Framingham Study. Arthritis Rheum. 1999, 42: 33-38. 10.1002/1529-0131(199901)42:1<33::AID-ANR4>3.0.CO;2-I.
Fontana L, Neel S, Claise JM, Ughetto S, Catilina P: Osteoarthritis of the thumb carpometacarpal joint in women and occupational risk factors: a case-control study. J Hand Surg [Am]. 2007, 32: 459-465. 10.1016/j.jhsa.2007.01.014.
Jones G, Cooley HM, Stankovich JM: A cross sectional study of the association between sex, smoking, and other lifestyle factors and osteoarthritis of the hand. J Rheumatol. 2002, 29: 1719-1724.
MacGregor AJ, Antoniades L, Matson M, Andrew T, Spector TD: The genetic contribution to radiographic hip osteoarthritis in women: results of a classic twin study. Arthritis Rheum. 2000, 43: 2410-2416. 10.1002/1529-0131(200011)43:11<2410::AID-ANR6>3.0.CO;2-E.
Peach CA, Carr AJ, Loughlin J: Recent advances in the genetic investigation of osteoarthritis. Trends Mol Med. 2005, 11: 186-191. 10.1016/j.molmed.2005.02.005.
Smith AJP, Keen LJ, Billingham MJ, Perry MJ, Elson CJ, Kirwan JR, Sims JE, Doherty M, Spector TD, Bidwell JL: Extended haplotypes and linkage disequilibrium in the IL1R1-IL1A-IL1B-IL1RN gene cluster: association with knee osteoarthritis. Genes Immun. 2004, 5: 451-460. 10.1038/sj.gene.6364107.
Loughlin J, Dowling B, Mustafa Z, Chapman K: Association of the interleukin-1 gene cluster on chromosome 2q13 with knee osteoarthritis. Arthritis Rheum. 2002, 46: 1519-1527. 10.1002/art.10260.
Loughlin J, Dowling B, Chapman K, Marcelline L, Mustafa Z, Southam L, Ferreira A, Ciesielski C, Carson DA, Corr M: Functional variants within the secreted frizzled-related protein 3 gene are associated with hip osteoarthritis in females. Proc Natl Acad Sci USA. 2004, 101: 9757-9762. 10.1073/pnas.0403456101.
Meulenbelt I, Bijkerk C, Miedema HS, Breedveld FC, Hofman A, Valkenburg HA, Pols HA, Slagboom PE, van Duijn CM: A genetic association study of the IGF-1 gene and radiological osteoarthritis in a population-based cohort study (the Rotterdam study). Ann Rheum Dis. 1998, 57: 371-374. 10.1136/ard.57.6.371.
Bergink AP, van Meurs JB, Loughlin J, Arp PP, Fang Y, Hofman A, van Leeuwen JP, van Duijn CM, Uitterlinden AG, Pols HA: Estrogen receptor alpha gene haplotype is associated with radiographic osteoarthritis of the knee in elderly men and women. Arthritis Rheum. 2003, 48: 1913-1922. 10.1002/art.11046.
Valdes AM, Van OM, Hart DJ, Surdulescu GL, Loughlin J, Doherty M, Spector TD: Reproducible genetic associations between candidate genes and clinical knee osteoarthritis in men and women. Arthritis Rheum. 2006, 54: 533-539. 10.1002/art.21621.
Valdes AM, Loughlin J, Timms KM, van Meurs JJ, Southam L, Wilson SG, Doherty S, Lories RJ, Luyten FP, Gutin A, Abkevich V, Ge D, Hofman A, Uitterlinden AG, Hart DJ, Zhang F, Zhai G, Egli RJ, Doherty M, Lanchbury J, Spector TD: Genome-wide association scan identifies a prostaglandin-endoperoxide synthase 2 variant involved in risk of knee osteoarthritis. Am J Hum Genet. 2008, 82: 1231-1240. 10.1016/j.ajhg.2008.04.006.
Meulenbelt I, Min JL, Bos S, Riyazi N, Houwing-Duistermaat JJ, Wijk van der HJ, Kroon HM, Nakajima M, Ikegawa S, Uitterlinden AG, van Meurs JB, Deure van der WM, Visser TJ, Seymour AB, Lakenberg N, Breggen van der R, Kremer D, van Duijn CM, Kloppenburg M, Loughlin J, Slagboom PE: Identification of DIO2 as a new susceptibility locus for symptomatic osteoarthritis. Hum Mol Genet. 2008, 17: 1867-1875. 10.1093/hmg/ddn082.
