
Abstract
Background
A simple and rapid stability-indicating, reversed phase ultra-performance liquid chromatography (UPLC) method was developed for the quantitative determination of lafutidine and its four potential impurities.
Results
Separation was achieved on Acquity BEH-shield RP18 UPLC column (3.0 mm × 100 mm, 1.7 μm) under the gradient mode of elution by using mobile phase A (0.02M diammonium hydrogen phosphate/acetonitrile, 80:20 v/v) and mobile phase B (0.02M diammonium hydrogen phosphate/acetonitrile, 30:70 v/v). The flow rate was maintained at 0.5 mL min−1. UV detection was carried out at 276 nm.
Conclusions
Stability-indicating capability of the developed method is established by analyzing forced degradation samples in which the spectral purity of lafutidine is ascertained along with the separation of degradation products from analyte peak. The developed UPLC method is validated as per International Conference on Harmonization guidelines with respect to system suitability, specificity, precision, sensitivity, accuracy, linearity, and robustness.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Lafutidine belongs to the class of H2-receptor antagonists and is used in the treatment of peptic ulcer and gastro-esophageal reflux disease or GERD [1]. It is available in the market under the brand name Lafaxid® in the form of tablets for oral administration containing 5 or 10 mg of lafutidine with daily dose of 5 to 10 mg twice daily [2]. Being a histamine H(2)-receptor antagonist, it inhibits daytime (i.e., postprandial) as well as night time gastric acid secretion. It also has gastroprotective activity that particularly affects the mucosal blood flow. It elevates the postprandial intragastric pH and increases plasma calcitonin gene-related peptide and somatostatin concentrations in humans. From the clinical studies, it is proven that lafutidine is equal or superior to conventional H2 antagonists in antiulcer potency and it is useful for the prevention of ulcer relapse and or treatment of NSAID-induced gastroduodenal damage [3].
The analytical method for determination of lafutidine and its impurities in bulk and pharmaceutical dosage form is highly essential to monitor and control the impurities in the drug substances. Although many analytical techniques like gas chromatography (GC), capillary electrophoresis (CE), and liquid chromatography (LC) can be employed for the analysis, the most widely used technique is the reversed-phase liquid chromatography employing a non-polar stationary phase [4, 5]. An extensive literature survey revealed that there are several bioanalytical high performance liquid chromatography (HPLC) methods reported in the literature for the analysis of lafutidine in human plasma using HPLC-ESI-MS [6, 7], in tablet dosage form by reversed-phase high performance liquid chromatography [8] and in human serum by HPLC with fluorescence detection [9]. To the best of our knowledge, none of the currently available analytical methods can separate all the known related compounds and degradation impurities of lafutidine drug substance. There is no pharmacopoieal reference available for analysis of bulk API or solid dosage form of lafutidine. Furthermore, there is no cost- and time-effective method reported in the literature that can adequately separate lafutidine from its impurities and degradents in bulk and pharmaceutical dosage form. Moreover, the analysis using LCMS, GC/MSD, and LC-MS is very expensive and delicate as compared to ultra-performance liquid chromatography (UPLC) for routine quality control analysis. Hence, we focused on developing a selective, fast, cost-effective, and stability-indicating method using this advanced technique (UPLC) for the determination of lafutidine and its impurities in bulk and pharmaceutical dosage form. Hence, a reproducible stability-indicating UPLC method was developed which is time- and cost-effective as compared to the reported methods. The method was successfully validated according to the USP<1225> Validation of Compendial Procedures and International Conference on Harmonization (ICH) guidelines Q2 (R1) for validation of analytical procedures: text and methodology [10, 11].
Methods
Materials and reagents
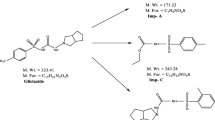
The samples of lafutidine and its four impurities viz., Imp A, Imp B, Imp C, and Imp D as shown in Figure 1 were received from synthetic laboratory, Emcure Pharmaceutical Ltd, Pune, India. Diammonium hydrogen orthophosphate of HPLC grade and liquid ammonia of AR grade were purchased from Merck, Mumbai, India. Acetonitrile of gradient grade was purchased from J.T. Baker, Mumbai, India. De-ionized water of high purity was obtained from Millipore, Milli-Q (Bedford, MA, USA) purification system.

Process-related impurities and degradation-related impurities of lafutidine. (a) Blank preparation. (b) Standard preparation.
