
Abstract
Context
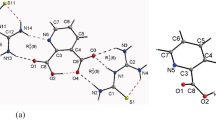
The electronic, discrete water solvation, and vibrational properties of zwitterionic sulfanilic acid were thoroughly investigated using periodic and non-periodic DFT approaches. The periodic-DFT results, obtained by employing the PBE-TS functional (Perdew-Burke-Ernzerhof (PBE) functional with the Tkatchenko and Scheffler (TS) dispersion correction) were first presented in order to analyze the band structures of the studied crystal. An attentive reading of the predicted band structures has shown three lowest gap energies calculated at 4.23, 4.24, and 4.29 eV arising from the Γ→Γ, Γ→Z, and Γ→S transitions, respectively. Then, non-periodic calculations were carried out, at the B3LYP-D3 level of theory (B3LYP functional with the D3 Grimme dispersion correction) in order to optimize the sulfanilic acid-(H2O)10 complex. Starting from the optimized structure, non-covalent interaction calculations were performed and the H-bonding, van der Waals, and steric effect interactions were identified. Finally, the PBE-TS calculations were strengthened by conducting anharmonic B3LYP-D3 calculations in order to achieve a complete decryption of the experimental IR spectrum of sulfanilic acid. The spectral analysis is not limited only to the interpretation of both the NH/CH stretching and fingerprint regions but also extended to the 1800–2600 cm−1 region, which is characterized by a strong anharmonic effect. In the latter wavenumber region, the large experimental IR band centered at 1937 cm−1 is reproduced theoretically employing the anharmonic B3LYP-D3 calculations. The similarity of this band with those usually considered as a fingerprint of zwitterionic amino acids is observed, and its origin is elucidated theoretically. In the vibrational spectroscopy field, the calculations presented in this study are probably the most appropriate for achieving vast analysis and accurate assignments of vibrational spectra of hydrogen bonding compounds recorded in the solid state.
Method
The periodic and non-periodic calculations were conducted within the Density Functional Theory (DFT) using the Generalized Gradient Approximation (GGA) at the PBE-TS level of theory and B3LYP-D3 functional with the 6-311++G(d,p) basis set, respectively. The PBE-TS and B3LYP-D3/6-311++G(d,p) calculations were performed using the CASTEP and Gaussian 09 programs, respectively. In addition, The non-covalent interactions were calculated by the Multiwfn 3.8 software. The obtained results for different calculations were visualized by employing the visualization tools in Materials Studio, GaussView, VMD, and Gnuplot programs.












Similar content being viewed by others

Data availability
Data will be available on reasonable request.
References
Hübschle CB, Messerschmidt M, Luger P (2004) Crystal structure of DL-Tryptophan at 173K. Cryst Res Technol 39:274–278. https://doi.org/10.1002/crat.200310182
Frey MN, Koetzle TF, Lehmann MS, Hamilton WC (1973) Precision neutron diffraction structure determination of protein and nucleic acid components. X. A comparison between the crystal and molecular structures of L-tyrosine and L-tyrosine hydrochloride. J Chem Phys 58:2547–2556. https://doi.org/10.1063/1.1679537
Edington P, Harding MM (1974) The crystal structure of DL-histidine. Acta Cryst B 30:204–206
