
Abstract
In this study, we have carried out the calculation for the interaction of fluorouracil anticancer drug with carbon nanotube using Hartree-Fock theory with 3-21G basis set and molecular mechanic/Monte Carlo simulation (with MM+, AMBER, and OPLS force field) in the gas phase, methanol, and water solvents at various temperatures (298 up to 312 K). The obtained results of potential energy, Gibbs free energy, enthalpy, entropy, and dipole moment value changes are compared in gas phase, methanol, and water, and the temperature effects on the stability of the interaction between fluorouracil anticancer drug and carbon nanotube in gas phase, methanol, and water solvents.
Similar content being viewed by others
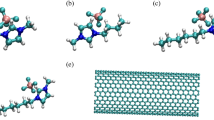

Background
Fluorouracil anticancer drug belongs to a bunch of medicines distinguished as antineoplastics, and it is sorted as an antimetabolite. It acts by preventing the growth of cancer cells and finally demolishing them. Fluorouracil is utilized for the treatment of the cancers such as the breast, rectum, colon, stomach, or the pancreas [1] in 1957 by Charles Heidelberger; 5-FU was designed, synthesized, and patented [2]. 5-Fluoro-2,4(1H, 3H)-pyrimidinedione is chemically named fluorouracil [3]. Three possible mechanisms of action of fluorouracil existed [4]. The first mechanism, the fluorouracil metabolite fluorodeoxyuridine monophosphate contests with uracil to bind with thymidylate synthetase and the folate cofactor [5], these consequences in declined thymidine production and hence declined DNA synthesis and repair, and eventually declined cell proliferation. Leucovorin (formyltetrahydrofolate, formyl-FH4) increases fluorouracil by stabilizing the binding of fluorodeoxyuridine monophosphate to thymidylate synthetase. The second mechanism, the fluorouracil metabolite fluorodeoxyuridine triphosphate is incorporated into DNA, thus interfering with DNA replication [4]. The final mechanism, the fluorouracil metabolite fluorouridine-5-triphosphate is being incorporated into RNA in place of uridine triphosphate, causing a fraudulent RNA and interfering with RNA processing and protein synthesis [6]. It is cell-cycle special [5]. Besides, the carbon nanotube (CNT) is a promising nanomaterial with rare physical characteristics [7]. Its geometry and chemical characteristics are ideal for an electron emitter with a low threshold voltage and long lifetime [8], and a wide range of usages have been proposed, such as gas sensors [9], transistors [10, 11], thermal sensors [12, 13], and pressure sensors [14]. In addition, CNTs are nanostructures which are members of the fullerene group. They are one of the most promising materials for the fabrication of nanotechnological devices in the future. Substantial study efforts are focused on the study of their structure, as well as their mechanical and electric characteristics [15, 16]. Different methods have developed for the production of CNTs, for example, laser vaporization [17] or catalytic decomposition of hydrocarbons [18]. In recent times, a new production way based on the thermal decomposition of C60 in the presence of transition metals was presented [19, 20]. Different conformations of carbon nanotube are three structures that the nanotube is either of the armchairs (n = m), zigzag (n = 0 or m = 0), or chiral (any other n and m) variety, and each of these forms has its own characteristics [21, 22]. Single-wall nanotubes (SWNTs) are molecular scale wires with fascinating and potentially useful characteristics [15]. It is well established of which the diameter and chirality of a SWNT determine the electronic structures of the nanotube [23]. In order to achieve homogeneous nanotube materials with special physical characteristics, it is necessary to control the structures of nanotubes by chemical synthesis techniques. The prerequisite for such control is a detailed understanding of the growth mechanism of nanotubes. A double-wall carbon nanotube, which consists of two concentric cylindrical graphene layers, has been successfully synthesized by catalytic chemical vapor deposition [23–25]. Double-wall carbon nanotubes are the topic of extreme investigations versus their promising usages. In particular, the double-wall carbon nanotube with a metallic inner tube and an insulating outer tube is a model system for a molecular conductive wire covered by an insulator. In our work, the interaction of fluorouracil with CNT was studied, applying molecular mechanic/Monte Carlo (MC) simulation in several force fields. Furthermore, the influences of different environments such as gas phase, methanol, and water on this interaction by quantum mechanic (QM) have been studied too. For the simulation of the solvent influences by QM method, self-consistent reaction field (SCRF) method is mainly used. In this place, solvent effects on the potential energy changes and thermodynamic value changes of this interaction with employ QM/MM methods are depicted.
Results and discussion
All the theoretical results present in this paper were obtained by two methods, firstly, molecular mechanic (Monte Carlo simulation) and secondly, quantum mechanic.
Fluorouracil inhibits the development of cancer cells and finally destroying them, and carbon nanotube is a carbon with cylindrical outline material and a nanometric-level diameter which has a spacious range of usages such as drug delivery; thus, our purpose of this article is to survey the interaction of a nanotube with a fluorouracil anticancer drug according to reaction 1. In this work, we see the image of the combination of fluorouracil and CNT in Figure 1.