Chapman K, Takahashi A, Meulenbelt I, Watson C, Rodriguez-Lopez J, Egli R, Tsezou A, Malizos KN, Kloppenburg M, Shi D, Southam L, Breggen van der R, Donn R, Qin J, Doherty M, Slagboom PE, Wallis G, Kamatani N, Jiang Q, Gonzalez A, Loughlin J, Ikegawa S: A meta-analysis of European and Asian cohorts reveals a global role of a functional SNP in the 5' UTR of GDF5 with osteoarthritis susceptibility. Hum Mol Genet. 2008, 17: 1497-1504. 10.1093/hmg/ddn038.
Loughlin J: The genetic epidemiology of human primary osteoarthritis: current status. Expert Rev Mol Med. 2005, 7: 1-12. 10.1017/S1462399405009257.
UK scientists in world's first major genome screen for osteoarthritis. [http://www.arc.org.uk/news/pressreleases/18030.asp]
Riyazi N, Meulenbelt I, Kroon HM, Ronday KH, Hellio le Graverand MP, Rosendaal FR, Breedveld FC, Slagboom PE, Kloppenburg M: Evidence for familial aggregation of hand, hip, and spine but not knee osteoarthritis in siblings with multiple joint involvement: the GARP study. Ann Rheum Dis. 2005, 64: 438-443. 10.1136/ard.2004.024661.
Jonsson H, Manolescu I, Stefansson SE, Ingvarsson T, Jonsson HH, Manolescu A, Gulcher J, Stefansson K: The inheritance of hand osteoarthritis in Iceland. Arthritis Rheum. 2003, 48: 391-395. 10.1002/art.10785.
Livshits G, Kato BS, Zhai GJ, Hart DJ, Hunter D, MacGregor AJ, Williams FM, Spector TD: Genomewide linkage scan of hand osteoarthritis in female twin pairs showing replication of quantitative trait loci on chromosomes 2 and 19. Ann Rheum Dis. 2007, 66: 623-627. 10.1136/ard.2006.060236.
Solovieva S, Hirvonen A, Siivola P, Vehmas T, Luoma K, Riihimäki H, Leino-Arjas P: Vitamin D receptor gene polymorphisms and susceptibility of hand osteoarthritis in Finnish women. Arthritis Res Ther. 2006, 8: R20-10.1186/ar1874.
Hunter DJ, Demissie S, Cupples LA, Aliabadi P, Felson DT: A genome scan for joint-specific hand osteoarthritis susceptibility: the Framingham study. Arthritis Rheum. 2004, 50: 2489-2496. 10.1002/art.20445.
Stefánsson SE, Jónsson H, Ingvarsson T, Manolescu I, Jónsson HH, Olafsdóttir G, Pálsdóttir E, Stefánsdóttir G, Sveinbjörnsdóttir G, Frigge ML, Kong A, Gulcher JR, Stefánsson K: Genomewide scan for hand osteoarthritis: a novel mutation in matrilin-3. Am J Hum Genet. 2003, 72: 1448-1459. 10.1086/375556.
Arromdee E, Michet CJ, Crowson CS, O'Fallon WM, Gabriel SE: Epidemiology of gout: is the incidence rising?. J Rheumatol. 2002, 29: 2403-2406.
Mikuls TR, Saag KG: New insights into gout epidemiology. Curr Opin Rheumatol. 2006, 18: 199-203. 10.1097/01.bor.0000209435.89720.7c.
Choi HK, Ford ES, Li C, Curhan G: Prevalence of the metabolic syndrome in patients with gout: the Third National Health and Nutrition Examination Survey. Arthritis Rheum. 2007, 57: 109-115. 10.1002/art.22466.
Zaka R, Williams CJ: New developments in the epidemiology and genetics of gout. Curr Rheumatol Rep. 2006, 8: 215-223. 10.1007/s11926-996-0028-0.
Choi HK, Atkinson K, Karlson EW, Willett W, Curhan G: Alcohol intake and risk of incident gout in men: a prospective study. Lancet. 2004, 363: 1277-1281. 10.1016/S0140-6736(04)16000-5.
Choi HK, Curhan G: Gout: epidemiology and lifestyle choices. Curr Opin Rheumatol. 2005, 17: 341-345. 10.1097/01.cco.0000152664.87204.3c.