Krishnan RS, Sankaranarayanan VN, Krishnan K (1973) Raman and Infra-red Spectra of Amino Acids. J Indian I Sci 55:66–116 (https://citeseerx.ist.psu.edu/document?repid=rep1&type=pdf&doi=0b6fe793565f1be98a971d8447051b2167baa2ed)
Boldyreva E (2008) Crystalline amino acids. In: Boldyreva E (ed) A link between chemistry, materials science and biology. Springer, Dordrecht, pp 167–192
Leiske MN, Mazrad ZA, Zelcak A, Wahi K, Davis TP, McCarroll JA, Holst J, Kempe K (2022) Zwitterionic amino acid-derived polyacrylates as smart materials exhibiting cellular specificity and therapeutic activity. Biomacromolecules 23:2374–2387. https://doi.org/10.1021/acs.biomac.2c00143
Araújo RL, Vasconcelos MS, Barboza CA, Neto JXL, Albuquerque EL, Fulco UL (2019) DFT calculations of the structural, electronic, optical and vibrational properties of anhydrous orthorhombic L-threonine crystals. Comput Theor Chem 1170:112621. https://doi.org/10.1016/j.comptc.2019.112621
Flores MZS, Freire VN, Dos Santos RP, Farias GA, Caetano EWS et al (2008) Optical absorption and electronic band structure first-principles calculations of α-glycine crystals. Phys Rev B 77:115104. https://doi.org/10.1103/PhysRevB.77.115104
Dague Y, Koyambo-Konzapa SJ, Nose H, Minguirbara A, Nsangou M, Amolo G (2024) DFT investigation on the structural and vibrational behaviours of the non-protein amino acids in hybrid explicit/continuum solvent: a case of the zwitterions γ-aminobutyric and α− aminoisobutyric acids. J Mol Model 30:17. https://doi.org/10.3390/ijms241814035
Low JN, Glidewell C (2002) A quasi-diamondoid hydrogen-bonded framework in anhydrous sulfanilic acid. Acta Crystallogr C 58:o209–o211. https://doi.org/10.1107/S0108270102003025
Habli H, Mejrissi L, Issaoui N, Yaghmour SJ, Oujia B, Gadéa FX (2015) Ab initio calculation of the electronic structure of the strontium hydride ion (SrH+). Int J Quantum Chem 115:172–186. https://doi.org/10.1002/qua.24813
Mhadhbi N, Issaoui N, Hamadou WS, Alam JM, Elhadi AS, Adnan M, Naϊli H, Badraoui R (2022) Physico-Chemical Properties, Pharmacokinetics, Molecular Docking and In-Vitro Pharmacological Study of a Cobalt (II) Complex Based on 2-Aminopyridine. Chemistry Select 7:e202103592. https://doi.org/10.1002/slct.202103592
Medimagh M, Mleh CB, Issaoui N, Kazachenko AS, Roisnel T, Al-Dossary OM, Marouani H, Bousiakoug LG (2023) DFT and molecular docking study of the effect of a green solvent (water and DMSO) on the structure, MEP, and FMOs of the 1-ethylpiperazine-1, 4-diium bis (hydrogenoxalate) compound. J Mol Liq 369:120851
Kazachenko AS, Medimagh M, Issaoui N, Al-Dossary O, Wojcik MJ, Kazachenko AS, Miroshnokova AV, Malyar YN (2022) Sulfamic acid/water complexes (SAA-H2O (1–8)) intermolecular hydrogen bond interactions: FTIR, X-ray, DFT and AIM analysis. J Mol Struct 1265:133394. https://doi.org/10.1016/j.molstruc.2022.133394
Srivastav G, Yadav RK, Yadav B, Yadav RA (2023) Vibrational spectra and molecular structure of sulfanilic acid: IR and low temperature Raman studies and DFT investigation of monomeric and dimeric forms. J Mol Struct 1272:134143. https://doi.org/10.1016/j.molstruc.2022.134143
Fatima A, Khanum G, Savita S, Pooja K, Verma I, Siddiqui N, Javed S (2022) Quantum computational, spectroscopic, Hirshfeld surface, electronic state and molecular docking studies on sulfanilic acid: An anti-bacterial drug. J Mol Liq 346:117150. https://doi.org/10.1016/j.molliq.2021.117150