Studied system of fluorouracil with CNT.
If the following reaction is considered:
we can calculate the potential energy and thermodynamic value changes on the interaction of fluorouracil with CNT for reaction 1 by the following equation:
where M is equal to μ, G, H, E, S, and EPOT parameters. Therefore, first, we investigated the different force fields to determine the potential energy and other kinds of geometrical parameters on the interaction of fluorouracil and CNT. Since the three different force fields have been utilized, the calculated potential energy of the molecules will not be the same. Thus, comparing the computed potential energy of one molecule using a particular force field with the potential energy of another molecule, which is calculated by another force field, is neither rational nor possible. In this article, the difference in force fields is illustrated by comparing the computed potential energy changes using various force fields like: MM+, AMBER, and OPLS (Table 1 and Figures 2, 3, and 4). The theoretical potential energy value changes are attained using different force fields. Then, we see that water solvent has the lowest value of potential energy changes. And also, by comparing ΔEPOT in different force fields such as MM+, AMBER, and OPLS in water solvent, we observe that AMBER force field is best when reaction 1 occurs in the water solvent (Figure 5).

The potential energy changes versus temperature in different solvents and MM+ force field for reaction 1.

The potential energy changes versus temperature in different solvents and AMBER force field for reaction 1.

The potential energy changes versus temperature in different solvents and OPLS field for reaction 1.

The potential energy changes versus temperature in water solvent and same force field for reaction 1.
In Table 2, the dipole moment values of the interaction of fluorouracil with CNT according to reaction 1 based on Equation 1 in gas phase, water, and methanol at different temperatures have been observed. Dipole moment values in gas phase, water, and methanol exhibit increasing dielectric constant; dipole moment will be increased that is demonstrated in Table 2. In the SCRF model, the solvent is signified by a continuous dielectric, recognized by a given dielectric constant. The solute supposed to be fixed into a cavity, with radius a0 in the molecule, and the electric field which is related to the solute by the solvent dipole will interact with the molecular dipole and direct to net stabilization. It must be noted which salvation energy computed using the SCRF method is the electrostatic distribution to the free energy of salvation. In this investigation, the cavity radius of fluorouracil, CNT, and fluorouracil/CNT system (a0) was calculated and substituted efficiency was improving. The volume of fluorouracil, CNT, and fluorouracil/open-end of CNT system was achieved that values of a0 for fluorouracil, CNT, and fluorouracil/CNT system were 3.76, 5.64, and 10.26 Ǻ, respectively; thus, according to Equation 1, we calculate the potential energy and thermodynamic value changes. For in the survey, you can see that the dipole moment changes Δμ (Debye) in (Table 2 and Figure 6) that by the increase of the solvent dielectric constant, the dipole moment for the interaction of fluorouracil with CNT in different temperatures such as 298, 301, 304, 308, and 312 K will be increased.

