
Abstract
Systemic lupus erythematosus and its murine equivalent, modelled in the New Zealand Black and New Zealand White (NZB × NZW)F1 hybrid strain, are polygenic inflammatory diseases, probably reflecting an autoimmune response to debris from cells undergoing programmed cell death. Several human and murine loci contributing to disease have been defined. The present study asks whether the proinflammatory purinergic receptor P2X7, an initiator of a form of programmed cell death known as aponecrosis, is a candidate product of murine and human lupus susceptibility loci. One such locus in (NZB × NZW)F1 mice is lbw3, which is situated at the distal end of NZW chromosome 5. We first assess whether NZB mice and NZW mice carry distinct alleles of the P2RX7 gene as expressed by common laboratory strains, which differ in sensitivity to ATP stimulation. We then compare the responses of NZB lymphocytes, NZW lymphocytes and (NZB × NZW)F1 lymphocytes to P2X7 stimulation. NZB and NZW parental strains express the distinct P2X7-L and P2X7-P alleles of P2RX7, respectively, while lymphocytes from these and (NZB × NZW)F1 mice differ markedly in their responses to P2X7 receptor stimulation. NZB mice and NZW mice express functionally distinct alleles of the proinflammatory receptor, P2X7. We show that current mapping suggests that murine and human P2RX7 receptor genes lie within lupus susceptibility loci lbw3 and SLEB4, and we argue that these encode a product with the functional characteristics consistent with a role in lupus. Furthermore, we argue that aponecrosis as induced by P2X7 is a cell death mechanism with characteristics that potentially have particular relevance to disease pathogenesis.
Similar content being viewed by others

Introduction
Systemic lupus erythematosus (SLE) is a polygenic disease, although the genes contributing towards the disease are unknown. Several human susceptibility loci have been identified, with eight of the strongest candidates mapping to 1q23, 1q25-31, 1q41-42, 2q35-37, 4p16-15.2, 6p11-21, 12q24 and 16q12 [1]. Of the murine models, the New Zealand Black and New Zealand White (NZB × NZW)F1 hybrid strain is widely studied due to its similarity to human disease and its female preponderance. As with human SLE, the disorder of (NZB × NZW)F1 mice is polygenic with a contribution from both parents. In a study of (NZB × NZW)F2 mice, eight susceptibility loci were identified [2]. In the case of the locus lbw3, at the distal region of chromosome 5, homozygosity for the NZW-derived locus was associated with increased mortality at 12 months. Although originally mapped to 88 cM on murine chromosome 5 [2], more recent data locate the microsatellite used to define lbw3 at 81 cM (discussed later).
We have studied the properties of the proinflammatory purinergic receptor P2X7, encoded by a gene within the human SLE locus SLEB4 [3] at 12q24 (Ensembl Genome Browser: http://www.ensembl.org/Homo_sapiens/contigview?chr=12&vc_start=119982631&vc_end=1200highlight=ENSG00000089041) and by the murine lbw3 region (Ensembl Genome Browser: http://http:www.ensembl.org/Mus_musculus/contigview?&chr=5&vc_start=119870152&vc_end=11987), and discuss its potential role in disease. The P2X7 receptor belongs to a family of ion channels gated by extracellular ATP, but unlike other P2X receptors it is largely restricted to haematopoietic cells. The P2X7 receptor has been proposed to play a role in a variety of immune functions including the secretion of leaderless cytokines and the shedding of the lymphocyte homing receptor CD62L [4]. As P2X7-deficient mice exhibit resistance to antibody-induced arthritis and impaired CD62L shedding and IL-1β secretion [5], stimulation of this receptor is proinflammatory – suggesting a potential role in autoimmune disease.