ThayalaSanker R, Arunpandian M, VelayuthamPillai M, Arunachalam S, Muneeswaran G, Babu B, Ho M, Sivakumar R (2021) Spectroscopic and density functional theory (DFT) approach of zwitterionic 4-aminobenzenesulfonic acid for optoelectronic applications. J Mater Sci-mater El 32:4982–4997. https://doi.org/10.1007/s10854-021-05236-7
Popoola SA, Almohammedi AR, Haruna K (2021) Spectroscopic and DFT evaluation of the positional effect of amino group on the properties of aminobenzenesulphonic acid: Solvents interactions. Chem Pap 75:2775–2789. https://doi.org/10.1007/s11696-020-01497-4
Hammami F, Issaoui N, Nasr S (2021) Investigation of hydrogen bonded structure of urea-water mixtures through Infra-red spectroscopy and non-covalent interaction (NCI) theoretical approach. Comput Theor Chem 1199:113218. https://doi.org/10.1016/j.comptc.2021.113218
Gatfaoui S, Issaoui N, Roisnel T, Marouani H (2021) Synthesis, experimental and computational study of a non-centrosymmetric material 3-methylbenzylammonium trioxonitrate. J Mol Struct 1225:129132. https://doi.org/10.1016/j.molstruc.2020.129132
Daghar C, Issaoui N, Roisnel T, Dorcet V, Marouani H (2021) Empirical and computational studies on newly synthesis cyclohexylammonium perchlorate. J Mol Struct 1230:129820. https://doi.org/10.1016/j.molstruc.2020.129820
Hasnip PJ, Refson K, Probert MIJ, Yates JR, Clark SJ, Pickard CJ (2014) Density functional theory in the solid state. Phil Trans R Soc A 372:20130270. https://doi.org/10.1098/rsta.2013.0270
Caetano EW, Silva JB, Bruno CH, Albuquerque EL, eSilva BP, dos Santos RC, Teixeira AMR, Freire VN (2024) Investigating the molecular crystals of L-Alanine, DL-Alanine, β-Alanine, and Alanine hydrogen chloride: Experimental and DFT analysis of structural and optoelectronic properties. J Mol Struct 1300:137228. https://doi.org/10.1016/j.molstruc.2023.137228
Chaudhry AR, Irfan A, Muhammad S, Al-Sehemi AG, Ahmed R, Jingping Z (2017) Computational study of structural, optoelectronic and nonlinear optical properties of dynamic solid-state chalcone derivatives. J Mol Graph Model 75:355–364. https://doi.org/10.1016/j.jmgm.2017.05.012
Giester G, Ghazaryan VV, AL, Zatikyan, AM, Petrosyan (2024) Polyiodides of amino acids L-Proline triiodides. Struct Chem 9:1–11. https://doi.org/10.1007/s11224-024-02291-8
Boukaoud A, Chiba Y, Sebbar D, Dehbaoui M, Guechi N (2021) A Theoretical Study of Vibrational and Optical Properties of Isatin. Braz J Phys 51:1207–1214. https://doi.org/10.1007/s13538-021-00924-5
Benaissa M, Boukaoud A, Sebbar D, Chiba Y, Krid A (2024) Periodic and non-periodic DFT studies of an organic semiconductor material: Structural, electronic, optical, and vibrational properties of ninhydrin. Spectrochim Acta A 307:123636. https://doi.org/10.1016/j.saa.2023.123636
Sahana R, Mounica P, Dineshkumar P, Elangovan A, Shanmugam R, Arivazhagan G (2023) Conformers of 1, 4-dioxane and their hydrogen bond complexation with methanol. Vib Spectrosc 126:103519
Wang C, Danovich D, Shaik S, Mo Y (2017) A unified theory for the blue-and red-shifting phenomena in hydrogen and halogen bonds. J Chem Theory Comput 13:1626–1637 (http://pubs.acs.org on March 5, 2017 )
Galkina YA, Kryuchkova NA, Vershinin MA, Kolesov BA (2017) Features of strong O-H⋯ O and N–H⋯ O hydrogen bond manifestation in vibrational spectra. J Struct Chem 58:911–918. https://doi.org/10.1134/S0022476617050080
