
Abstract
Regulatory/suppressor T cells (Tregs) maintain immunologic homeo-stasis and prevent autoimmunity. In this article, past studies and recent studies of Tregs in mouse models for lupus and of human systemic lupus erythematosus are reviewed concentrating on CD4+CD25+Foxp3+ Tregs. These cells consist of thymus-derived, natural Tregs and peripherally induced Tregs that are similar phenotypically and functionally. These Tregs are decreased in young lupus-prone mice, but are present in normal numbers in mice with established disease. In humans, most workers report CD4+Tregs are decreased in subjects with active systemic lupus erythematosus, but the cells increase with treatment and clinical improvement. The role of immunogenic and tolerogenic dendritic cells in controlling Tregs is discussed, along with new strategies to normalize Treg function in systemic lupus erythematosus.
Similar content being viewed by others

Introduction
Systemic lupus erythematosus (SLE) is a disorder of immune regulation characterized by the breakdown of tolerance to self-nuclear, cytoplasmic and cell surface molecules and by the production of autoantibodies to these elements. The result is generalized autoimmunity manifested by multisystem chronic inflammatory disease. Many T-cell and B-cell abnormalities have been described, and these include defects in the regulatory/suppressor T cells (Tregs) that normally prevent pathologic self-reactivity. In the present article, we shall review the literature on this topic in both human lupus and animal models of this disease written before and after the resurgence of interest in suppressor T cells in the past decade. Treg abnormalities could contribute to T-cell and B-cell hyperactivity in SLE for various reasons. These include decreased numbers and/or inhibitory function of these cells, increased resistance of effector T cells to suppression, or greater expansion of effector T cells relative to normal Tregs. Alternatively, the principal effect of Tregs on T-cell function could be indirect by altering the properties of antigen-presenting cells. Evidence for each of these mechanisms will be discussed.
T cells with the ability to control autoantibody production were first described by Teague and Friou in 1969. These workers reported that the transfer of thymus cells from young mice to old mice prevented the development of anti-nucleoprotein antibodies, and also blocked their appearance after immunization [1]. When the mitogen concanavalin A was found to induce T cells to develop suppressive activity, many workers reported decreased concanavalin A suppressive activity in human SLE and mouse models [2, 3]. Interest in this topic diminished, however, until its renaissance in the past decade. In 1996 Sakaguchi and coworkers noted that 3-day-thymectomized mice developed organ-specific auto-immune disease [4]. This was because suppressor T cells were depleted by neonatal thymectomy. Subsequently the T cells were identified as CD4+ cells that expressed CD25, the α-chain of the IL-2 receptor. Similar multiorgan auto-immune disease could also be produced by transferring CD4+CD25-cells to immunodeficient mice, but this was prevented by cotransfer of CD4+CD25+ cells [5].
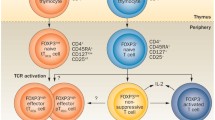
It is now evident that Tregs consist of heterogeneous populations of CD4 cells, CD8 cells and even natural killer T cells [6]. Conveniently, the cells can be divided into those that express the forkhead/winged helix transcription factor, Foxp3, and those that do not. The latter include T regulatory 1 cells that produce predominantly IL-10, and or T helper 3 cells that produce predominantly transforming growth factor beta (TGFβ). Foxp3+ Tregs are crucial for preventing auto-immunity and keeping the immune system in homeostatic balance. This transcription factor not only is responsible for Treg differentiation, but also prevents these cells from becoming Th17 proinflammatory effector cells. Depletion of only Foxp3+Tregs in neonatal or adult mice results in massive lymphoproliferation and rapidly fatal multisystem autoimmunity [7]. Mutations of Foxp3 also result in severe autoimmune syndromes in humans [8]. The present review will concentrate on Tregs that express Foxp3 since information about T regulatory 1 cells and T helper 3 cells in SLE is very limited. Information on invariant natural killer T cells in SLE has recently been reviewed [9]. These cells also have an important role in immune surveillance.
In the mouse approximately 5% of CD4+ cells are Tregs that express Foxp3 [5]. In humans only 2% of CD4+ cells express Foxp3, and these are the most brightly staining CD25+ cells [10]. Foxp3 is unfortunately not a reliable marker of human Tregs because activated CD4+ cells can transiently co-express this transcription factor [11, 12]. Besides naturally occurring, thymus-derived CD4+CD25+Foxp3+ cells (nTregs), it is known that IL-2 and TGFβ can induce peripheral CD4+ cells to become Foxp3+ suppressor cells [13]. These suppressor cells are adaptive CD4+CD25+Foxp3+ cells (iTregs), induced in peripheral lymphoid tissues [14]. It is now apparent that both nTregs and Foxp3+ iTregs have a similar phenotype and similar functional properties. The CD4+CD25+Foxp3+ Tregs that circulate in the blood are probably a mixture of both subsets since a marker to distinguish these subsets is not available. Similarities and differences between Foxp3+ nTregs and Foxp3+ iTregs are reviewed elsewhere [15]. Importantly, both IL-2 and TGFβ are required for the maintenance of Foxp3 expression and for the survival of both subsets [16, 17]. Since production of both of these cytokines is decreased in SLE [3], these defects probably contribute to abnormalities of Foxp3+ Tregs as described in the present review.
Regulatory T cells in mouse models of lupus
Natural Tregs have protective effects in lupus-prone mice (Table 1). In the lupus-prone New Zealand mixed 2328 mice, 3-day thymectomy results in an early-onset, fatal glomerulo-nephritis in females. The nephritis, however, largely regressed in males, thereby revealing a checkpoint in lupus glomerulo-nephritis progression that depends on gender. The transfer of CD25+ nTregs from 6-week-old asymptomatic donors effectively suppressed dsDNA autoantibody, but did not protect the mice from the proliferative lupus glomerulo-nephritis and sialoadenitis. Therefore, although nTregs have some protective effects in New Zealand mixed 2328 mice, these cells by themselves cannot control the disease. Other Tregs are apparently needed for full protection [18].