The dipole moment changes calculated versus temperature for reaction 1.
After computing using the SCRF model, it is demonstrated which ΔG, ΔE, ΔH and ΔS values of the interaction of fluorouracil with CNT for reaction 1 are sensitive to polarity of the surrounding solvent. Thus, in Table 3, you observe that ΔG, ΔE, ΔH and ΔS values depend on the dielectric constant (ϵ), and Table 3 indicates which increasing temperature from 298 to 312 K and values of ΔG of interaction of fluorouracil with CNT for reaction 1 in gas phase, water, and methanol will be increased. As a result, the lowest amount of ΔG is in 298 K, and we have the highest resistance for reaction 1 in 298 K (Figure 7). Also, as the solvent dielectric constant increases, the attained ΔG values of interaction of fluorouracil with CNT decrease. So, the most negative ΔG value of this interaction is in water solvent. The water solvent is the best environment for reaction 1 as shown in Figure 7. Also, in Table 3, we can observe the attained ΔG, ΔE, ΔH, and ΔS values in various environments such as in gas phase, methanol, and water versus temperature alteration from 298 to 312 K. Thereupon, by increasing the temperature from 298 to 312 K, the most positive value of ΔE, ΔH, and ΔS is for 298 K. The other actuality that is confirmed by the calculated values of ΔE, ΔH, and ΔS versus solvent dielectric constant increase in temperature of 298 to 312 K is which of the values of ΔE and ΔH will shift toward negative values in which you can observe the most negative of these parameters in 312 K. Also, in Table 3, with the increase of solvent dielectric constant in temperature from 298 to 312 K, ΔS values increase. So, Table 3 proves which of the value of ΔS in water solvent has the most positive value (Figure 8). Then, water solvent is the best for reaction 1. So, when the studied system of interaction of fluorouracil with CNT is placed in water solvent, the lowest values of enthalpy take place (Figure 9).

Calculated Δ G versus temperature for reaction 1 at different dielectric constants.

Calculated Δ S versus temperature for reaction 1 at different dielectric constants.