Instrumentation and operating conditions
Waters’ Acquity UPLC system (Waters, Milford, MA, USA) equipped with quaternary gradient pump, auto sampler, column oven, and photodiode array detector was used for method development and method validation. The output signal was monitored and processed using Empower software version 3. The UPLC columns used during method development were Acquity UPLC BEH-C18 (2.1 mm × 50 mm, 1.7 μm), Acquity CSH phenyl-hexyl (2.1 mm × 100 mm, 1.7 μm), and Acquity UPLC BEH-shield RP18 (3.0 mm × 100 mm, 1.7 μm).
Acquity UPLC BEH-shield RP18 (3.0 mm × 100 mm, 1.7 μm) column thermostated at 35°C was used for the separation. Mobile phase A was prepared by mixing the phosphate buffer (aqueous solution of 0.02 M diammonium hydrogen phosphate, adjusted the pH to 9 with liquid ammonia) and acetonitrile in the ratio of 80:20 v/v, whereas mobile phase B was prepared by mixing the phosphate buffer and acetonitrile in the ratio of 30:70 v/v. The flow rate and injection volume was 0.5 mL min−1 and 4 μL, respectively. The analysis was carried out under the gradient conditions as time (min)/A (v/v)/B (v/v); T0.01/100:0, T2.0/75:25, T3.0/60:40, T10/30:70, T12/30:70, T12.5/100:0, and T16/100:0. The data were acquired at 276 nm for 16 min and processed by using Empower software version 3.0. For the analysis of forced degradation study, the photo diode array detector was set to scan from 200 to 400 nm. The peak homogeneity was calculated in terms of peak purity.
Preparation of solutions
The diluent used for the analysis was prepared by preparing homogenous mixture of 700 ml of water and 300 ml acetonitrile. A solution of lafutidine was prepared at concentration of 1,000 μg mL−1 in diluent. Stock solutions of each impurity (Imp-A, Imp-B, Imp-C, and Imp-D) were prepared at concentration about 150 μg mL−1 in diluent. These solutions were further diluted to prepare the standard solution for quantification of impurities according to the specification limits (0.15%) of each impurity.
The sample of the formulation drug was prepared by powdering 20 tablets of lafutidine. An accurately weighed quantity of tablet powder was taken to prepare the solution equivalent to 1,000 μg mL−1 of lafutidine in diluent. After adding about 50% of the total volume of diluent and ultrasonicating for about 15 to 20 min, the solution was made up to the mark with the diluent and filtered through Merck nylon syringe filter (Merck KGaA, Darmstadt, Germany) having pore size 0.45 μm. The clear liquid was collected and used for the determination of related substances in the pharmaceutical dosage forms. This solution was diluted further volumetrically to 100 μg mL−1 with diluent for assay determination in pharmaceutical dosage forms.
Analytical procedure
Lafutidine with concentration of 1,000 μg mL−1 was spiked with Imp-A, Imp-B, Imp-C, and Imp-D at specification level, i.e., of 0.15% and this solution was used as a SSM. Using 4 μL each of blank preparation as SSM, six replicates of standard solution and test solution are injected and separately chromatographed under the given chromatographic conditions. Resolution condition of not less than 2.0 between lafutidine and related compound D was set as system suitability parameter for SSM. The relative standard deviation of not more than 5.0% for areas obtained from six replicate injections of all impurities was used to verify the system precision.
Procedure for forced degradation study
The forced degradation study was planned to investigate specificity and stability-indicating property of the established method. The API was deliberately stressed in a solution state at acidic and alkaline pH and under oxidation conditions, in solid state at thermal and photolytic conditions. Solutions for degradation were prepared by dissolving the API in diluent at specification level, i.e., 1,000 μg mL−1 in diluent and then treating with 0.1 N hydrochloric acid (1 h heating at 90°C), with 0.1 N sodium hydroxide (1 h heating at 90°C), and with 30% aqueous hydrogen peroxide. The solutions after treatment were diluted up to the mark of desired concentration of 1,000 μg mL−1 and were injected in the established method. For thermal degradation, the solid API was heated at 60°C for 24 h in calibrated oven. For photolytic degradation, the sample was exposed to UV source as per ICH guidelines for 7 days. The treated samples were diluted with diluent to the desired concentration of 1,000 μg mL−1 and injected in the established method. All the stressed samples are quantified for lafutidine and its impurities. A photodiode array detector was used to establish peak purity and thus the homogeneity of lafutidine peaks under every stress condition. Mass balance of the degraded samples was carried out to correlate the loss of a parent drug and the increase in the amount of degradation products.
Results and discussion
Method development
Development and optimization of UPLC chromatographic conditions
Initially, the method was developed on HPLC for determination of lafutidine and its related impurities in bulk and pharmaceutical dosage forms. Due to the longer run time of about 60 min, the HPLC method was not validated but further migrated to UPLC wherein the run time was reduced about four times from 60 to 16 min.