The protective effects of endogenous CD4+CD25+ Tregs in other mouse models are complex. Depletion of CD4+CD25+ cells in (NZB × NZW)F1 hybrid mice accelerated the onset of glomerulonephritis [19]. In young BWF1 and/or SNF1 mice that spontaneously develop a lupus-like disease, CD4+CD25+ nTregs are decreased before they develop glomerulonephritis [20–22]. One group estimated that the pool of CD4+CD25+ cells in BWF1 mice is 40% to 50% that of phenotypically normal mice [21]. This is important since all strains of lupus-prone mice have B-cell abnormalities with hyperactive B cells [23] and may also have hyperactive T cells with a low threshold for activation [24]. On the other hand, the total number of CD4+CD25+ cells is not reduced in older mice that have developed lupus [20], and several groups have found that the suppressor cell activity in vitro is intact in lupus-prone mice [20, 21, 25]. Another group has reported that T cells from MLR/lpr mice are resistant to T-cell suppression [25]. These cells may not be able to overcome the strength of T-cell stimulation or the inhibitory effects of proinflammatory cytokines produced by other cells. It is known that strong Toll-like receptor stimulation triggered by microbial infections results in IL-6 and other cytokines that block Treg function [26]. nTregs may therefore not be able to function normally in inflamed tissues.
The best evidence for the importance of Tregs in the pathogenesis of SLE is that increasing the numbers of Foxp3+ Tregs can alter the disease course. The adoptive transfer of expanded CD4+CD25+ cells to BWF1 mice delayed onset of the disease [20]. iTregs induced ex vivo with IL-2 and TGFβ also have protective effects in lupus-like syndromes. The transfer of parental DBA/2 spleen cells into (DBA/2 × C57/BL6)F1 mice results in a chronic graft versus host disease with high titers of anti-dsDNA autoantibodies and a fatal immune complex glomerulonephritis. A single dose of anti-donor iTregs prevented this syndrome and doubled the survival of mice that had already developed anti-dsDNA antibodies [27]. Polyclonal iTregs induced by stimulating T cells with anti-CD3/CD28-coated beads with IL-2 and TGFβ have also been recently found to have protective effects in this model and other mouse models of autoimmune disease [28]; in a collaboration with Antonio La Cava and Bevra Hahn at UCLA, we have found these polyclonal iTregs also have protective effects in the BWF1 model (unpublished observations).
SLE is characterized by high levels of IL-6; this cytokine can interfere with the function of Tregs [26], and can even convert nTregs to IL-17-producing cells [29]. IL-17 levels have been reported to be increased in SLE [30]. iTregs induced by IL-2 and TGFβ, however, are resistant to this effect of IL-6. This cytokine combination downregulates IL-6 receptor expression and signaling [31].
Immunization with peptides derived from nuclear autoantigens or VH complementarity determining region sequences derived from pathogenic anti-DNA antibodies can suppress the development of lupus [32–35]. One group reported that high-dose intravenous administration of a small ribonucleoprotein peptide to BWF1 mice resulted in IL-10-producing Tregs that delayed the onset of lupus [36]. Two groups have shown therapeutic effects following nasal or subcutaneous immunization of BWF1 and/or SNF1 mice with a tolerogenic histone H4 peptide 471-94, described by Datta and colleagues [37]. Wu and Staines reported that intranasal immunization increased the numbers of CD4+CD25+ cells and decreased the T-cell proliferative response to this peptide [33]. Datta's group reported that low-dose peptide subcutaneous immunization induces SNF1 mice to develop Foxp3+CD4+ and Foxp3+CD8+ Tregs that produce TGFβ, resulting in a delay of glomerulonephritis and prolonged survival [32]. Similarly, mice immunized with low doses of an artificial V(H) peptide that contains T-cell determinants (called pConsensus) results in CD4 and CD8 Tregs that each expressed Foxp3. These Tregs blocked production of anti-DNA antibodies and prolonged survival [38, 39].
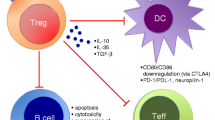
Overproduction of type I interferons in SLE matures dendritic cells (DCs) into immunogenic antigen-presenting cells that strongly contribute to the T-cell and B-cell hyperactivity characteristic of this disease [40]. An imbalance between immunogenic and tolerogenic DCs in SLE probably limits the expansion of Tregs and can account for the decreased numbers of these cells. The remaining Tregs cannot overcome the strong T-cell stimulation provided by immunogenic DCs. This imbalance, however, is reversible. Before SNF1 mice were given tolerogenic peptides, plasmacytoid DCs from these mice produced high levels of IL-6 and their T cells produced IL-17. Following immunization with tolerogenic H4 471-94 peptides, however, plasmacytoid DCs instead produced high amounts of TGFβ, and autoantigen-specific T cells no longer produced IL-17 [32]. The balance between tolerogenic and immunogenic DCs can therefore be restored in a mouse model of SLE (see Figure 1).

T-cell/dendritic cell interactions in health and in systemic lupus erythematosus. Regulatory T cells (Tregs) (red) and effector T cells, shown as potentially pathogenic anti-self T cells (blue), can affect the maturation of immature dendritic cells (DCs) to immunogenic or tolerogenic antigen-presenting cells. Transforming growth factor beta (TGFβ) and IL-10 produced by Tregs promote tolerogenic DCs, and IFNγ or IL-17 produced by effector T cells promotes immunogenic DCs. Toll-like receptor (TLR) 7 and TLR9 stimulation by apoptotic bodies in systemic lupus erythematosus (SLE) results in type 1 interferon production, which promotes immunogenic DCs that activate potentially pathogenic self-reactive T cells. The feedback loop shown sustains immunogenic DCs and, secondarily, results in decreased Tregs.
Regulatory T cells and dendritic cells in human SLE
In the past it had been generally believed that decreased numbers and/or function of Tregs contribute to the T-cell and B-cell hyperactivity characteristic of SLE. During the 1970s and 1980s when suppressor cells were considered to be principally CD8+ cells, many groups reported decreased functional activity [3]. A recent study has also documented impaired CD8+cell suppressive function in SLE [41]. With the shift in attention from CD8+ Tregs to CD4+ Tregs, published studies of CD4+CD25+cells and CD4+Foxp3+ cells in human SLE have been apparently contradictory.