In this respect, SLE is of particular interest. Not only is SLE an inflammatory disorder, but it probably reflects, at least in part, an immune response to debris of cells undergoing programmed cell death (PCD). As P2X7 stimulation is proinflammatory and induces PCD, functional polymorphisms in this gene would be predicted to affect lupus susceptibility. Moreover, PCD stimulated through the P2X7 receptor belongs to a category that bears many of the hallmarks of 'classic' caspase-dependent apoptosis, but also to other categories such as cytoplasmic vacuolization more often associated with necrosis. Such cell death has sometimes been termed 'aponecrosis' [6]. Whereas removal of 'classic' apoptotic cells is believed to be immunologically silent, necrotic cell debris is proinflammatory [7]. The effect of intermediate forms of PCD such as aponecrosis, for which clearance mechanisms have not been defined, is unknown, yet such material potentially plays a significant role in the pathogenesis of SLE (discussed later). Finally, P2X7 stimulation results in rapid translocation of phosphatidylserine (PS) from the inner to the outer leaflet of the plasma membrane, which is reversible if stimulation is brief (and thus independent of cell death). As PS and associated proteins are major targets of autoantibodies in SLE [8], cells stimulated via the P2X7 receptor may be a significant source of autoantigen in this disease.
An allelic variation (P451L) of the cytoplasmic domain of the P2X7 receptor in commonly used mouse strains is associated with significant differences in its sensitivity to the ATP ligand [9]. These allelic forms with proline (P2X7-P) and leucine (P2X7-L) at position 451 confer high sensitivity and low sensitivity to stimulation by ATP, respectively. While the NZW strain has been shown to express the more responsive allele of P2X7 (P2X-P) [9], that expressed by the NZB strain is unknown. We show in the present article that NZB mice and NZW mice express different alleles of the proinflammatory receptor P2X7, and furthermore that NZW lymphocytes are markedly more responsive to P2X7 stimulation than those from NZB mice. Lymphocytes from (NZB × NZW)F1 mice exhibited intermediate sensitivities to P2X7-induced PS translocation and to PCD, but were as sensitive to induction of CD62L shedding as those from NZW mice, indicating a comparatively complex phenotypic penetration. The results indicate that P2X7 is a strong candidate for being the product of the murine lbw3 locus. As the human P2RX7 gene maps close to SLEB4, we hypothesize that similar polymorphisms may also contribute towards human disease.
Methods
Mice
Male mice were purchased from Harlan-Olac (Bicester, UK) and used at between 8 and 14 weeks. Institute guidelines for care of laboratory animals were followed. All studies received ethical review approval.
P2X7PCRs
PCR amplification of the NZB mouse genomic sequence encompassing the T1352C polymorphism [9] was performed using the forward and reverse primers CCTGTCTAGGCTGTCCCTAT and GCTTATGGAAGAGCTTGGAG for 30 cycles. PCR products were cloned using the TOPO TA cloning system (Invitrogen, Paisley, UK). Forty-three independent clones were sequenced using an ABI PRISM Big Dye terminator ready reaction kit (Applied Biosystems, Warrington, UK) and were analysed on a 3700 DNA Analyser (Applied Biosystems). Nucleotide and amino acid substitutions were numbered using the cDNA sequence accession number NM-011027.
Reagents
Matrix metalloproteinase inhibitor III was from Calbiochem (Nottingham, UK). Other reagents were from Sigma (Poole, UK), unless stated. Diluents had no effect in any assay used.
Flow cytometry
Mesenteric lymphocyte cells (107/ml) in phenol red-free DMEM were stained with a combination of CD4APC, CD4CYCHROME, CD4PE, CD8APC, CD8CYCHROME, CD8PE, CD8FITC and CD62LFITC antibodies (Becton Dickinson, Oxford, UK) as indicated, washed and resuspended in DMEM. Cells were equilibrated with annexin VFITC or annexin VCY5 (Becton Dickinson) to assess cell surface PS exposure, or with propidium iodide for 4 min to assess cell death, and were analysed by flow cytometry on a FACScalibur machine using CellQuest (Becton Dickinson) or Flowjo (Tree Star, Ashland, OR, USA) software. Baseline fluorescence was established for approximately 1 min prior to addition of 150 μM (unless otherwise stated) 2'-3'-O-(4-benzoylbenzoyl)-adenosine 5'-triphosphate (BzATP). Cells were monitored for PS exposure or CD62L shedding continuously in real time for up to 9 min or were monitored for uptake of propidium iodide, as indicated. All results are representative of at least three independent experiments.