The core objective of developing the UPLC method was to detect and determine the potential impurities present in lafutidine bulk and pharmaceutical dosage form and to achieve the separation of all four impurities (Imp-A, Imp-B, Imp-C, and Imp-D) along with the degradation products generated during stress studies.
UPLC column selection
Various UPLC stationary phases like Acquity UPLC BEH-C18 (2.1 mm × 50 mm, 1.7 μm), Acquity CSH phenyl-hexyl (2.1 mm × 100 mm, 1.7 μm), and Acquity UPLC BEH-shield RP18 (3.0 mm × 100 mm, 1.7 μm) were used during development. Based on parameter-like resolution, tailing factor, and selectivity especially for impurity A which is a phenolic impurity, Acquity UPLC BEH-shield RP18 (3.0 mm × 100 mm, 1.7 μm) was finalized for analysis.
Buffer selection
Various buffers like phosphate, sulfate, and acetate with pH ranging from 3.0 to 10.0 were tried for development. It was observed that at basic pH of about 8.0 which is equivalent to the pKa of the phosphate buffer, the separation and the tailing factors are satisfactory. Therefore, diammonium hydrogen phosphate buffer was selected for analysis.
Organic modifier selection
Organic modifiers like acetonitrile and methanol were evaluated as mobile phase B. Satisfactory chromatographic separation was achieved when acetonitrile was used as mobile phase B in terms of better resolution and tailing factor. When methanol was tried as an organic modifier, the tailing factor for all the peaks was not within the system suitability criteria. Therefore, acetonitrile was finalized as organic modifier for analysis.
Optimization of chromatographic conditions
Premixed buffer and organic modifier combinations were prepared for mobile phase A and mobile phase B to improve the baseline interference. Various combinations of gradient time programs were tried and then finalized wherein all impurities and degradents are well separated from the main peak due to lafutidine. The wavelength for analysis was selected as 276 nm which is UV maxima of lafutidine. The temperature selection of 35°C was optimized so as to attain maximum separation between the critical pair which is Imp-D and lafutidine. The rationale for selecting the flow rate of 0.5 ml/min for 3-mm ID column was that the increase in flow rate reduced the resolution between Imp-D and lafutidine to less than 2.0 which was not acceptable. The effect of sub-2μm particle size was not considered while calculating the total flow rate because from Van Deemter curve, at around 1.7 μm particle size, one can attain the lowest height equivalent of theoretical plates (or highest resolution) that is constant over a wide range of flow rates.
The typical retention times of Imp-A, Imp-B, Imp-C, Imp-D, and lafutidine were about 1.37, 1.75, 3.64, 4.79, and 5.04 min, respectively (Figures 2 and 3), and the developed UPLC method was found to be specific for determination of lafutidine and its impurities in bulk and pharmaceutical dosage form.

Typical UPLC chromatograms of blank preparation and standard preparation. At specification level (Imp-A, Imp-B, Imp-C, and Imp-D at 0.15% and lafutidine at 0.10% of the test concentration). (a) Unspiked test sample. (b) Spiked test preparation.

Typical UPLC chromatograms of lafutidine unspiked test preparation and lafutidine spiked test preparation with known impurities. Imp-A, Imp-B, Imp-C, and Imp-D. (a) Acid-treated test sample. (b) Alkali-treated test sample. (c) Peroxide-treated test sample.
Identification of impurities
Impurities A, B, C, and D are spiked in lafutidine at desired concentrations of 0.15% with respect to test concentration of 1,000 μg mL−1 to confirm the retention times under the chosen chromatographic conditions. All the impurities were well resolved from each other. Among the identified impurities, Imp-A and Imp-B are the process as well as degradation impurities; Imp-C is the key starting material in the process as well as degradation impurity; Imp-D is a process related impurity (Figure 1).
System suitability criteria
A system suitability test was defined based on the results obtained in several representative chromatograms. The column efficiency determined from the analyte peak >20,000, the tailing factor <2.0, and relative standard deviation (RSD) for impurity areas in six replicate injection of system suitability solution was <5.0%. A resolution between lafutidine and Imp-D was >2.0 in SST solution. All the system suitability criteria during the validation of the study and the batch analysis study were within the acceptance limit. The results of system suitability are depicted in Table 1.