Early reports of CD4+CD25+ T cells in human SLE revealed decreased numbers of this subset (Table 2) [42–45]. Foxp3 expression and suppressive activity was then revealed to be concentrated in the CD4 fraction staining brightly for CD25 [10]. Eight groups subsequently reported decreased percentages of CD4+CD25highFoxp3+ Tregs in SLE [22, 46–52]. Four of these groups reported an inverse correlation between percentages of CD4+CD25+ cells and disease activity [47, 49–51]. Two groups published sequential studies that revealed an increase in CD4+CD25high cells in patients when the disease became inactive [49]. CD4+CD25high cells also increased following various treatments that included corticosteroid therapy [53], therapeutic plasmapheresis [54], or B-cell depletion with Rituximab therapy [55, 56]. Since the total number of T cells in patients with active SLE is generally decreased, the absolute numbers of circulating CD4+CD25high cells would also be decreased.
Two contrary reports have recently appeared documenting that CD4+CD25high cells in SLE are similar to those of healthy donors [57, 58]. A third group reported that although the percentage of CD4+CD25high cells in SLE in patients with active disease was normal, the relative number of these Tregs was actually decreased because of a greater expansion of effector T cells [59].
As with the numbers of CD4+CD25+ cells, most workers have found suppressor function in human SLE to be abnormal. Several groups have reported in addition to decreased CD4+CD25high Tregs that the suppressive activity in vitro was also decreased [47, 48, 50, 57]. Most of these groups attributed the defects to the Tregs, but one group reported increased resistance to suppression [57]. This resistance positively correlated with disease activity. Although this group attributed this resistance to decreased sensitivity of responder T cells to suppression, they did not exclude the possibility that excessive costimulation by autologous SLE accessory cells included in the cultures was responsible for this finding. Consistent with this explanation, another group reported that SLE responder cells can be inhibited by Tregs in an assay that did not contain accessory cells [50].
Some discrepant reports of suppressive activity of Tregs in SLE have appeared. A group that reported decreased Treg percentages in SLE found that the inhibitory function of these cells was normal [49]. Another group reported normal percentages of Tregs in SLE but found decreased suppressive function in approximately one-third of the patients [60]. The reasons for discrepant results include heterogeneous patient populations, the effects of treatment on the subjects studied, and technical differences such as the presence or absence of accessory cells in the assays (see above).
Studies of CD4+Foxp3+ cells in SLE have been even more variable that those on CD4+CD25+ cells. Two groups have reported decreased CD4+CD25+Foxp3+ cells that correlated inversely with disease activity [48, 50]. One other group reported decreased CD4+CD25+Foxp3+ cells in SLE without any correlation with disease activity [46]. Three other studies have reported normal values for CD4+CD25+Foxp3+ cells [57–59]. Surprisingly, during the past year four separate groups have reported increased percentages of CD4+Foxp3+ in lupus and found that this result correlated with disease activity [47, 61–63]. Here Foxp3 was not only expressed by CD4+CD25+cells, but also by CD4+CD25-Foxp3+ cells. In our studies of untreated SLE patients admitted to the Los Angeles County/University of Southern California Medical Center for active SLE, we have generally found normal or increased percentages of CD4+CD25highFoxp3+ cells and have found that CD25-Foxp3+ cells are also increased.
Although Foxp3 is a reliable marker of Tregs in mice, it has become evident that conventional human CD4+ cells can transiently express this transcription factor when they are activated [11]. Thus, the controversial results concerning percentages of Foxp3+ CD4+ cells and correlations with disease activity in SLE may be explained by a variability of disease activity in the patients studied. Investigators who reported decreased percentages probably studied patients with chronic, moderately active disease and the percentages of these Foxp3+ CD4+ Tregs increased with treatment and disease improvement. By contrast, investigators who reported increased Foxp3+ CD4+ cells probably studied patients with more acutely active disease. These cells included activation-induced Foxp3+ CD4+ nonTregs which became Foxp3- as the disease became less severe. The CD4+CD25-Foxp3+ cells remain to be characterized.
CD127, the α-chain of the IL-7 receptor, has been found a useful marker to discriminate between CD4+ regulatory and effector cells [64]. Both naïve and most previously activated CD4+cells stain brightly for CD127. Although most human CD4+CD25highFoxp3+ Tregs are previously activated memory cells, this marker has been downregulated and these cells have become CD127dim. Two groups have studied CD4+CD127dim cells in SLE. One study confirmed that suppressive activity in CD4+CD25+ cells in SLE was predominantly contained in the CD127dim subset [57]. The other group reported that although the percentage of CD127dim cells in SLE was similar to that in healthy controls this subset was relatively decreased because of expansion of activated CD4+CD25+ effector cells [59].
Another problem with measurements of Tregs in humans is that the numbers circulating in the blood may not correlate with the numbers and function of these cells in the tissues. There is very limited information concerning Foxp3+ Treg numbers in lymphoid organs and in the tissues of patients with SLE. One group has reported decreased Foxp3+ cells in a lymph node from a patient with active SLE and in mRNA isolated from the kidneys of five patients [49]. Another group found decreased numbers of Foxp3+ cells in the skin of patients with cutaneous lupus erythematosus. The number of circulating Foxp3+ cells in these patients was normal [65].
Dysfunctional DCs in human SLE have also been described. Monocyte-derived DCs from lupus patients displayed an abnormal phenotype characterized by accelerated differentiation, maturation and secretion of proinflammatory cyto-kines, suggesting they are in a preactivated state [66, 67] – although not all workers agree [68]. The numbers of plasmacytoid DCs in the blood were reduced, but these cells accumulated in the kidney suggesting increased migration to the tissues [69]. Yan and coworkers have evidence that suggests antigen-presenting cells are responsible for Treg defects in SLE [62]. These authors reported increased circulating CD4+CD25+Foxp3+ cells that positively correlated with disease activity, and the suppressive function of these cells was intact in cultures without antigen-presenting cells. The functional activity of both patient and healthy control Tregs, however, was decreased in the presence of the patient's antigen-presenting cells. Moreover, IFNα produced by these antigen-presenting cells strongly contributed to the defective function. These findings are consistent with the evidence of the important role of type I interferons in the pathogenesis of SLE. The suppressor cell defects described were therefore probably secondary to the resulting imbalance between immunogenic DCs and tolerogenic DCs.
