
Abstract
Background
Diacerein and aceclofenac drug product combination is used for the pain management in osteoarthritis. The main aspects of the present study is to develop stability indicating, sensitive reverse phase high-performance liquid chromatographic method for the simultaneous determination of diacerein and ceclofenac in tablet dosage form. Were, the chromatographic separation was achieved on Kromasil C-18, 150 × 4.6 mm, 3.5-μm analytical column using mobile phase double distilled water (pH 2.7 with glacial acetic acid)-acetonitrile (45:55 v/v). Detector was set at 256 nm.
Results
The described method shows excellent linearity over a range of 75–2.5 μg/mL for diacerein and 150–5 μg/mL for aceclofenac. The correlation coefficient for diacerein is 0.9999 and for aceclofenac is 0.9997.
Conclusions
The proposed method was found to be suitable for simultaneous determination and stability study of diacerein and aceclofenac in tablet dosage form.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Diacerein is chemically 1,8-diacetoxy-3-carboxyanthraquinone and is also known as diacetylrhein. This drug is used in the treatment of osteoarthritis. After absorption, the drug is metabolized to its active metabolite rhein [1]. Diacerein and rhein are anthraquinone compounds that ameliorate the course of osteoarthritis [2–4]. Aceclofenac is chemically (2-[(2,6-dichlorophenyl)amino] phenyl acetoxyacetic acid) [5–7]. It has analgesic properties and a good tolerability profile in a variety of painful conditions.
A combined fixed dose of 50 mg diacerein and 100 mg aceclofenac is available commercially as tablets and are widely used for the treatment of osteoarthritis. The two main aspects of drug product analysis that play an important role in shelf life determinations are assay of active drug and degradants generated during the stability study. However, most of reference HPLC methods are less sensitive and not stability indicating [8–12]. Comparison of the proposed HPLC method with the reference HPLC methods is shown in Table 1.
Stability indicating RP-HPLC method for the simultaneous determination of aceclofenac and diacerein in tablet dosage form is not available in any pharmacopeia. Hence, it was felt essential to develop and validate a sensitive, accurate and stability indicating RP-HPLC method for the simultaneous determination of aceclofenac and diacerein in tablet dosage form. The chemical structures of diacerein and aceclofenac used in this work are shown in Figure 1. Overlain UV spectra of diacerein and aceclofenac are shown in Figure 2.

The chemical structures of diacerein and aceclofenac.

Overlain UV spectra of diacerein and aceclofenac.
Methods
Chemicals
Diacerein and aceclofenac working standards were obtained from Sir Sayyed College Aurangabad, India. Glacial acetic acid, HPLC grade acetonitrile, sodium hydroxide, hydrochloric acid and hydrogen peroxide were obtained from Merck Ltd., Mumbai, India. The 0.45-μm nylon filter was obtained from Advanced Micro Devices Pvt Ltd. (Ambala Cantt, India). The combination product of diacerein and aceclofenac label claim (diacerein 50 mg and aceclofenac 100 mg) branded tablets were purchased from the Indian market. Double distilled water was used throughout the experiment.
Equipment
The chromatographic system used was an Agilent-1100 series (Bangalore, India) comprised of degasser, quaternary pump, auto injector, column compartment and photodiode array detector. The system was controlled through Empower Software (Orlando, Florida).
Chromatographic conditions
Kromasil C-18, 150 × 4.6 mm, 3.5-μm column was used. The mobile phase consists of double distilled water (pH 2.7 with glacial acetic acid)-acetonitrile (45:55 v/v). Isocratic elution was at a flow rate of 1 mL/min and column temperature at 25°C. Detector wavelength was kept at 256 nm using a photodiode array detector.
Preparation of standard solution
The standard stock solution was prepared by dissolving 25 mg diacerein and 50 mg of aceclofenac. Working standard with 10 mL of dimethyl acetamide and dilution with the mobile phase up to 100 mL. Standard solution containing 25 μg/mL of diacerein and 50 μg/mL of aceclofenac was prepared by transferring 5 mL of standard stock solution to 50-mL volumetric flask and diluting with the mobile phase.
Sample preparation
The average fill weight of 20 capsules was determined. Accurately weighed sample powder (100 mg of aceclofenac and 50 mg of diacerein) was transferred into a 200-mL volumetric flask. Dimethyl acetamide (20 mL) was added and sonicated for about 20 min to dissolve; then, mobile phase was added to obtain a volume of 200 mL. The centrifuged solution was filtered through 0.45-μm filter. From the filtered solution, 5 mL of solution was transferred into a 50-mL volumetric flask and diluted to volume with mobile phase.
System suitability studies
The resolution, number of theoretical plates and peak asymmetry were calculated for the standard solutions and are shown in Table 2. The values obtained demonstrated the suitability of the system for the analysis of these drugs in combination. The typical chromatogram of the standard solution is shown in Figure 3.