Experimental
Specificity (selectivity)
The data on forced degradation studies revealed that the degradation products were well separated from lafutidine and the peak purity data (purity angle less than purity threshold) of lafutidine proved the homogeneity of the peak. Significant degradation was observed in acid, alkali, and peroxide solution as shown in Figure 4. The mass balance is a process of adding together the assay value and the levels of degradation products to see how closely these add up to 100% of the initial value with due consideration of the margin of analytical error [12, 13]. The mass balance of stressed samples was close to 100%. Forced degradation study results are given in Table 2.

Typical UPLC chromatograms of forced degradation study. (a) Acid-treated test sample, (b) base-treated test sample, and (c) peroxide-treated test sample.
Linearity
Linearity test solutions for related substance were prepared by diluting the impurity stock solution (as described in the section of ‘Analytical procedure’) to the required concentrations. The solutions were prepared at six concentration levels from limit of quantitations (LOQ) to 250% (LOQ, 0.75, 1.2, 1.5, 2.25, and 3.75 μg mL−1) with respect to the specification level of impurities. The data were subjected to statistical analysis using a linear regression model; the regression equations and correlation coefficients are given in Table 3.
Limits of detection and quantitation
The limit of detection and limit of quantitation (LOD and LOQ) were determined for lafutidine and for each of the related substances as per ICH Q2R1 guideline from the standard deviation of the peak areas and slope of linearity data. The values of LOD and LOQ for impurities A, B, C, and D were found to be in the range of 0.007% to 0.022% and 0.008% to 0.023%, respectively. The calculated LOQ concentrations of all the components were verified for precision by injecting six individual preparations of Imp-A, Imp-B, Imp-C, Imp-D, and lafutidine. The RSD of LOQ precision was in the range of 1.22% to 9.78%. These limits of quantification levels of the impurities were helpful for the research work in controlling the impurities at the accepted level during the optimization of the process. The results were depicted in Table 3.
Precision
System precision
System precision for related substances determination was verified by system suitability mixture (SSM), which was analyzed for six times and RSD of Lafutidine and all impurities peak areas were evaluated and found to be between 0.36% to 1.30%.
Method precision and Intermediate precision
Precision of the method was studied for method precision and intermediate precision. Related substances method precision was demonstrated by analyzing six separate Lafutidine sample solutions that were prepared by spiking the related substances viz., Imp-A, Imp-B, Imp-C and Imp-D at specification level. The RSD (n = 6) for each related substance was evaluated. In the intermediate precision study, the similar procedure of method precision was carried out by a different analyst, using different mobile phase and diluent preparations and instrument on a different day with different lot of same brand column. The percentage relative standard deviation of the results for related substances method was evaluated and found to be 0.4-1.5%. The results were reported in Table 3.
Accuracy (recovery)
Accuracy of the method for all the related substances was determined by analyzing lafutidine sample solutions spiked with all the related substances at seven different concentration levels of LOQ, 20 40,50,80,100 and 150% of each in triplicate at the specified limit. The recovery of all these related substances were found to be in-between the predefined acceptance criterion of 80.0–120.0% and the data is given in Table 3.
Stability of analytical solution
To determine the stability of sample solution, the sample solutions of lafutidine spiked with related substances at specified level were prepared and analyzed immediately after preparation and after different time intervals up to 48 hrs, while maintaining the sample cooler temperature at about 25°C The results from these studies indicated that the sample solutions were stable at room temperature upto 48 hrs.
Robustness
To evaluate the robustness of the developed method, the chromatographic conditions were deliberately altered and the resolution between closely eluting peak pair i.e. Imp-D and Lafutidine was evaluated. The flow rate of the mobile phase was 0.5 mL min-1. To study the effect of flow rate on the resolution, the same was altered by 0.1 units i.e. from 0.4 to 0.6 mL min-1. The effect of column temperature on resolution was studied at 32°C and 38°C instead of 35°. All the other mobile phase components were held constant as described above. In all the deliberate varied chromatographic conditions (flow rate and column temperature), the tailing factor of Lafutidine was less than 1.30 and the resolution between any two peaks was greater than 2.0. There was a very minor variation in the resolution and tailing factor results observed in all the robustness conditions illustrating the robustness of the method.
Application of the method
The analysis of commercial formulation sample and bulk drug sample indicated that the method is specific and selective for determination of related substances in the formulation and bulk drug samples (Table 4). The developed method is capable for quantitative analysis of lafutidine in the bulk drug and in a pharmaceutical dosage form.
Conclusions
The method presented in this communication describes the development of a rapid, simple, accurate, and selective gradient UPLC method that separates all related substances of lafutidine with good resolution. The developed method was validated to ensure the compliance in accordance with ICH guidelines. The method was found to be simple, selective, precise, accurate, and robust. Therefore, this method can be used for routine testing as well as stability analysis of lafutidine drug substance and drug products. All statistical results (percentage, mean, RSD, percentage difference, and recovery percentage) were within the acceptance criteria. The method could be of use for routine evaluation of the quality of lafutidine in bulk drug manufacturing unit.