Conclusion and future directions
The studies reviewed above are consistent with the view that decreased numbers and/or function of Foxp3+ Tregs contribute to the pathogenesis of SLE. In mouse lupus, this evidence included decreased numbers of Foxp3+ Tregs in the early stage. Although Tregs are present in adequate numbers in the tissues of animals with established disease, they are unable to terminate chronic inflammation for reasons that may include increased levels of IL-6. Nonetheless, the adoptive transfer of nTregs or iTregs alters the SLE disease course, and the administration of tolerogenic peptides to young lupus-prone mice results in increased numbers of Foxp3+CD4+ and Foxp3+CD8+ iTregs that have protective effects.
In human lupus the evidence that Treg defects play a major role in the perpetuation of this disease is only suggestive. Studies of Tregs in SLE are mostly limited to blood lymphocytes and although there is considerable evidence that that CD4+CD25high cells are decreased, not all workers agree.
This is probably because of contaminating activated effector cells in the fraction of CD4+CD25+ cells measured. Two groups, however, have reported normal percentages of CD4+CD127dim cells, a subset that probably excludes most of these contaminating cells [57, 58]. The possibility remains that Tregs are relatively decreased due to expansion of effector T cells [59]. Most workers have found decreased suppressor T-cell activity in SLE, but here also contradictory reports have been published. Rather than a true functional defect, defective suppressive activity could reflect increased resistance of responder T cells to suppression or increased costimulatory activity of antigen-presenting cells (Table 2). Even those who report decreased suppressor T-cell function have observed some activity of these cells in SLE because the T-cell response to stimulation was augmented when Tregs were removed [58].
Further information is needed in several areas. While in mice it has been well documented that IL-2 and TGFβ can convert naïve CD4+CD25- T cells to CD25+Foxp3+ suppressor cells, similar conversion in humans is more complex. In both mice and humans, suppressive nTregs can become proinflam-matory IL-17-producing cells [29, 70]. iTregs in mice induced ex vivo with IL-2 and TGFβ are resistant to this conversion [31], but in humans this has yet to be demonstrated. The cytokine and gene expression profile of Foxp3+ Tregs in SLE in comparison with healthy subjects has yet to be defined. The functional properties of nTegs and iTregs need to be better characterized, and more information is also needed about CD8+ suppressor cells in SLE. It is difficult to draw meaningful conclusions about the Treg functional activity from assays based upon the suppression of T-cell proliferation in vitro. Tregs also inhibit T-cell migration, differentiation and apoptosis. Moreover, the principal target of Treg activity is probably antigen-presenting DCs rather than T cells [71, 72]. In addition, Tregs may have direct suppressive effects on B cells [73], on natural killer cells [74, 75] and on osteoclasts [76, 77].
The contact-dependent mechanism of action of CD4+Foxp3+ Tregs also remains poorly understood. TGFβ has an essential role since effector T cells that cannot respond to this cytokine fail to be inhibited [78]. How human Tregs convert the latent precursor to its biologically active form remains to be clarified. Other molecules that regulate Treg function include cytotoxic T-lymphocyte antigen 4 [79], B7 family molecules expressed on the cell surface [80], TNF family proteins [81], and adenosine and its receptors [82, 83].
It is evident in SLE that normal immunologic homeostasis has been disrupted, with the balance strongly weighted towards sustained T-cell reactivity. The two principal external mechanisms that control T-cell reactivity, Treg suppression and activation-induced apoptosis have failed. There is, however, an important difference that distinguishes human lupus from mouse lupus. In mice the course of the disease is steadily downhill and fatal, whereas human disease is cyclic and characterized by exacerbations and remission. Moreover when patients enter remission, many of the abnormalities of T cells and B cells improve [3]. This difference implies that at least some feedback regulatory mechanisms in humans become functional again as disease activity remits.
Other manifestations of disrupted immunologic homeostasis in active SLE include impaired host defense. This abnormality is probably the secondary sequelae of failed attempts to control self-reactive cells. Finally, lymphocyte production of IL-2 and the mature form of TGFβ is decreased in SLE (reviewed in [3]), and these are the cytokines required for the growth and functional activity of Tregs.
An important goal in the management of human SLE is to restore the balance between immunogenic and tolerogenic DCs, and thereby to correct Treg numbers and function. In active lupus, the products of the apoptotic cells that bind to Toll-like receptor 7, Toll-like receptor 8, and Toll-like receptor 9 [84] expressed by DCs, and the proinflammatory cytokines produced by activated T cells, continuously stimulate immature DC to become immunogenic antigen-presenting cells. These cells, in turn, activate more self-reactive T cells to produce proinflammatory cytokines which sustain this pathologic circuit (Figure 1). Strategies are needed to interrupt this cycle and to drive the maturation of immature DCs towards tolerogenic DCs that induce Tregs. Possible approaches include treatment with anti-CD3 antibodies, immunization with tolerogenic peptides, or the transfer of autologous Tregs generated and/or expanded ex vivo. As stated above, the latter approach has been successful in mice. Transfer of ex vivo expanded nTregs or of polyclonal iTregs induced with IL-2 and TGFβ has had beneficial effects. Besides controlling the activity of other T cells and B cells, evidence has been obtained that these Tregs also induce tolerogenic DCs in transplant tolerance [85]. In SLE once the DC balance has been shifted back to tolerogenic predominance, further stimulation of these tolerogenic DCs with peptide autoantigens should expand or induce new iTregs. Normalization of Treg numbers and function in individuals with SLE has the potential to lead to remission of this autoimmune disease.
Abbreviations
- DC:
-
dendritic cell
- IFN:
-
interferon
- IL:
-
interleukin
- iTreg:
-
adaptive or induced CD4+CD25+Foxp3+ cell
- nTreg:
-
natural or thymus-derived CD4+CD25+Foxp3+ cell
- SLE:
-
systemic lupus erythematosus
- TGFβ:
-
transforming growth factor beta
- Treg:
-
regulatory T cell.