IL-1β secretion
Spleens from NZW mice and NZB mice were disaggregated and erythrocytes were lysed (Puregene RBC lysis solution; Gentra Ltd, Minneapolis, MN, USA). Splenocytes (5 × 106/ml) were then resuspended in DMEM supplemented with 10% FCS (Helena Biosciences, Sunderland, UK), and were stimulated with 2 μg/ml lipopolysaccharide. After 6 hours at 37°C the medium was removed and replaced with DMEM. Cells were then incubated at 37°C for 30 min with BzATP added as indicated. The supernatant was then collected and the cells and particulate matter were removed by centrifugation. Supernatants were then frozen at -20°C. IL-1β was quantified by ELISA (Quantikine mouse IL-1β kit; R&D Systems, Minneapolis, MN, USA) in accordance with the manufacturer's instructions. Statistical significance was measured by Student's t test.
Results
Real-time comparison of P2X7-stimulated PS translocation and CD62L shedding by NZW, (NZB × NZW)F1and NZB lymphocytes
We initially confirmed that NZW mice are homozygous for the P2X-P allele of P2RX7 [9] associated with high sensitivity to stimulation, and we showed that NZB mice are homozygous for the low sensitivity allele P2X-L (data not shown). These forms differ at a single amino acid (451). P2X7 activation, stimulated by BzATP (Fig. 1), results in rapid externalization of PS and shedding of CD62L. We therefore developed real-time flow cytometric assays to directly compare the responses to P2X7 simulation of NZB lymphocytes, NZW lymphocytes and (NZB × NZW)F1 lymphocytes.

P2X7-stimulated phosphatidylserine (PS) exposure on lymphocytes. P2X7-dependent exposure of PS on New Zealand Black (NZB) lymphocytes, New Zealand White (NZW) lymphocytes and (NZB × NZW)F1 (NZB/W) lymphocytes. To enable the direct comparison of responses of cells from NZB mice, NZW mice and (NZB × NZW)F1 mice in a single tube, lymphocytes from these strains were stained with anti-CD4CYCHROME, anti-CD4PE and anti-CD4APC, respectively, mixed and equilibrated with annexin VFITC. Thus labelled, cells could subsequently be distinguished by flow cytometric gating. Cells were stimulated with the P2X7 agonist 2'-3'-O-(4-benzoylbenzoyl)-adenosine 5'-triphosphate (BzATP) at the time indicated by the arrow in (a). (a) Density plots of the rate of extracellular PS exposure in each cell population, as indicated by increased binding of annexin VFITC. (b) Corresponding percentage of cells bearing exposed PS in each population at a single timepoint (indicated by boxes in (a)).
PS translocation
PS is largely confined to the inner leaflet of the plasma membrane in healthy cells. Loss of lipid asymmetry, as evidenced by surface exposure of PS occurring prior to membrane breakdown, is generally assumed to be a marker of PCD. To enable the direct comparison of responses of cells from NZB mice, NZW mice and (NZB × NZW)F1 mice in a single tube, lymphocytes from these three strains were stained with anti-CD4CYCHROME, anti-CD4PE and anti-CD4APC, respectively, mixed and equilibrated with annexin VFITC (to detect exposed PS). Labelled cells could thus subsequently be distinguished by flow cytometric gating. The rates of P2X7-stimulated PS exposure by lymphocyte subsets derived from the three mouse strains were directly compared by real-time flow cytometry (Fig. 1). Baseline fluorescence was established, and cells were stimulated with the P2X7 agonist BzATP at the time indicated. The order of responsiveness to P2X7 stimulation, as evidenced by the percentage of cells translocating PS, was consistently NZW > (NZB × NZW)F1 > NZB. Therefore, consistent with the greater sensitivity of P2X7-P, lymphocytes from NZW mice show greater sensitivity to P2X7 stimulation, and show co-dominance with the NZB-derived allele (P2X7-L) in F1 hybrid mice.