Calculated Δ H versus temperature for reaction 1 at different dielectric constants.
Conclusion
In this work, two calculations were performed, one by HyperChem program and second by the ab initio computations which were carried out with Hartree-Fock (HF) functional, utilizing 3-21G basis set and the Gaussian 98. According to achieved consequences, water solvent is the best environment because the studied system of the interaction of fluorouracil with CNT according to reaction 1 has the most negative values of ΔG. Also, using the SCRF model, the obtained consequences have seemed quite sensitive to the polarity of the enclosing solvent, and we can see that with the increase of solvent dielectric constant, ΔS of studied system of the interaction of fluorouracil with CNT has the most positive value. Thus, we can conclude that water solvent is best. Besides, the achieved consequences of potential energy changes from force fields MM+, AMBER, and OPLS were compared, and we observe which AMBER force field is the best for the survey of reaction 1.
Methods
The theoretical results demonstrated in this article were achieved by means of the ab initio and molecular mechanic/Monte Carlo simulation (MM+, AMBER, and OPLS). Geometry optimization and structural characteristics computations were done in Gaussian 98 program, Wallingford, CT, USA. Also, Monte Carlo computations were carried out by HyperChem program, Gainesville, FL, USA.
Ab initio
The most ordinary form of ab initio computation is named Hartree-Fock computation (abbreviated HF), as the initial approximation. This means that the coulombic electron–electron repulsion is not clearly taken into account, though its average influence is contained in the computation [26]. Quantum Monte Carlo is a technique which prevents the HF from making mistakes. There are some sorts of QMC: variation, diffusion, and Green's functions. These techniques work with an openly correlated wave function and compute integrals numerically using a Monte Carlo integration. These computations are very time-consuming, but they are the most correct techniques that are known up to now. On the whole, ab initio computations prepare perfect quantitative consequences and can give increasingly accurate quantitative consequences as the molecules under consideration become smaller [27].
Monte Carlo simulation
In the fields of chemistry, biology, physics, and engineering, Monte Carlo simulations are widely utilized to determine the structural and thermodynamic characteristics of complex systems at the atomic level. By Monte Carlo techniques, we can determine not only the thermodynamic averages of molecular characteristics but also minimum energy. Structures [28] in this technique [29] and a sequence of points in phase space are created from an initial geometry by adding a random ‘kick’ to the coordinates of a fortuitously chosen particle (atom or molecule). If the energy declines, the new computation is accepted, with a probability of e-E/KT if the energy heightens. The metropolis [30] procedure ensures which of the configurations in the ensemble obey a Boltzmann distribution. The probability of accepting higher energy computations permits MS technique to heighten and go over from a local minimum. In MC simulation, you need only the capability to evaluate the energy of the system if computing the first derivative is difficult or time-consuming; this capability maybe advantageous. Additionally, since only a single particle is moved in each step, the only thing that must be configurated are the energy alterations accompanied with this move, not the total energy for the complete system. The lack of the time dimension and atomic velocities are some disadvantages of MC techniques. And so, they are not appropriate for spending time and investigating characteristics depending on time-dependent phenomena or momentum [31].
In the present article, differences in force fields are demonstrated by comparing the computed potential energy changes by applying force fields MM+, AMBER, and OPLS. In this article, HyperChem program is utilized for the quantum chemical computations. We have done geometry optimization and Monte Carlo simulation by this software [32]. The QM computations on the interaction of fluorouracil with carbon nanotube have been done by Gaussian 98 program [33] utilizing the standard 3-21G basis set. Thus, solvent effects have been studied on the values of thermodynamic parameters and dipole moment of water and methanol encircling fluorouracil/CNT system within the SCRF model utilizing a Hartree-Fock method and the temperature effects on fluorouracil/CNT system. Therefore, achieved thermodynamic data of fluorouracil/CNT system are compared in gas phase and various solvent environments such as methanol and water.
References
Dorr RT, Von-Hoff DD: Cancer Chemotherapy Handbook. 2nd edition. Norwalk: Appleton and Lange; 1994:27–28.
Chu E: Clinical colorectal cancer: ode to 5-fluorouracil. Clin. Colorectal Cancer 2007,6(9):609. 10.3816/CCC.2007.n.029
Longley DB, Harkin DP, Johnston PG: 5-fluorouracil: mechanisms of action and clinical strategies. Nat. Rev. Cancer May 2003,3(5):330–338. 10.1038/nrc1074
van der Wilt CL, Marinelli A, Pinedo HM, Cloos J, Smid K, van de Velde CJ, Peters GJ: The effects of different routes of administration of 5-fluorouracil on thymidylase synthase inhibition in the rat. Eur. J. Cancer 1995,31A(5):754–760.
McEvoy GK (Ed): AHFS 2006 Drug Information. Bethesda: American Society of Health-System Pharmacists; 2006:1052–1056.
McEvoy GK (Ed): AHFS 2005 Drug Information. Bethesda: American Society of Health-System Pharmacists; 2005:1020–1025.