Abbreviations
- ICH:
-
International Conference on Harmonization
- LC-MS:
-
Liquid chromatography-mass spectrometry
- UPLC:
-
Ultra performance liquid chromatography
- UV:
-
Ultraviolet-visible.
References
Nakano M, Kitano S, Nanri M, Kiniwa M: Lafutidine, a unique histamine H2-receptor antagonist, inhibits distention-induced gastric acid secretion through an H2 receptor-independent mechanism. Eur J Pharmacol 2011,658(2–3):236–241. 10.1016/j.ejphar.2011.02.007
Dewan B, Philipose N: Lafutidine 10 mg versus rabeprazole 20 mg in the treatment of patients with heartburn-dominant uninvestigated dyspepsia: a randomized, multicentric trial. Gastroenterology Res Pract 2011. Article ID 640685):8 doi:10.1155/2011/640685
Shimatani T, Inoue M, Kuroiwa T, Xu J, Nakamura M, Tazuma S, Ikawa K, Morikawa N: Lafutidine, a newly developed antiulcer drug, elevates postprandial intragastric pH and increases plasma calcitonin gene-related peptide and somatostatin concentrations in humans: comparisons with famotidine. Dig Dis Sci 2006,51(1):114–120. 10.1007/s10620-006-3094-2
Snyder LR, Kirkland JJ, Dolan JW: Introduction to modern liquid chromatography. Hoboken: Wiley; 1997.
Snyder LR, Kirkland JJ, Glajch JL: Practical HPLC method development. 2nd edition. Hoboken: Wiley; 1997.
Wu L, Zhang Z, Tian Y, Li W, Xu F, Chen Y, Wei H: Determination of lafutidine in human plasma by high-performance liquid chromatography-electrospray ionization mass spectrometry: application to a bioequivalence study. J Mass Spectrom 2005,40(12):1637–1643. 10.1002/jms.942
Chena W, Lianga Y, Lia H, Xionga Y, Liua X, Wanga G, Xi L: Simple, sensitive and rapid LC-ESI-MS method for the quantitation of lafutidine in human plasma-application to pharmacokinetic studies. J Pharm Biomed Anal 2006,41(1):256–260. 10.1016/j.jpba.2005.10.008
Sumithra M, Sundaram PS, Srinivasulu K: Analytical method development and validation of lafutidine in tablet dosage form by RP-HPLC. Int J Chem Tech Res 2011,3(3):1403–1407.
Xiang-lin Z, Yun-di Q, Kai-peng L, Qingming D, Xiao L: Determination of lafutidine in human serum by HPLC-FD method and study of pharmacokinetics. Chin J Hospital Pharm CNKI:SUN:ZGYZ.0.2005–12–01712 CNKI:SUN:ZGYZ.0.2005-12-01712
The United States Pharmacopeia: Validation of compendial methods. 32nd edition. 2009. USP 32 Section <1225> http://www.pharmacopeia.cn/v29240/usp29nf24s0_c1225.html
International Federation of Pharmaceutical Manufactures & Associations (IFPMA): Validation of Analytical Procedure. In International conference on harmonization (ICH), methodology Q2(R1). Switzerland: Geneva; 1996. 6 November 1996
Ruan J, Tattersall P, Lozano R, Shah P: The role of forced degradation studies in stability indicating HPLC method development. Am Pharm Rev 2006,9(1):46–53.
Bakshi M, Singh S: Development of validated stability-indicating assay methods-critical review. J Pharm Biomed Anal 2002, 28: 1011–1040. 10.1016/S0731-7085(02)00047-X
Acknowledgements
The authors wish to thank the management of Emcure pharmaceuticals Ltd. for supporting this work. The author is also thankful to his colleagues in the division of Research and Development of Emcure Pharma (P) Ltd for their cooperation in carrying out this work.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
ASJ carried out designing of analytical method development. NW carried out analytical method development and analytical method validation. SAJ and SRB drafted the manuscript and participated in the design and coordination of manuscript. SD reviewed the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Joshi, A.S., Warghude, N., Deshmukh, S. et al. Development and validation of stability-indicating UPLC method for the determination of lafutidine and its impurities in bulk and pharmaceutical dosage form. Int J Ind Chem 4, 32 (2013). https://doi.org/10.1186/2228-5547-4-32
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2228-5547-4-32