CD62L shedding
Shedding of CD62L from T cells is a key event in lymphocyte migration to inflammatory sites [10] and is known to be induced by P2X7 stimulation. Lymphocytes from NZB mice, NZW mice and (NZB × NZW)F1 mice were differentially stained as already stated but were labelled with FITC-conjugated anti-CD62L in place of annexin VFITC to allow direct comparison of the rate of CD62L shedding in a single tube. P2X7-stimulated CD62L shedding was apparent as a decrease in fluorescence in the FL-1 channel. While the rates of P2X7-stimulated CD62L shedding were high and low in NZW lymphocytes and NZB lymphocytes, respectively (Fig. 2), interestingly the rate of CD62L shedding by (NZB × NZW)F1 lymphocytes was indistinguishable from that by NZW cells. The high NZW response of P2X7 thus appears dominant with respect to CD62L shedding, indicating that factors downstream of P2X7 stimulation contribute to this phenotype. That loss of CD62L reflects shedding and not decreased cell surface expression through other mechanisms is evidenced by its blockade by an inhibitor of matrix metalloproteinase [11] (Fig. 2c).

P2X7-stimulated shedding of CD62L by lymphocytes. To enable the direct comparison of responses of cells from New Zealand Black (NZB) mice, New Zealand White (NZW) mice and (NZB × NZW)F1 (NZB/W) mice in a single tube, lymphocytes from these strains were stained with anti-CD4PE, anti-CD4CYCHROME and anti-CD4APC, respectively, mixed and stained with anti-CD62LFITC. Thus labelled, cells could subsequently be distinguished by flow cytometric gating. Cells were stimulated with the P2X7 agonist 2'-3'-O-(4-benzoylbenzoyl)-adenosine 5'-triphosphate (BzATP) at the time indicated by the arrow in (a). (a) Density plots of the rate of CD62L shedding in each cell population, as indicated by decreased binding of anti-CD62LFITC. (b) Corresponding levels of cell surface CD62L in each population (NZB, red line; NZW, green line; NZB/W, black line) immediately preceding P2X7 stimulation (indicated by left-hand gates in (a)) or 7 min after P2X7 stimulation (indicated by right-hand gates in (a)). (c) Effect of a broad inhibitor of metalloproteinases on loss of CD62L. Lymphocytes from NZW mice were stained with anti-CD4CYCHROME and anti-CD62LPE, and the rate of loss of CD62L was assessed by flow cytometry. Cells were stimulated with BzATP in the presence or absence of 10 μM metalloproteinase inhibitor at the time indicated by an arrow. Shedding of CD62L is indicated by decreased binding of anti-CD62LPE. MMP, matrix metallopoteinase.
P2X7-induced secretion of IL-1β
Stimulation of the P2X7 receptor on lipopolysaccharide-stimulated monocytes and macrophages promotes secretion of the proinflammatory cytokine IL-1β [5, 12], which may therefore be expected to differ between mice bearing P2X7-L or P2X7-P receptors. Indeed IL-1β secretion by NZW splenocytes stimulated in vitro with lipopolysaccharide and BzATP exceeded that by cells from NZB mice (P < 0.05 at 50, 100 and 150 μM BzATP; Fig. 3). Elevated IL-1β secretion by NZW splenocytes was apparent even in the absence of BzATP (although slightly below statistical significance), suggesting that inadvertent stimulation of the high (but not low) sensitivity P2X7 receptor may have occurred through cell death and the consequent release of ATP during cell preparation (spleen disaggregation and erythrocyte lysis). In one NZW splenocyte preparation exhibiting particularly high IL-1β secretion, cells were refractory to further stimulation of the P2X7 receptor in vitro.

P2X7-stimulated secretion of IL-1β. Splenocytes were primed in vitro with lipopolysaccharide and were then stimulated with 2'-3'-O-(4-benzoylbenzoyl)-adenosine 5'-triphosphate (BzATP) as indicated. The graph shows IL-1β secretion by cells from New Zealand Black mice (open squares) and New Zealand White mice (filled squares). Each line represents IL-1β secretion by cells from a single mouse.
P2X7-induced PCD of NZW and NZB lymphocytes
Several lines of evidence indicate that lupus reflects an autoimmune response to debris from cells undergoing PCD. Although stimulation of P2X7 results in rapid PS translocation, the effects are reversible if exposure to the agonist is brief [4]. Only prolonged treatment with agonist results in PCD. Translocation of PS following P2X7 activation cannot therefore be used as a direct measure of irreversible commitment to PCD. To measure PCD following prolonged P2X7 stimulation, we therefore compared the rate of terminal membrane breakdown (indicated by propidium iodide uptake) following BzATP treatment of NZW lymphocytes, NZB lymphocytes and (NZB × NZW)F1 lymphocytes (Fig. 4). P2X7 stimulation resulted in significant PCD in all populations tested, with the order of sensitivity NZW > (NZB × NZW)F1 > NZB, consistent with the high responder status of the NZW cells and the dominance of the NZW-derived P2RX7 allele in this response.