Boukaoud A, Chiba Y, Sebbar D (2021) A periodic DFT study of IR spectra of amino acids: An approach toward a better understanding of the NH and OH stretching regions. Vib Spectrosc 116:103280. https://doi.org/10.1016/j.vibspec.2021.103280
Cao X, Fischer G (1999) Infrared spectral, structural, and conformational studies of zwitterionic L-tryptophan. J Phys Chem A 103:9995–10003. https://doi.org/10.1021/jp992421c
Cao X, Fischer G (2000) The infrared spectra and molecular structure of zwitterionic L-β-phenylalanine. J Mol Struct 519:153–163. https://doi.org/10.1016/S0022-2860(99)00291-4
Carbonnière P, Erba A, Richter F, Dovesi R, Rérat M (2020) Calculation of anharmonic IR and Raman intensities for periodic systems from DFT calculations: Implementation and validation. J Chem Theory Comput 16:3343–3351. https://doi.org/10.1021/acs.jctc.9b01061
Clark SJ, Segall MD, Pickard CJ, Hasnip PJ, Probert MI, Refson K, Payne MC (2005) First principles methods using CASTEP. Z Kristallogr 220:567–570. https://doi.org/10.1524/zkri.220.5.567.65075
Perdew JP, Yue W (1986) Accurate and simple density functional for the electronic exchange energy: Generalized gradient approximation. Phys Rev B 33:8800–8802. https://doi.org/10.1103/PhysRevB.33.8800
Perdew JP, Burke K, Ernzerhof M (1996) Generalized gradient approximation made simple. Phys Rev Lett 77:3865–3868. https://doi.org/10.1103/PhysRevLett.77.3865
Tkatchenko A, Scheffler M (2009) Accurate molecular van der Waals interactions from ground-state electron density and free-atom reference data. Phys Rev Lett 102:073005. https://doi.org/10.1103/PhysRevLett.102.073005
Refson K, Tulip PR, Clark SJ (2006) Variational density-functional perturbation theory for dielectrics and lattice dynamics. Phys Rev B 73:155114. https://doi.org/10.1103/PhysRevB.73.155114
Tulip PR, Clark SJ (2004) Dielectric and vibrational properties of amino acids. J Chem Phys 121:5201–5210. https://doi.org/10.1063/1.1781615
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA,Cheeseman JR, Scalmani G, Barone V, Petersson GA, Nakatsuji H, Li X, Caricato M, Marenich AV, Bloino J, Janesko BG, Gomperts R, Mennucci B, Hratchian HP, Ortiz JV, Izmaylov AF,Sonnenberg JL, Williams-Young D, Ding F, Lipparini F, Egidi F, Goings J, Peng B, Petrone A, Henderson T, Ranasinghe D, Zakrzewski VG, Gao J, Rega N, Zheng G, Liang W, Hada M,Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Throssell K, Montgomery JA Jr, Peralta JE, Ogliaro F, Bearpark MJ, Heyd JJ, Brothers EN, Kudin KN, Staroverov VN, Keith TA, Kobayashi R, Normand J, Raghavachari K, Rendell AP, Burant JC, Iyengar SS, Tomasi J, Cossi M, Millam JM, Klene M, Adamo C, Cammi R, Ochterski JW, Martin RL, Morokuma K, Farkas O, Foresman JB, Fox DJ (2010) Gaussian 09, Revision B.01. Gaussian Inc, Wallingford 33. Dennington R, Keith TA, Millam JM (2016) GaussView 6.0. 16.Semichem Inc, Shawnee Mission, KS, USA, pp 143–150
Lee C, Yang W, Parr RG (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37:785–789. https://doi.org/10.1103/PhysRevB.37.785
Benalia A, Boukaoud A, Amrani R, Krid A (2024) A B3LYP-D3 computational study of electronic, structural and torsional dynamic properties of mono-substituted naphthalenes: the effect of the nature and position of substituent. J Mol Model 30:88. https://doi.org/10.1007/s00894-024-05884-6