References
Teague PO, Friou GJ: Antinuclear antibodies in mice. II. Transmission with spleen cells; inhibition or prevention with thymus or spleen cells. Immunology. 1969, 17: 665-675.
Krakauer RS, Strober W, Rippeon DL, Waldmann TA: Prevention of autoimmunity in experimental lupus erythematosus by soluble immune response suppressor. Science. 1977, 196: 56-59. 10.1126/science.300174.
Horwitz DA, Gray JD, editors: The interaction of T cells with cells of the innate immune system and B cells in the pathogenesis of SLE. Dubois' lupus erythematosus. 2007, New York: Lippincott Williams & Wilkins, 133-160. 7
Asano M, Toda M, Sakaguchi N, Sakaguchi S: Autoimmune disease as a consequence of developmental abnormality of a T cell subpopulation. J Exp Med. 1996, 184: 387-396. 10.1084/jem.184.2.387.
Sakaguchi S, Ono M, Setoguchi R, Yagi H, Hori S, Fehervari Z, Shimizu J, Takahashi T, Nomura T: Foxp3+ CD25+CD4+ natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol Rev. 2006, 212: 8-27. 10.1111/j.0105-2896.2006.00427.x.
Lan RY, Ansari AA, Lian ZX, Gershwin ME: Regulatory T cells: development, function and role in autoimmunity. Autoimmun Rev. 2005, 4: 351-363. 10.1016/j.autrev.2005.01.007.
Kim JM, Rasmussen JP, Rudensky AY: Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol. 2007, 8: 191-197. 10.1038/ni1428.
Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova JL, Buist N, Levy-Lahad E, Mazzella M, Goulet O, Perroni L, Bricarelli FD, Byrne G, McEuen M, Proll S, Appleby M, Brunkow ME: X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet. 2001, 27: 18-20. 10.1038/83707.
Godo M, Sessler T, Hamar P: Role of invariant natural killer T (iNKT) cells in systemic lupus erythematosus. Curr Med Chem. 2008, 15: 1778-1787. 10.2174/092986708785132988.
Baecher-Allan C, Brown JA, Freeman GJ, Hafler DA: CD4+CD25high regulatory cells in human peripheral blood. J Immunol. 2001, 167: 1245-1253.
Gavin MA, Torgerson TR, Houston E, DeRoos P, Ho WY, Stray-Pedersen A, Ocheltree EL, Greenberg PD, Ochs HD, Rudensky AY: Single-cell analysis of normal and FOXP3-mutant human T cells: FOXP3 expression without regulatory T cell development. Proc Natl Acad Sci USA. 2006, 103: 6659-6664. 10.1073/pnas.0509484103.
Pillai V, Ortega SB, Wang CK, Karandikar NJ: Transient regulatory T-cells: a state attained by all activated human T-cells. Clin Immunol. 2007, 123: 18-29. 10.1016/j.clim.2006.10.014.
Horwitz DA, Zheng SG, Wang J, Gray JD: Critical role of IL-2 and TGF-β in generation, function and stabilization of Foxp3(+)CD4(+) Treg. Eur J Immunol. 2008, 38: 912-915. 10.1002/eji.200738109.
Bluestone JA, Abbas AK: Natural versus adaptive regulatory T cells. Nat Rev Immunol. 2003, 3: 253-257. 10.1038/nri1032.
Horwitz DA, Zheng SG, Gray JD: Natural and TGF-β-induced Foxp3(+)CD4(+)CD25(+) regulatory T cells are not mirror images of each other. Trends Immunol. 2008, 29: 429-435. 10.1016/j.it.2008.06.005.
Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY: A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat Immunol. 2005, 6: 1142-1151. 10.1038/ni1263.
Marie JC, Letterio JJ, Gavin M, Rudensky AY: TGF-β1 maintains suppressor function and Foxp3 expression in CD4+CD25+ regulatory T cells. J Exp Med. 2005, 201: 1061-1067. 10.1084/jem.20042276.
Bagavant H, Tung KS: Failure of CD25+ T cells from lupus-prone mice to suppress lupus glomerulonephritis and sialoadenitis. J Immunol. 2005, 175: 944-950.
Hayashi T, Hasegawa K, Adachi C: Elimination of CD4(+)CD25(+) T cell accelerates the development of glomerulonephritis during the preactive phase in autoimmune-prone female NZB × NZW F mice. Int J Exp Pathol. 2005, 86: 289-296. 10.1111/j.0959-9673.2005.00438.x.
Scalapino KJ, Tang Q, Bluestone JA, Bonyhadi ML, Daikh DI: Suppression of disease in New Zealand Black/New Zealand White lupus-prone mice by adoptive transfer of ex vivo expanded regulatory T cells. J Immunol. 2006, 177: 1451-1459.
Hsu WT, Suen JL, Chiang BL: The role of CD4CD25 T cells in autoantibody production in murine lupus. Clin Exp Immunol. 2006, 145: 513-519. 10.1111/j.1365-2249.2006.03173.x.
Liu MF, Wang CR, Fung LL, Wu CR: Decreased CD4+CD25+ T cells in peripheral blood of patients with systemic lupus erythematosus. Scand J Immunol. 2004, 59: 198-202. 10.1111/j.0300-9475.2004.01370.x.
Theofilopoulos AN, Shawler DL, Eisenberg RA, Dixon FJ: Splenic immunoglobulin-secreting cells and their regulation in autoimmune mice. J Exp Med. 1980, 151: 446-466. 10.1084/jem.151.2.446.
Zielinski CE, Jacob SN, Bouzahzah F, Ehrlich BE, Craft J: Naive CD4+ T cells from lupus-prone Fas-intact MRL mice display TCR-mediated hyperproliferation due to intrinsic threshold defects in activation. J Immunol. 2005, 174: 5100-5109.