P2X7-stimulated lymphocyte programmed cell death (PCD). Lymphocytes from New Zealand Black (NZB) mice, New Zealand White (NZW) mice and (NZB × NZW)F1 (NZB/W) mice were stained with anti-CD4APC and anti-CD8FITC, and were equilibrated with propidium iodide (PI). Panels show the mean percentage (± standard deviation, n = 5) of dead cells (those taking up PI) as assessed by flow cytometry before (t = 0), and at 15-min intervals subsequent to, stimulation of the P2X7 receptor with 2'-3'-O-(4-benzoylbenzoyl)-adenosine 5'-triphosphate. (a) CD4+ cells, and (b) CD8+ cells. NZB, open squares; NZW, open circles; NZB/W, open diamonds.
Discussion
Stimulation of the proinflammatory haematopoietic P2X7 receptor [5] results in IL-1β secretion, in high rates of PCD [4] and in CD62L shedding [13], each of which is associated with human SLE [14–16]. The P2X7 receptor therefore has the characteristics of a candidate lupus susceptibility gene product. Moreover, the gene encoding human P2X7 is located within a region (12q24; Ensembl Genome Browser: http://www.ensembl.org/Homo_sapiens/contigview?chr=12&vc_start=119982631&vc_end=1200highlight=ENSG00000089041) recently identified and confirmed in Hispanic and European-American Families as a lupus susceptibility locus, designated SLEB4 [3].
A polymorphism in the cytoplasmic domain of the P2X7 receptor of common mouse strains is associated with differential responsiveness [9]. While most strains, including NZW mice [9], possess proline in amino acid position 451, we showed that NZB mice express P2X7 with lysine at this position and that the variant confers markedly decreased sensitivity to P2X7 stimulation. Notably, the murine P2RX7 gene is encoded by a gene on chromosome 5 within a region designated lbw3 due to the identification of a NZW-derived susceptibility locus conferring increased mortality at 12 months [2]. While susceptibility regions are broad, the microsatellite marker D5Mit101 (defining lbw3 [2]) was in the original study mapped to 88 cM on chromosome 5, which may have discouraged identification of P2RX7 as a candidate susceptibility gene. Current mapping data, however, show this marker located at 81 cM – Mouse Genome Informatics http://www.informatics.jax.org/javawi2/servlet/WIFetch?page=markerDetail&key=6077a and Ensembl Genome Browser http://www.ensembl.org/Mus_musculus/markerview?marker=D5Mit101. As the marker D5Mit118 that is adjacent to P2RX7 is located at 67 cM – Ensembl Genome Browser http://www.ensembl.org/Mus_musculus/markerview?marker=D5Mit101 and Mouse Genome Informatics http://www.informatics.jax.org/javawi2/servlet/WIFetch?page=markil&key=6095 – the two are approximately 14 cM (or 19 Mb) apart (120 Mb versus 139 Mb), easily within the 20 cM distance used by Kono and colleagues [2] to define coverage by markers in their study.
Although gene polymorphisms may have unpredicted effects, other than P2RX7 there appear to be few candidate susceptibility genes (based on lymphoid expression and protein activity) in the region described by lbw3. However, other candidates might include those encoding: lnk, an adaptor protein in T-cell signalling (65.0 cM) [17]; P2X4, a purinergic receptor (65.0 cM) whose activity is assumed primarily to be neuronal, but which is also expressed (at least at the level of mRNA) in lymphocytes [18]; shp2, a tyrosine phosphatase [19] (~66 cM [118.6 Mb]); and FLT3 (CD135, 82.0 cM), a tyrosine kinase expressed in haemaotopoietic cells [20]. None of these has been reported to be polymorphic between NZB mice and NZW mice.