A typical chromatogram of the tablet: diacerein and aceclofenac.
Forced degradation study of drug product
Transferred sample powder equivalent to 100 mg of aceclofenac and 50 mg of diacerein into a 200-mL volumetric flask was added with 20 mL of dimethyl acetamide and sonicated for about 20 min to dissolve and was treated with acid, alkali and peroxide as described in Table 3. The stress studies were performed as per International Conference on Harmonisation (ICH) recommendation. Results of stressed studies are summarized in Table 3. Figures 4, 5 and 6 show degradation products formed during the degradation study.

Chromatogram of the acid hydrolysis-degraded tablet solution.

Chromatogram of the base hydrolysis-degraded tablet solution.

Chromatogram of the peroxide-degraded tablet solution.
Results and discussion
Optimization of the chromatographic conditions
The main criteria of a successful HPLC method development for determination of diacerein and aceclofenac in tablet dosage form was the method should be able to determine the assay of both drugs in single run. The method should be robust, stability indicating, free of interference from degradation products and useful for routine use in the quality control laboratory.
Our objective of the chromatographic method development was to achieve a peak tailing factor that is <2, run time up to 10 min, along with a resolution that is >5 between diacerein and aceclofenac. In order to optimize the LC separation of aceclofenac and diacerein, initially, the retention behavior of both the components was studied in the pH range of 2.5-6.8 using mobile phases of buffer (pH 2.5-6.8) and acetonitrile, methanol as organic modifier.
To ensure the resolution between diacerein and aceclofenac not to less than 5, the method was as fast as possible, an isocratic run was optimized using the mobile phase consisted of double distilled water (pH adjusted 2.7 with glacial acetic acid)-acetonitrile (45:55 v/v), isocratic elution at flow rate of 1 mL/min and column temperature at 25°C. Detector wavelength was kept at 256 nm using a photodiode array detector. This was selected as an appropriate chromatographic condition, which gave good resolution and acceptable peak parameters for both diacerein and aceclofenac. The analytes of this combination had adequate retentions, peak shape, less tailing and more resolution. The chromatographic analysis time was 10 min. In optimized conditions, diacerein, aceclofenac and their degradants were well separated.
Experimental
Method validation
As per ICH guidelines [13] the method validation parameters were checked for linearity, precision, accuracy, limit of detection, limit of quantitation and robustness.
Specificity
Photodiode array detection was used as an evidence of the specificity of the method. Complete separation of diacerein and aceclofenac was noticed in the presence of tablet excipients. In addition, there was no interference at the retention time of diacerein and aceclofenac in the chromatogram of placebo solution. In peak purity analysis with photo diode detector, purity angle was less than purity threshold for both the analytes. This shows that the peak of analytes was pure, and excipients in the formulation did not interfere with the peak of analytes.
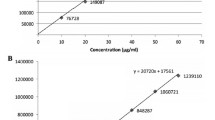
Linearity
Linearity of the method was tested from 10% to 300% of the targeted level of the assay concentration (diacerein 25 μg/mL, aceclofenac 50 μg/mL) for both analytes. Linearity solutions were injected. The calibration graphs were obtained by plotting the peak area ratio against the concentration of the drugs. The equations of the calibration curves for diacerein and aceclofenac obtained were and , respectively. In the simultaneous determination, the calibration graphs were found to be linear in the aforementioned concentrations with correlation coefficient of 0.9999 for diacerein and 0.9997 for aceclofenac.
Precision
The repeatability of the analytical method was evaluated by assaying six sample solutions of diacerein 25 μg/mL and aceclofenac 50 μg/mL during the same day, under the same experimental conditions. Intermediate precision was evaluated by assaying solutions on different days. Peak areas were determined and compared. Precision was expressed as percentage relative standard deviation (RSD% < 2). From the data obtained, the developed RP-HPLC method was found to be precise, and results are reported in Table 4. The results obtained by the proposed method for repeatability were compared with those of the literature HPLC method (7) and found that %difference between %mean assay (n = 6) is 0.2% as shown in Table 6.
Accuracy (recovery test)
Accuracy of the method was studied by recovery experiments. The recovery experiments were performed by spiking solution of known amounts of the drugs in the placebo. The recovery was performed at three levels: 50%, 100% and 150% of the label claim of the tablet (100 mg of aceclofenac and 50 mg of diacerein). Placebo equivalent to one tablet was transferred into a 200-mL volumetric flask, and the amounts of aceclofenac and diacerein at 50%, 100% and 150% of the label claim of the tablet were added. Three samples were prepared for each recovery level. The solutions were then analyzed, and the percentage recoveries were calculated. The recovery values for diacerein and aceclofenac are shown in Table 4.