Monk CR, Spachidou M, Rovis F, Leung E, Botto M, Lechler RI, Garden OA: MRL/Mp CD4+, CD25- T cells show reduced sensitivity to suppression by CD4+, CD25+regulatory T cells in vitro: a novel defect of T cell regulation in systemic lupus erythe-matosus. Arthritis Rheum. 2005, 52: 1180-1184. 10.1002/art.20976.
Pasare C, Medzhitov R: Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science. 2003, 299: 1033-1036. 10.1126/science.1078231.
Zheng SG, Wang JH, Koss MN, Quismorio F, Gray JD, Horwitz DA: CD4+ and CD8+ regulatory T cells generated ex vivo with IL-2 and TGF-β suppress a stimulatory graft-versus-host disease with a lupus-like syndrome. J Immunol. 2004, 172: 1531-1539.
Godebu E, Summers-Torres D, Lin MM, Baaten BJ, Bradley LM: Polyclonal adaptive regulatory CD4 cells that can reverse type I diabetes become oligoclonal long-term protective memory cells. J Immunol. 2008, 181: 1798-1805.
Xu L, Kitani A, Fuss I, Strober W: Cutting edge: regulatory T cells induce CD4+CD25-Foxp3- T cells or are self-induced to become Th17 cells in the absence of exogenous TGF-β. J Immunol. 2007, 178: 6725-6729.
Wong CK, Lit LC, Tam LS, Li EK, Wong PT, Lam CW: Hyperproduction of IL-23 and IL-17 in patients with systemic lupus erythematosus: Implications for Th17-mediated inflammation in auto-immunity. Clin Immunol. 2008, 127: 385-393. 10.1016/j.clim.2008.01.019.
Zheng SG, Wang J, Horwitz DA: Foxp3+ CD4+CD25+ regulatory T cells induced by IL-2 and TGF-β are resistant to Th17 conversion by IL-6. J Immunol. 2008, 180: 7112-7116.
Kang HK, Liu M, Datta SK: Low-dose peptide tolerance therapy of lupus generates plasmacytoid dendritic cells that cause expansion of autoantigen-specific regulatory T cells and contraction of inflammatory Th17 cells. J Immunol. 2007, 178: 7849-7858.
Wu HY, Staines NA: A deficiency of CD4+CD25+T cells permits the development of spontaneous lupus-like disease in mice, and can be reversed by induction of mucosal tolerance to histone peptide autoantigen. Lupus. 2004, 13: 192-200. 10.1191/0961203303lu1002oa.
Eilat E, Zinger H, Nyska A, Mozes E: Prevention of systemic lupus erythematosus-like disease in (NZBxNZW)F1 mice by treating with CDR1- and CDR3-based peptides of a pathogenic autoantibody. J Clin Immunol. 2000, 20: 268-278. 10.1023/A:1006663519132.
Hahn BH, Singh RP, La Cava A, Ebling FM: Tolerogenic treatment of lupus mice with consensus peptide induces Foxp3-expressing, apoptosis-resistant, TGFβ-secreting CD8+ T cell suppressors. J Immunol. 2005, 175: 7728-7737.
Riemekasten G, Langnickel D, Enghard P, Undeutsch R, Humrich J, Ebling FM, Hocher B, Humaljoki T, Neumayer H, Burmester GR, Hahn BH, Radbruch A, Hiepe F: Intravenous injection of a D1 protein of the Smith proteins postpones murine lupus and induces type 1 regulatory T cells. J Immunol. 2004, 173: 5835-5842.
Kaliyaperumal A, Michaels MA, Datta SK: Naturally processed chromatin peptides reveal a major autoepitope that primes pathogenic T and B cells of lupus. J Immunol. 2002, 168: 2530-2537.
Singh RP, La Cava A, Hahn BH: pConsensus peptide induces tolerogenic CD8+ T cells in lupus-prone (NZB × NZW)F1 mice by differentially regulating Foxp3 and PD1 molecules. J Immunol. 2008, 180: 2069-2080.
Singh RP, La Cava A, Wong M, Ebling F, Hahn BH: CD8+T cell-mediated suppression of autoimmunity in a murine lupus model of peptide-induced immune tolerance depends on Foxp3 expression. J Immunol. 2007, 178: 7649-7657.
Banchereau J, Pascual V: Type I interferon in systemic lupus erythematosus and other autoimmune diseases. Immunity. 2006, 25: 383-392. 10.1016/j.immuni.2006.08.010.
Filaci G, Bacilieri S, Fravega M, Monetti M, Contini P, Ghio M, Setti M, Puppo F, Indiveri F: Impairment of CD8+ T suppressor cell function in patients with active systemic lupus erythematosus. J Immunol. 2001, 166: 6452-6457.
Crispin JC, Martinez A, Alcocer-Varela J: Quantification of regulatory T cells in patients with systemic lupus erythematosus. J Autoimmun. 2003, 21: 273-276. 10.1016/S0896-8411(03)00121-5.
Fathy A, Mohamed RW, Tawfik GA, Omar AS: Diminished CD4+CD25+ T-lymphocytes in peripheral blood of patients with systemic lupus erythematosus. Egypt J Immunol. 2005, 12: 25-31.
Lee JH, Wang LC, Lin YT, Yang YH, Lin DT, Chiang BL: Inverse correlation between CD4+ regulatory T-cell population and autoantibody levels in paediatric patients with systemic lupus erythematosus. Immunology. 2006, 117: 280-286. 10.1111/j.1365-2567.2005.02306.x.
Yang XY, Lu XY, Xu DH, Lu QH, Wang QH, Wu HX: Clinical significance of CD4+CD25+ T cells in peripheral blood of patients in systemic lupus erythematosus. Zhonghua Nei Ke Za Zhi. 2005, 44: 570-572.
Barath S, Aleksza M, Tarr T, Sipka S, Szegedi G, Kiss E: Measurement of natural (CD4+CD25high) and inducible (CD4+IL-10+) regulatory T cells in patients with systemic lupus erythematosus. Lupus. 2007, 16: 489-496. 10.1177/0961203307080226.
Bonelli M, von Dalwigk K, Savitskaya A, Smolen JS, Scheinecker C: Foxp3 expression in CD4+ T cells of patients with systemic lupus erythematosus: a comparative phenotypic analysis. Ann Rheum Dis. 2008, 67: 664-671. 10.1136/ard.2007.074690.