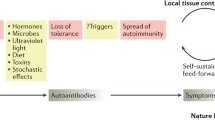
It is widely thought that lupus reflects an autoimmune response to cells undergoing PCD. Aberrant responses to such debris may reflect qualitative or quantitative abnormalities; for example, if its handling is defective and/or following exposure to increased levels of 'apoptotic' material. Both have been reported to contribute to human SLE [15, 16]. That prolonged stimulation of the P2X7 receptor induces PCD is therefore of particular note. However, multiple pathways of PCD exist. While 'apoptosis' and 'PCD' are frequently used as synonyms, 'apoptosis' is often used to imply caspase-dependent cell death. Nevertheless, caspase involvement is not a good indicator of the physiologic importance, or 'programming', of a cell death pathway, and consequently classic 'apoptosis' may describe one end of a continuum of active PCD mechanisms [21]. Hence, in principle, a defect in one of many PCD pathways, rather than increased susceptibility to PCD per se, may be sufficient to increased the burden of cellular debris and hence the susceptibility to lupus. Indeed, that SLE may reflect an autoimmune response to debris from 'apoptotic' cells, despite clearance of such material being thought generally immunologically silent [7], has been a conundrum.
To reconcile these findings it has been suggested that, in SLE, mechanisms for removing apoptotic debris are overloaded, with remaining cells undergoing secondary necrosis, and/or that apoptotic cells have some immunostimulatory properties [7]. We suggest the additional possibility that different forms of PCD may give rise to debris with different degrees of immunogenicity. It is therefore necessary to dissect distinct PCD pathways to assess the potential effects that defects have on the disease process. It is attractive to speculate that P2X7-induced aponecrotic debris, perhaps due to the catastrophic nature of its generation or the apparent differences in cell dismantling, may be more necrotic than apoptotic in character and thus be immunostimulatory. Such material may either promote responses to surrounding 'apoptotic' cells and/or directly stimulate autoimmune responses to itself (if lupus autoantigens are appropriately packaged in P2X7-induced PCD). P2X7 receptor-induced PCD is therefore potentially a source of lupus autoantigens or may represent a catastrophic form of cell death that overwhelms the host's ability to clear such material.
We therefore suggest the following involvement of the P2X7 receptor in SLE. ATP exists at very high concentrations in normal cells (5–10 mM), and is released upon cell death before its rapid breakdown by ATPases. Consequently, extracellular concentrations of ATP, although normally low, are transiently increased at sites of tissue damage. Stimulation of P2X7 occurs at sufficient concentrations of ATP, resulting in secretion of IL-1β and in CD62L shedding within minutes. P2X7 stimulation thus acts to promote the inflammatory response. The resulting lymphoid infiltration leads to additional lymphocyte-mediated cell death, and to consequent ATP release, exacerbating the P2X7-driven inflammatory cycle. Indeed, given sufficient tissue damage, prolonged stimulation of P2X7 itself induces PCD, further adding to the cycle of ATP release and destruction. Release of autoantigens within P2X7-stimulated aponecrotic debris may also contribute to a breakdown in self-tolerance and initiation of autoimmunity.
While one must make the proviso that little is known at the moment about the level of ATP released at sites of tissue damage, its rate of decay and how these may vary between pathological conditions including SLE, we suggest it is reasonable to hypothesize that polymorphisms within P2X7 can influence the pathogenesis of lupus. Importantly, there are a number of polymorphisms within P2X7 that affect its activity. The Ile-568 to Asn [22], Arg307 to Gln [23], and Glu496 to Ala [24] polymorphisms therefore all result in reduced function of human P2X7, and might each be hypothesized to result in decreased severity of SLE.
Conclusions
In summary, we have shown that polymorphism of the P2X7 receptor between NZW and NZB strains is associated with marked differences in P2X7-stimulated proinflammatory responses, consistent with high responsiveness and low responsiveness previously reported for the two alleles. We also show that current genetic mapping indicates that the P2RX7 gene is located within the region defined as lbw3 and is a therefore a strong candidate for being the product of this lupus susceptibility locus. Furthermore, as the human gene maps very close to SLEB4, we hypothesize that polymorphisms within P2RX7 may also contribute to human disease. Stimulation of the P2X7 receptor is proinflammatory and induces a form of cell death known as aponecrosis, which exhibits several characteristics of apoptosis. We therefore suggest that the P2X7 receptor and gene have the functional and positional characteristics suggestive of a role in the pathogenesis in SLE, and that the potential of the cell death mechanism aponecrosis to contribute to disease warrants study.