LOD and LOQ
The LOD and LOQ for diacerein and aceclofenac was determined at a signal-to-noise ratio of 3:1 and 10:1, respectively, by injecting a series of dilute solutions with known concentrations. The LOD and LOQ are as shown in Table 4.
Robustness
The robustness of a method is the ability to remain unaffected by small changes in parameters. To determine the robustness of the method, experimental conditions were purposely altered, and system suitability parameters were evaluated. The flow rate of the mobile phase was 1.0 mL/min. To study the effect of flow rate on system suitability parameters, it was changed to 0.2 units from 1.0 mL/min to1.2 mL/min and 0.8 mL/min. The effect of column temperature on system suitability parameters was studied at 20°C and 30°C instead of 25°C. The effect of pH variation of buffer on system suitability parameters was studied at pH 2.5 and pH 2.9 instead of pH 2.7. The effect of organic variation (acetonitrile) in mobile phase on system suitability parameters was studied at 90% and 110% instead of 100%. Robustness results are shown in Table 5.
Conclusions
The developed simple LC method for assay determination of diacerein and aceclofenac is linear, precise, accurate and specific. The method was validated to the requirements of ICH, and the results were satisfactory. The developed stability-indicating analytical method can be used for the routine analysis of production samples, where sample load is higher and high throughput is essential for faster delivery of results. Overall, the method provides a high throughput solution for determination of diacerein and aceclofenac.
References
Tamura T, Ohmori K: Rhein, an Active Metabolite of Diacerein, Suppresses the Interleukin-1α-Induced Proteoglycan Degradation in Cultured Rabbit Articular Chondrocytes. Jpn J Pharmacol 2001, 85: 101–104. 10.1254/jjp.85.101
Mahajan A, Singh V, Tandon VR, Kumar S, Kumar H: Diacerein : A New Symptomatic Slow Acting Drug for Osteoarthritis. JK Sci 2006, 8: 173–175.
Mendes AF, Caramona MM, Carvalho AP, Lopes MC: Diacerhein and Rhein Prevent Interleukin-1β-Induced Nuclear Factor-κB Activation by Inhibiting the Degradation of Inhibitor κB-α. Pharmacol Toxicol 2002, 91: 22–28. 10.1034/j.1600-0773.2002.910104
Borgmann SHM, Parcianello L, Arend MZ, Bajerski L, Cardoso G: Development and Validation of a Dissolution Method with Spectrophotometric Analysis for Diacerhein Capsules. Sci Pharm 2008, 76: 541–554. 10.3797/scipharm.0804-17
Rhim SY, Park JH, Park YS, Lee MH, Shaw LM, Kang JS: Bioequivalence and pharmacokinetic evaluation of two branded formulations of aceclofenac 100 mg: a single-dose, randomized, open-label, two-period crossover comparison in healthy Korean adult volunteers. Clin Ther 2008, 30: 633–640. 10.1016/j.clinthera.2008.04.008
Ojha A, Rathod R, Padh H: Simultaneous HPLC–UV determination of rhein and aceclofenac in human plasma. J Chromatogr B 2009, 877: 1145–1148. 10.1016/j.jchromb.2009.02.061
Hinz B, Auge D, Rau T, Rietbrock S, Brune K, Werner U: Simultaneous determination of aceclofenac and three of its metabolites in human plasma by high-performance liquid chromatography. Biomed Chromatogr 2003, 17: 268–275. 10.1002/bmc.243
Bhinge JR, Kumar RV, Sinha VR: A simple and sensitive stability-indicating RP-HPLC assay method for the determination of aceclofenac. J Chromatogr Sci 2008, 46: 440–444.
Godse VP, Deodhar MN, Bhosale AV, Sonawane RA, Sakpal PS, Borkar DD, Bafana YS: Reverse Phase HPLC Method for Determination of Aceclofenac and Paracetamol in Tablet Dosage Form. Asian J Research Chem 2009, 2: 37–40.
Momin MY, Yeole PG, Puranik MP, Wadher SJ: Reverse phase HPLC method for determination of aceclofenac and paracetamol in tablet dosage form. Indian J Pharmaceutic Sci 2006, 68: 387–389. 10.4103/0250-474X.26672
Karthikeyan V, Yuvaraj V, Nema RK: Simultaneous estimation of paracetamol, chlorzoxazone and aceclofenac in pharmaceutical formulation by HPLC method. Int J ChemTech Res 2009, 1: 457–460.
Kachhadia PK, Doshi AS, Ram VR, Joshi HS: Validated LC Method for Simultaneous Analysis of Tramadol Hydrochloride and Aceclofenac in a Commercial Tablet. Chromatographia 2008, 68: 997–252. 10.1365/s10337-008-0829-6 0009-5893/08/12
Validation of analytical procedures: text and methodology: 254 In: ICH Harmonized Tripartite Guidelines Q2 (R1). ICH, Swtizerland; 2005.
Acknowledgments
The authors would like to thank Sir Sayyed College Aurangabad, for providing working standard, Chemicals used for research work.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
KS carried out designing Analytical method development, coordination and reviewing the manuscript. AP carried out Analytical method development, method validation and drafted the manuscript, AI participated in forced degradation study. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Shaikh, K.A., Patil, A.T. & Ingole, A.B. Sensitive LC method for the simultaneous determination of diacerein and aceclofenac in tablet dosage form. Int J Ind Chem 3, 3 (2012). https://doi.org/10.1186/2228-5547-3-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2228-5547-3-3