Choi HK, Atkinson K, Karlson EW, Curhan G: Obesity, weight change, hypertension, diuretic use, and risk of gout in men: the health professionals follow-up study. Arch Intern Med. 2005, 165: 742-748. 10.1001/archinte.165.7.742.
Choi HK, Curhan G: Soft drinks, fructose consumption, and the risk of gout in men: prospective cohort study. BMJ. 2008, 336: 309-312. 10.1136/bmj.39449.819271.BE.
Choi HK, Willett W, Curhan G: Coffee consumption and risk of incident gout in men: a prospective study. Arthritis Rheum. 2007, 56: 2049-2055. 10.1002/art.22712.
Vitart V, Rudan I, Hayward C, Gray NK, Floyd J, Palmer CN, Knott SA, Kolcic I, Polasek O, Graessler J, Wilson JF, Marinaki A, Riches PL, Shu X, Janicijevic B, Smolej-Narancic N, Gorgoni B, Morgan J, Campbell S, Biloglav Z, Barac-Lauc L, Pericic M, Klaric IM, Zgaga L, Skaric-Juric T, Wild SH, Richardson WA, Hohenstein P, Kimber CH, Tenesa A, Donnelly LA, et al: SLC2A9 is a newly identified urate transporter influencing serum urate concentration, urate excretion and gout. Nat Genet. 2008, 40: 437-442. 10.1038/ng.106.
Chang SJ, Tsai PC, Chen CJ, Lai HM, Ko YC: The polymorphism-863C/A in tumour necrosis factor-alpha gene contributes an independent association to gout. Rheumatology. 2007, 46: 1662-1666. 10.1093/rheumatology/kem235.
Al-Allaf AW, Dunbar KL, Hallum NS, Nosratzadeh B, Templeton KD, Pullar T: A case-control study examining the role of physical trauma in the onset of fibromyalgia syndrome. Rheumatology (Oxford). 2002, 41: 450-453. 10.1093/rheumatology/41.4.450.
Buskila D, Neumann L, Vaisberg G, Alkalay D, Wolfe F: Increased rates of fibromyalgia following cervical spine injury: a controlled study of 161 cases of traumatic injury. Arthritis Rheum. 1997, 40: 446-452. 10.1002/art.1780400310.
Gupta A, Silman AJ, Ray D, Morriss R, Dickens C, MacFarlane GJ, Chiu YH, Nicholl B, McBeth J: The role of psychosocial factors in predicting the onset of chronic widespread pain: results from a prospective population-based study. Rheumatology. 2007, 46: 666-671. 10.1093/rheumatology/kel363.
McBeth J, Silman AJ, Gupta A, Chiu YH, Ray D, Morriss R, Dickens C, King Y, Macfarlane GJ: Moderation of psychosocial risk factors through dysfunction of the hypothalamic-pituitary-adrenal stress axis in the onset of chronic widespread musculoskeletal pain: findings of a population-based prospective cohort study. Arthritis Rheum. 2007, 56: 360-371. 10.1002/art.22336.
Arnold LM, Hudson JI, Hess EV, Ware AE, Fritz DA, Auchenbach MB, Starck LO, Keck PE: Family study of fibromyalgia. Arthritis Rheum. 2004, 50: 944-952. 10.1002/art.20042.
Offenbaecher M, Bondy B, de Jonge S, Glatzeder K, Kruger M, Schoeps P, Ackenheil M: Possible association of fibromyalgia with a polymorphism in the serotonin transporter gene regulatory region. Arthritis Rheum. 1999, 42: 2482-2488. 10.1002/1529-0131(199911)42:11<2482::AID-ANR27>3.0.CO;2-B.
Yunus MB, Khan MA, Rawlings KK, Green JR, Olson JM, Shah S: Genetic linkage analysis of multicase families with fibromyalgia syndrome. J Rheumatol. 1999, 26: 408-412.
Gursoy S, Erdal E, Herken H, Madenci E, Alasehirli B, Erdal N: Significance of catechol-O-methyltransferase gene polymorphism in fibromyalgia syndrome. Rheumatol Int. 2003, 23: 104-107.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Rights and permissions
About this article
Cite this article
Oliver, J.E., Silman, A.J. What epidemiology has told us about risk factors and aetiopathogenesis in rheumatic diseases. Arthritis Res Ther 11, 223 (2009). https://doi.org/10.1186/ar2585
Published:
DOI: https://doi.org/10.1186/ar2585