Abbreviations
- BzATP:
-
2'-3'-O-(4-benzoylbenzoyl)-adenosine 5'-triphosphate
- DMEM:
-
Dulbecco's modified Eagle's medium
- ELISA:
-
enzyme-linked immunosorbent assay
- FCS:
-
foetal calf serum
- FITC:
-
fluorescein isothiocyanate
- IL:
-
interleukin
- NZB:
-
New Zealand Black
- NZW:
-
New Zealand White
- PCD:
-
programmed cell death
- PCR:
-
polymerase chain reaction
- PS:
-
phosphatidylserine
- SLE:
-
systemic lupus erythematosus.
References
Tsao BP: Update on human systemic lupus erythematosus genetics. Curr Opin Rheumatol. 2004, 16: 513-521. 10.1097/01.bor.0000132648.62680.81.
Kono DH, Burlingame RW, Owens DG, Kuramochi A, Balderas RS, Balomenos D, Theofilopoulos AN: Lupus susceptibility loci in New Zealand mice. Proc Natl Acad Sci. 1994, 91: 10168-10172.
Nath SK, Quintero-Del-Rio AI, Kilpatrick J, Feo L, Ballesteros M, Harley JB: Linkage at 12q24 with systemic lupus erythematosus (SLE) is established and confirmed in Hispanic and European American families. Am J Hum Genet. 2004, 74: 73-82. 10.1086/380913.
MacKenzie A, Wilson HL, Kiss-Toth E, Dower SK, North RA, Surprenant A: Rapid secretion of interleukin-1beta by microvesicle shedding. Immunity. 2001, 15: 825-835. 10.1016/S1074-7613(01)00229-1.
Labasi JM, Petrushova N, Donovan C, McCurdy S, Lira P, Payette MM, Brissette W, Wicks JR, Audoly L, Gabel CA: Absence of the P2X7 receptor alters leukocyte function and attenuates an inflammatory response. J Immunol. 2002, 168: 6436-6445.
Formigli L, Papucci L, Tani A, Schiavone N, Tempestini A, Orlandini GE, Capaccioli S, Orlandini SZ: Aponecrosis: morphological and biochemical exploration of a syncretic process of cell death sharing apoptosis and necrosis. J Cell Physiol. 2000, 182: 41-49. 10.1002/(SICI)1097-4652(200001)182:1<41::AID-JCP5>3.0.CO;2-7.
Savill J, Dransfield I, Gregory C, Haslett C: A blast from the past: clearance of apoptotic cells regulates immune responses. Nat Rev Immunol. 2002, 2: 965-975. 10.1038/nri957.
McClain MT, Arbuckle MR, Heinlen LD, Dennis GJ, Roebuck J, Rubertone MV, Harley JB, James JA: The prevalence, onset, and clinical significance of antiphospholipid antibodies prior to diagnosis of systemic lupus erythematosus. Arthritis Rheum. 2004, 50: 1226-1232. 10.1002/art.20120.
Adriouch S, Dox C, Welge V, Seman M, Koch-Nolte F, Haag F: Cutting edge: a natural P451L mutation in the cytoplasmic domain impairs the function of the mouse P2X7 receptor. J Immunol. 2002, 169: 4108-4112.
Gallatin WM, Weissman IL, Butcher EC: A cell-surface molecule involved in organ-specific homing of lymphocytes. Nature. 1983, 304: 30-34. 10.1038/304030a0.
Preece G, Murphy G, Ager A: Metalloproteinase-mediated regulation of L-selectin levels on leucocytes. J Biol Chem. 1996, 271: 11634-11640. 10.1074/jbc.271.20.11634.
Grahames CB, Michel AD, Chessell IP, Humphrey PP: Pharmacological characterization of ATP- and LPS-induced IL-1beta release in human monocytes. Br J Pharmacol. 1999, 127: 1915-1921. 10.1038/sj.bjp.0702732.