Lyssuk EY, Torgashina AV, Soloviev SK, Nassonov EL, Bykovskaia SN: Reduced number and function of CD4+CD25highFoxP3+ regulatory T cells in patients with systemic lupus erythematosus. Adv Exp Med Biol. 2007, 601: 113-119.
Miyara M, Amoura Z, Parizot C, Badoual C, Dorgham K, Trad S, Nochy D, Debre P, Piette JC, Gorochov G: Global natural regulatory T cell depletion in active systemic lupus erythematosus. J Immunol. 2005, 175: 8392-8400.
Valencia X, Yarboro C, Illei G, Lipsky PE: Deficient CD4+CD25high T regulatory cell function in patients with active systemic lupus erythematosus. J Immunol. 2007, 178: 2579-2588.
Lee HY, Hong YK, Yun HJ, Kim YM, Kim JR, Yoo WH: Altered frequency and migration capacity of CD4+CD25+regulatory T cells in systemic lupus erythematosus. Rheumatology (Oxford). 2008, 47: 789-794. 10.1093/rheumatology/ken108.
Mellor-Pita S, Citores MJ, Castejon R, Tutor-Ureta P, Yebra-Bango M, Andreu JL, Vargas JA: Decrease of regulatory T cells in patients with systemic lupus erythematosus. Ann Rheum Dis. 2006, 65: 553-554. 10.1136/ard.2005.044974.
Suarez A, Lopez P, Gomez J, Gutierrez C: Enrichment of CD4+CD25high T cell population in patients with systemic lupus erythematosus treated with glucocorticoids. Ann Rheum Dis. 2006, 65: 1512-1517. 10.1136/ard.2005.049924.
Barath S, Soltesz P, Kiss E, Aleksza M, Zeher M, Szegedi G, Sipka S: The severity of systemic lupus erythematosus negatively correlates with the increasing number of CD4+CD25(high)FoxP3+ regulatory T cells during repeated plasmapheresis treatments of patients. Autoimmunity. 2007, 40: 521-528. 10.1080/08916930701610028.
Sfikakis PP, Souliotis VL, Fragiadaki KG, Moutsopoulos HM, Boletis JN, Theofilopoulos AN: Increased expression of the FoxP3 functional marker of regulatory T cells following B cell depletion with rituximab in patients with lupus nephritis. Clin Immunol. 2007, 123: 66-73. 10.1016/j.clim.2006.12.006.
Vallerskog T, Gunnarsson I, Widhe M, Risselada A, Klareskog L, van Vollenhoven R, Malmstrom V, Trollmo C: Treatment with rit-uximab affects both the cellular and the humoral arm of the immune system in patients with SLE. Clin Immunol. 2007, 122: 62-74. 10.1016/j.clim.2006.08.016.
Venigalla RK, Tretter T, Krienke S, Max R, Eckstein V, Blank N, Fiehn C, Ho AD, Lorenz HM: Reduced CD4+, CD25- T cell sensitivity to the suppressive function of CD4+, CD25high, CD127-/low regulatory T cells in patients with active systemic lupus erythematosus. Arthritis Rheum. 2008, 58: 2120-2130. 10.1002/art.23556.
Yates J, Whittington A, Mitchell P, Lechler RI, Lightstone L, Lombardi G: Natural regulatory T cells: number and function are normal in the majority of patients with lupus nephritis. Clin Exp Immunol. 2008, 153: 44-55. 10.1111/j.1365-2249.2008.03665.x.
Zhao SS, Li XM, Li XP, Zhai ZM, Chen Z, Ma Y, Zhang H, Zheng SG: Expression of CD4+ CD25+CD127(low/-) T cells in patients with systemic lupus erythematosus. Zhonghua Yi Xue Za Zhi. 2008, 88: 453-456.
Alvarado-Sanchez B, Hernandez-Castro B, Portales-Perez D, Baranda L, Layseca-Espinosa E, Abud-Mendoza C, Cubillas-Tejeda AC, Gonzalez-Amaro R: Regulatory T cells in patients with systemic lupus erythematosus. J Autoimmun. 2006, 27: 110-118. 10.1016/j.jaut.2006.06.005.
Lin SC, Chen KH, Lin CH, Kuo CC, Ling QD, Chan CH: The quantitative analysis of peripheral blood FOXP3-expressing T cells in systemic lupus erythematosus and rheumatoid arthritis patients. Eur J Clin Invest. 2007, 37: 987-996. 10.1111/j.1365-2362.2007.01882.x.
Yan B, Ye S, Chen G, Kuang M, Shen N, Chen S: Dysfunctional CD4+, CD25+ regulatory T cells in untreated active systemic lupus erythematosus secondary to interferon-alpha-producing antigen-presenting cells. Arthritis Rheum. 2008, 58: 801-812. 10.1002/art.23268.
Zhang B, Zhang X, Tang F, Zhu L, Liu Y, Chen W, Lipsky P: Clinical significance of increased CD4+CD25-Foxp3+T cells in patients with new-onset systemic lupus erythematosus. Ann Rheum Dis. 2008, 67: 1037-1040. 10.1136/ard.2007.083543.
Liu W, Putnam AL, Xu-Yu Z, Szot GL, Lee MR, Zhu S, Gottlieb PA, Kapranov P, Gingeras TR, Fazekas de St Groth B, Clayberger C, Soper DM, Ziegler SF, Bluestone JA: CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med. 2006, 203: 1701-1711. 10.1084/jem.20060772.
Franz B, Fritzsching B, Riehl A, Oberle N, Klemke CD, Sykora J, Quick S, Stumpf C, Hartmann M, Enk A, Ruzicka T, Krammer PH, Suri-Payer E, Kuhn A: Low number of regulatory T cells in skin lesions of patients with cutaneous lupus erythematosus. Arthritis Rheum. 2007, 56: 1910-1920. 10.1002/art.22699.
Ding D, Mehta H, McCune WJ, Kaplan MJ: Aberrant phenotype and function of myeloid dendritic cells in systemic lupus erythematosus. J Immunol. 2006, 177: 5878-5889.