Dekker C: Carbon nanotubes as molecular quantum wires. Physics Today. 1999,52(5):22–28. 10.1063/1.882658
Bonard JM, Kind H, Stöckli T, Nilsson LO: Field emission from carbon nanotubes: the first five years. Solid State Electron. 2001, 45: 893. 10.1016/S0038-1101(00)00213-6
Kong J, Franklin NR, Zhou C, Chapline M, Peng G, Cho SK, Dai H: Nanotube molecular wires as chemical sensors. Science 2000, 287: 622–625. 10.1126/science.287.5453.622
Artukovic E, Kaempgen M, Hecht DS, Roth S, Gruner G: Transparent and flexible carbon nanotube transistors. Nano Lett. 2005, 5: 757–760. 10.1021/nl050254o
Hines DR, Mezhenny S, Breban M, Williams ED, Ballarotto VW, Esen G, Southard A, Fuhrer MS: Nanotransfer printing of organic and carbon nanotube thin-film transistors on plastic substrates. Appl. Phys. Lett. 2005, 86: 163101. 10.1063/1.1901809
Selvarasah S, Chen C-L, Chao S-H, Makaram P, Busnaina A, Dokmeci MR: A three dimensional thermal sensor based on single-walled carbon nanotubes. In Proceedings of the 14th International Conference on Solid-State Sensors, Actuators and Microsystems (Transducers '07). Lyon; 2007:1023–1026. 10–14 Jun 10–14 Jun
Fung CKM, Wong VTS, Chan RHM, Li WJ: Dielectrophoretic batch fabrication of bundled carbon nanotube thermal sensors. IEEE Trans. Nanotechnology 2004, 3: 395–403. 10.1109/TNANO.2004.834156
Fung CKM: Fabrication of CNT-based MEMS piezoresistive pressure sensors using DEP nanoassembly. In Proceedings of the 5th International Conference on Nanotechnology (IEEENANO). Nagoya: Nagoya; 2005:199–202. 11–15 Jul 11–15 Jul
Dresselhaus MS, Dresselhaus G, Eklund PC: Science of Fullerenes and Carbon Nanotubes. San Diego: Academic Press; 1996.
Ebbesen TW (Ed): Carbon Nanotubes: Preparation and Properties. Boca Raton: CRC Press; 1997.
Thess A, Lee R, Nikolaev P, Dai H, Petit P, Robert J, Xu C, Lee YH, Kim SG, Rinzler AG, Colbert DT, Scuseria GE, Tomanek D, Fischer JE, Smalley RE: Crystalline ropes of metallic carbon nanotubes. Science 1996, 273: 483. 10.1126/science.273.5274.483
Ivanov V, Nagy JB, Lambin P, Lucas A, Zhang XB, Zhang XF, Bernaerts D, Van Tendeloo G, Amellinckx S, Van Landuyt J: The study of carbon nanotubules produced by catalytic method. Chem. Phys. Lett. 1994, 223: 329. 10.1016/0009-2614(94)00467-6
Ehlich R, Biro LP, Hertel IV: Growth of nanotubes by decomposition of C60 on transitional metal surfaces. Synth. Met. 1999, 103: 2486. 10.1016/S0379-6779(98)01069-8
Bir´o LP, Ehlich R, Tellgmann R, Gromov A, Krawetz N, Tschaplyguine M, Pohl M-M, Zsoldos E, V´ertesy Z, Horv´ath ZE, Campbell EEB: Growth of carbon nanotubes by fullerene decomposition in the presence of transition metals. Chem. Phys. Lett. 1999, 306: 155. 10.1016/S0009-2614(99)00433-9
Meng S, Maragakis P, Papaloukas C, Kaxiras E: DNA nucleoside interaction and identification with carbon nanotubes. Nano Lett. 2007, l7: 45. No1 No1
Cuniberti G, Yi J, Porto M: arXiv: Pure-carbon ring transistor: role of topology and structure. Appl. Phys. Lett. 2002, 81: 850. 10.1063/1.1497195
Flahaut E, Peigney A, Laurent CH, Rousset A: Synthesis of single-walled carbon nanotube-Co-MgO composite powders and extraction of the nanotubes. J. Mater. Chem. 2000, 10: 249. 10.1039/a908593i
Sugai T, Omote H, Bandow S, Tanaka N, Shinohara H: Carbon nanotubes by high-temperature pulsed arc discharge. J. Chem. Phys. 2000, 112: 6000. 10.1063/1.481172
Zhu H, Xu C, Wei B, Wu D: A new method for synthesizing double-walled carbon nanotubes. Carbon 2002, 40: 2021. 10.1016/S0008-6223(02)00040-4
Nsangou M, Jaidane NE, Ben Z: Lakhdar: Internet electronic. J. Molecular Design. 2006, 5: 89–101.
Young D: Cytoclonal Pharmaceutics Inc.: Introduction to computational chemistry. Chem. Aust. 1998, 11: 5.
Monajjemi M, Chahkandi B: Study of the hydrogen bond indifferent orientations of adenine-thymine base pairs: an ab initio study. J. Mol. Struct. (THEOCHEM) 2004, 714: 43.
Jorgensen WL: Biological reactions are slow in absence of a catalyst. Adv. Chem. Phys. 1988, 70: 469.
Metropolis NA, Rosenbluth AW, Teller AH, Teller E: Equation of state calculations by fast computing machines. J. Chem. Phys. 1953, 21: 10–87.
Haeri HH, Hashemianzadeh SM, Monajjemi M: Temperature effects on the stochastic gating of the IP3R calcium release channel: a numerical simulation study. J. Biol. Syst. 2009, 17: 817–852. 10.1142/S0218339009003058
HyperChem 7.0. Gainesville: Hypecube Inc; 2001.
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Zakrzewski VG, Montgomery JA Jr, Stratmann RE, Burant JC, Dapprich S, Millam JM, Daniels AD, Kudin KN, Strain MC, Farkas O, Tomasi J, Barone V, Cossi M, Cammi R, Mennucci B, Pomelli C, Adamo C, Clifford S, Ochterski J, Petersson GA, Ayala PY, Cui Q, Morokuma K, Malick DK, et al.: Gaussian 98 Revision A.6. Pittsburgh: Gaussian, Inc; 1998.
Acknowledgements
The authors would like to thank the Islamic Azad University for financial and other supports.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
FN carried out the computational simulations and the results analysis and participated in the manuscript elaboration. KZ coordinated in the project, discussed the results, and helped to draft the manuscript. HS and RSG examined the results and helped to draft the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Zare, K., Najafi, F., Sadegh, H. et al. Studies of ab initio and Monte Carlo simulation on interaction of fluorouracil anticancer drug with carbon nanotube. J Nanostruct Chem 3, 71 (2013). https://doi.org/10.1186/2193-8865-3-71
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2193-8865-3-71