Gu B, Bendall LJ, Wiley JS: Adenosine triphosphate-induced shedding of CD23 and L-selectin (CD62L) from lymphocytes is mediated by the same receptor but different metalloproteases. Blood. 1998, 92: 946-951.
Sfikakis PP, Charalambopoulos D, Vaiopoulos G, Mavrikakis M: Circulating P- and L-selectin and T-lymphocyte activation and patients with autoimmune rheumatic diseases. Clin Rheumatol. 1999, 18: 28-32. 10.1007/s100670050047.
Ren Y, Tang J, Mok MY, Chan AW, Wu A, Lau CS: Increased apoptotic neutrophils and macrophages and impaired macrophage phagocytic clearance of apoptotic neutrophils in systemic lupus erythematosus. Arthritis Rheum. 2003, 48: 2888-2897. 10.1002/art.11237.
Emlen W, Niebur J, Kadera R: Accelerated in vitro apoptosis of lymphocytes from patients with systemic lupus erythematosus. J Immunol. 1994, 152: 3685-3692.
Brown SB, Clarke MC, Magowan L, Sanderson H, Savill J: Constitutive death of platelets leading to scavenger receptor-mediated phagocytosis. A caspase-independent cell clearance program. J Biol Chem. 2000, 275: 5987-5996. 10.1074/jbc.275.8.5987.
Wang L, Jacobsen SE, Bengtsson A, Erlinge D: P2 receptor mRNA expression profiles in human lymphocytes, monocytes and CD34+stem and progenitor cells. BMC Immunol. 2004, 5: 16-10.1186/1471-2172-5-16.
Gjorloff-Wingren A, Saxena M, Han S, Wang X, Alonso A, Renedo M, Oh P, Williams S, Schnitzer J, Mustelin T: Subcellular localization of intracellular protein tyrosine phosphatases in T cells. Eur J Immunol. 2000, 30: 2412-2421. 10.1002/1521-4141(2000)30:8<2412::AID-IMMU2412>3.0.CO;2-J.
Masten BJ, Olson GK, Kusewitt DF, Lipscomb MF: Flt3 ligand preferentially increases the number of functionally active myeloid dendritic cells in the lungs of mice. J Immunol. 2004, 172: 4077-4083.
Jaattela M, Tschopp J: Caspase-independent cell death in T lymphocytes. Nat Immunol. 2003, 4: 416-423. 10.1038/ni0503-416.
Wiley JS, Dao-Ung LP, Li C, Shemon AN, Gu BJ, Smart ML, Fuller SJ, Barden JA, Petrou S, Sluyter R: An Ile-568 to Asn polymorphism prevents normal trafficking and function of the human P2X7 receptor. J Biol Chem. 2003, 278: 17108-17113. 10.1074/jbc.M212759200.
Gu BJ, Sluyter R, Skarratt KK, Shemon AN, Dao-Ung LP, Fuller SJ, Barden JA, Clarke AL, Petrou S, Wiley JS: An Arg307 to Gln polymorphism within the ATP-binding site causes loss of function of the human P2X7 receptor. J Biol Chem. 2004, 279: 31287-31295. 10.1074/jbc.M313902200.
Sluyter R, Dalitz JG, Wiley JS: P2X7 receptor polymorphism impairs extracellular adenosine 5' -triphosphate-induced interleukin-18 release from human monocytes. Genes Immun. 2004, 5: 588-591. 10.1038/sj.gene.6364127.
Acknowledgements
This work was supported by the Medical Research Council of Great Britain. The authors would like to thank Dr T Vyse for helpful discussions.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author(s) declare there are no competing interests.
Authors' contributions
JIE conceived the study, carried out the flow cytometric and IL-1β secretion experiments, and wrote the first draft of the manuscript. JHM designed allele-specific primers and typed the P2RX7 genes of NZB mice and NZW mice, and contributed to drafting of the manuscript. CFH contributed to the design of experiments and drafting of the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Elliott, J.I., McVey, J.H. & Higgins, C.F. The P2X7receptor is a candidate product of murine and human lupus susceptibility loci: a hypothesis and comparison of murine allelic products. Arthritis Res Ther 7, R468 (2005). https://doi.org/10.1186/ar1699
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/ar1699