Ahmad F, Alam MJ, Alam M, Azaz S, Parveen M, Park S, Ahmad S (2018) Synthesis, spectroscopic, computational (DFT/B3LYP), AChE inhibition and antioxidant studies of imidazole derivative. J Mol Struct 1151:327–342. https://doi.org/10.1016/j.molstruc.2017.09.056
Grossman EF, Daramola DA, Botte GG (2021) Comparing B3LYP and B97 Dispersion-corrected Functionals for Studying Adsorption and Vibrational Spectra in Nitrogen Reduction. ChemistryOpen 10:316–326. https://doi.org/10.1002/open.202000158
Dennington R, Keith TA, Millam JM (2016) GaussView 6.0. 16.Semichem Inc, Shawnee Mission, KS, USA, pp 143–150
Materials Studio (2019), Dassault Systemes BIOVIA, San Diego, CA
Lu T, Chen F (2012) Multiwfn: a multifunctional Wavefunction analyzer. J Comput Chem 33:580–592. https://doi.org/10.1002/jcc.2288
Schwieters CD, Clore GM (2001) The VMD-XPLOR visualization package for NMR structure refinement. J Magn Reson 149(2):239–244. https://doi.org/10.1006/jmre.2001.2300
Williams T, Kelley C, Merritt EA, Bersch C, Bröker HB, Campbell J, Cunningham R, Denholm D, Elber E, Fearick R. Gnuplot 5.4. 4: An Interactive Plotting Program; 2022
Chowdhry BZ, Dines TJ, Jabeen S, Withnall R (2008) Vibrational spectra of α-amino acids in the zwitterionic state in aqueous solution and the solid state: DFT calculations and the influence of hydrogen bonding. J Phys Chem A 112:10333–10347. https://doi.org/10.1021/jp8037945
Alam MJ, Ahmad S (2012) Anharmonic vibrational studies of L-aspartic acid using HF and DFT calculations. Spectrochim Acta A 96:992–1004. https://doi.org/10.1016/j.saa.2012.07.135
Tulip PR, Bates SP (2009) First principles determination of structural, electronic and lattice dynamical properties of a model dipeptide molecular crystal. Mol Phys 107:2201–2212. https://doi.org/10.1080/00268970903224955
Silva JB, Echeverry JP, dos Santos RCR et al (2023) Molecular γ-amino butyric acid and its crystals: Structural, electronic and optical properties. J Solid State Chem 321:123900. https://doi.org/10.1016/j.jssc.2023.123900
Caetano EWS, Fulco UL, Albuquerque EL, de Lima Costa AH, Costa SN, Silva AM, Sales FAM, Freire VN (2018) Anhydrous proline crystals: structural optimization, optoelectronic properties, effective masses and Frenkel exciton energy. J Phys Chem Solids 121:36–48. https://doi.org/10.1016/j.jpcs.2018.05.006
Silva AM, Silva BP, Sales FAM, Freire VN, Moreira E, Fulco UL, Albuquerque EL, Maia FF Jr, Caetano EWS (2012) Optical absorption and DFT calculations in L-aspartic acid anhydrous crystals: Charge carrier effective masses point to semiconducting behavior. Phys Rev B 86:195201. https://doi.org/10.1103/PhysRevB.86.195201
Silva BP, Lemes RP, Zanatta G, Rodrigues dos Santos RC, de Lima-Neto P, Caetano EW, Freire VN (2019) Solid state properties of hydroxyurea: Optical absorption measurement and DFT calculations. J Appl Phys 125:134901. https://doi.org/10.1063/1.5068773
Flores MZS, Freire VN, Dos Santos RP, Farias GA, Caetano EWS, De Oliveira MCF, Fernandez JRL, Scolfaro LMR, Bezerra MJB, Oliveira TM, Bezerra GA, Cavada BS, Leite Alves HW (2008) Optical absorption and electronic band structure first-principles calculations of α-glycine crystals. Phys Rev B 77:115104. https://doi.org/10.1103/PhysRevB.77.115104