Decker P, Kotter I, Klein R, Berner B, Rammensee HG: Monocyte-derived dendritic cells over-express CD86 in patients with systemic lupus erythematosus. Rheumatology (Oxford). 2006, 45: 1087-1095. 10.1093/rheumatology/kel061.
Koller M, Zwolfer B, Steiner G, Smolen JS, Scheinecker C: Phenotypic and functional deficiencies of monocyte-derived den-dritic cells in systemic lupus erythematosus (SLE) patients. Int Immunol. 2004, 16: 1595-1604. 10.1093/intimm/dxh160.
Tucci M, Quatraro C, Lombardi L, Pellegrino C, Dammacco F, Silvestris F: Glomerular accumulation of plasmacytoid dendritic cells in active lupus nephritis: role of interleukin-18. Arthritis Rheum. 2008, 58: 251-262. 10.1002/art.23186.
Koenen HJ, Smeets RL, Vink PM, Rijssen EV, Boots AM, Joosten I: Human CD25highFoxp3pos regulatory T-cells differentiate into IL-17 producing cells. Blood. 2008, 112: 2340-2352. 10.1182/blood-2008-01-133967.
Tadokoro CE, Shakhar G, Shen S, Ding Y, Lino AC, Maraver A, Lafaille JJ, Dustin ML: Regulatory T cells inhibit stable contacts between CD4+ T cells and dendritic cells in vivo. J Exp Med. 2006, 203: 505-511. 10.1084/jem.20050783.
Tang Q, Adams JY, Tooley AJ, Bi M, Fife BT, Serra P, Santamaria P, Locksley RM, Krummel MF, Bluestone JA: Visualizing regulatory T cell control of autoimmune responses in nonobese diabetic mice. Nat Immunol. 2006, 7: 83-92. 10.1038/ni1289.
Zhao DM, Thornton AM, DiPaolo RJ, Shevach EM: Activated CD4+CD25+ T cells selectively kill B lymphocytes. Blood. 2006, 107: 3925-3932. 10.1182/blood-2005-11-4502.
Giroux M, Yurchenko E, St-Pierre J, Piccirillo CA, Perreault C: T regulatory cells control numbers of NK cells and CD8α+ immature dendritic cells in the lymph node paracortex. J Immunol. 2007, 179: 4492-4502.
Terme M, Chaput N, Combadiere B, Ma A, Ohteki T, Zitvogel L: Regulatory T cells control dendritic cell/NK cell cross-talk in lymph nodes at the steady state by inhibiting CD4+self-reactive T cells. J Immunol. 2008, 180: 4679-4686.
Kim YG, Lee CK, Nah SS, Mun SH, Yoo B, Moon HB: Human CD4+CD25+ regulatory T cells inhibit the differentiation of osteoclasts from peripheral blood mononuclear cells. Biochem Biophys Res Commun. 2007, 357: 1046-1052. 10.1016/j.bbrc.2007.04.042.
Zaiss MM, Axmann R, Zwerina J, Polzer K, Guckel E, Skapenko A, Schulze-Koops H, Horwood N, Cope A, Schett G: Treg cells suppress osteoclast formation: a new link between the immune system and bone. Arthritis Rheum. 2007, 56: 4104-4112. 10.1002/art.23138.
Fahlen L, Read S, Gorelik L, Hurst SD, Coffman RL, Flavell RA, Powrie F: T cells that cannot respond to TGF-β escape control by CD4(+)CD25(+) regulatory T cells. J Exp Med. 2005, 201: 737-746. 10.1084/jem.20040685.
Zheng SG, Wang JH, Stohl W, Kim KS, Gray JD, Horwitz DA: TGF-β requires CTLA-4 early after T cell activation to induce FoxP3 and generate adaptive CD4+CD25+regulatory cells. J Immunol. 2006, 176: 3321-3329.
Alderson KL, Zhou Q, Berner V, Wilkins DE, Weiss JM, Blazar BR, Welniak LA, Wiltrout RH, Redelman D, Murphy WJ: Regulatory and conventional CD4+ T cells show differential effects correlating with PD-1 and B7-H1 expression after immunotherapy. J Immunol. 2008, 180: 2981-2988.
Ono M, Shimizu J, Miyachi Y, Sakaguchi S: Control of autoimmune myocarditis and multiorgan inflammation by glucocorti-coid-induced TNF receptor family-related protein(high), Foxp3-expressing CD25+and CD25- regulatory T cells. J Immunol. 2006, 176: 4748-4756.
Aswad F, Kawamura H, Dennert G: High sensitivity of CD4+CD25+ regulatory T cells to extracellular metabolites nicotinamide adenine dinucleotide and ATP: a role for P2X7 receptors. J Immunol. 2005, 175: 3075-3083.
Kobie JJ, Shah PR, Yang L, Rebhahn JA, Fowell DJ, Mosmann TR: T regulatory and primed uncommitted CD4 T cells express CD73, which suppresses effector CD4 T cells by converting 5'-adenosine monophosphate to adenosine. J Immunol. 2006, 177: 6780-6786.
Krieg AM, Vollmer J: Toll-like receptors 7, 8, and 9: linking innate immunity to autoimmunity. Immunol Rev. 2007, 220: 251-269. 10.1111/j.1600-065X.2007.00572.x.
Min WP, Zhou D, Ichim TE, Strejan GH, Xia X, Yang J, Huang X, Garcia B, White D, Dutartre P, Jevnikar AM, Zhong R: Inhibitory feedback loop between tolerogenic dendritic cells and regulatory T cells in transplant tolerance. J Immunol. 2003, 170: 1304-1312.
Acknowledgements
The author thanks Song Guo Zheng and J Dixon Gray for helpful comments, and is grateful for the help of Omar De La Cruz in preparing the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
DAH serves as a consultant for Becton Dickinson Biosciences (San Jose, CA, USA).
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
About this article
Cite this article
Horwitz, D.A. Regulatory T cells in systemic lupus erythematosus: past, present and future. Arthritis Res Ther 10, 227 (2008). https://doi.org/10.1186/ar2511
Published:
DOI: https://doi.org/10.1186/ar2511