Mythili P, Kanagasekaran T, Gopalakrishnan R (2007) Investigations on nucleation kinetics, growth and characterization of sulphanilic acid single crystals. Cryst Res Techno 42:791–799. https://doi.org/10.1002/crat.200710907
Sohail U, Ullah F, Mahmood T, Muhammad S, Ayub K (2022) Adsorption of industrial gases (CH4, CO2, and CO) on olympicene: a DFT and CCSD (T) investigation. ACS omega 7:18852–18860. https://doi.org/10.1021/acsomega.2c01796
Boukaoud A, Chiba Y, Dehbaoui M, Guechi N (2019) Vibrational analysis and hydrogen-bonding effects on the vibrational modes of zwitterionic DL-Tryptophan: IR spectroscopy and DFT calculations. Sigma J Eng & Nat Sci 37:1180–1198 (https://dergipark.org.tr/en/download/article-file/2029792)
Jarmelo S, Reva I, Carey PR, Fausto R (2007) Infrared and Raman spectroscopic characterization of the hydrogen-bonding network in L-serine crystal. Vib Spectrosc 43:395–404. https://doi.org/10.1016/j.vibspec.2006.04.025
Pawlukojć A, Hołderna-Natkaniec K, Bator G, Natkaniec I (2014) INS, IR, RAMAN, 1H NMR and DFT investigations on dynamical properties of l-asparagine. Vib Spectrosc 72:1–7. https://doi.org/10.1016/j.vibspec.2014.02.002
Parker SF (2013) Assignment of the vibrational spectrum of L-cysteine. Chem Phys 424:75–79. https://doi.org/10.1016/j.chemphys.2013.04.020
Chesalov YA, Chernobay GB, Boldyreva EV (2008) Temperature effects on the IR spectra of crystalline amino acids, dipeptides, and polyamino acids II. L-and DL-serines. J Struct Chem 49:627–638. https://doi.org/10.1007/s10947-008-0087-3
Uno T, Machida K, Hanai K (1968) Infrared spectra of benzene-and pentadeuterobenzenesulphonyl compounds. Spectrochim Acta A Mol Spectrosc 24:1705–1712. https://doi.org/10.1016/0584-8539(68)80225-9
Magdaline JD, Chithambarathanu T (2015) Vibrational spectra (FT-IR, FT-Raman), NBO and HOMO, LUMO studies of 2-thiophene carboxylic acid based on density functional method. IOSR J Appl Chem 8:6–14.
Boukaoud A, Meinnel J, Boudjada A, Juranyi F, Carlile CJ, Jeannin O (2011) Inelastic neutron scattering of methyl tunnelling in isotopic mixtures of dibromomesitylene. Chem Phys Lett 509:20–24. https://doi.org/10.1016/j.cplett.2011.04.069
Acknowledgements
This work covers a large part of the Algerian PRFU project (2021–2022: No B00L02UN260120220001). The financial support from the Algerian Directorate-General for Scientific Research and Technological Development (DGRSDT) is highly appreciated. The assistance provided by the Algerian Ministry of Higher Education and Scientific Research is also thanked.
The authors would like to thank Mr. Mouloud Sofiane for providing access to his laboratory.
Funding
This work is financed by the Algerian Directorate-General for Scientific Research and Technological Development.
Author information
Authors and Affiliations
Contributions
Y.S. writing and prepared figures; A.B. supervision, writing and Data curation; Y.C. investigation and resources; D.S. investigation and resources. M.A.A. investigation; A.A. investigation. All authors reviewed the manuscript
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary file1 (MP4 3631 KB)
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Slimani, Y., Boukaoud, A., Chiba, Y. et al. Exploration of electronic and vibrational properties of sulfanilic acid through periodic and non-periodic DFT calculations. J Mol Model 30, 121 (2024). https://doi.org/10.1007/s00894-024-05911-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-024-05911-6