
Abstract
Abstract
The morphology of porous silicon (p-Si) depends on several parameters such as the doping type and the carriers’ concentration of the crystalline silicon substrate. The electrolytes used in the p-Si fabrication also have an important role. The final structure determines if p-Si is luminescent or suitable for photonic applications. Experimental results on p-Si produced by electrochemical etching show that although the carriers are greatly reduced by the etching process, boron atoms remain in the bulk. The study of p-type porous silicon nanostructures by means of an ab initio computational simulation might help to understand how boron atoms influence the p-Si final structure. Here, we report electronic and topological properties of ten p-type porous silicon structures as an extension of our previous paper on p-type crystalline silicon. Our results suggest that the boron atoms can not remain bonded on the porous surface but do so in the bulk. The presence of impurities changes the bond distance of their neighbors within a radius of 5 Å. The energy of the models is essentially the same for all the boron positions in the silicon backbone. The high electronic density around the boron impurity could influence the trajectory of an HF ion entering a p-Si pore during the fabrication process.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
We have extensively studied porous silicon, both theoretically [1–3] and experimentally [4, 5], and successfully implemented different applications such as biosensors [6], luminescent structures [7], one-dimensional photonic crystals [5], mirrors for solar concentration devices [8], and filters [5]. These diverse applications are possible in varying the structure of the porous silicon samples by changing the crystalline silicon substrate and the electrochemical etching conditions.
In order to understand the role of boron in the structure of porous silicon (p-Si) fabricated from p-type silicon wafers, we have recently studied some properties of crystalline p-type silicon models by means of ab initio computational simulations [9]. This article is another step in this direction. Here, we simulate ten p-type porous silicon models (PSiHB), with the highest number of silicon atoms used until now on a periodic solid configuration by an ab initio code. Our models incorporate experimental conditions like the concentration of boron atoms (corresponding to the resistivity of the p-type silicon wafers used in the fabrication of p-Si for photonic applications (NR, JdR, and PPM, unpublished work)) and dangling bonds passivated with hydrogen. The technical features of the ab initio software employed in our work and the procedure used to generate the PSiHB models are described in the ‘Methods’ section. In the ‘Results and discussion’ section, we report the total radial distribution functions (RDFs), topological radius influence of boron atoms, plane angle distributions, and relative electronic densities (RelD(E)s) along planes which contain the first Si neighbors of the B impurities.
Our results suggest that boron atoms cannot remain bonded on the surface of the pore but are bound in the bulk. The boron impurities favor the crystallinity of the PSiHB models because they reduce the interatomic distances among second silicon neighbors. Also, we find a large electronic density around the boron atom with a short-range influence of about two atomic layers. We speculate that due to this electronic density, during the electrochemical fabrication of p-Si, the HF ion avoids the boron atoms. This ion movement could determine the p-Si final topology.
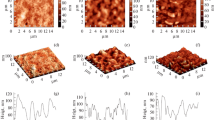
Experimental facts
There are some experimental facts that support our PSiHB theoretical models. It is known that the HF electrochemical attack of the silicon substrate begins at surface irregularities due to a tip-type effect [10]. Then, the porous formation continues vertically due to the presence of holes on the pore tips as an electrical current is applied. In our model, the porous silicon structure remains crystalline, as it happens in real samples (see Figure 1a, b). We use the fact that few boron atoms remain in the p-Si structure after the electrochemical etching of the silicon substrate [11]. Another important fact is that the final topology of the samples strongly depends on the silicon substrate electrical resistivity. In low-resistivity (10−3Ω cm−1) substrates, porous silicon shows a column-like structure, see Figure 2a, and photonic features (NR, JdR, and PPM, unpublished work). On the other hand, high-resistivity (10 Ω cm−1) substrates produce a sponge-like structure with luminescent properties, as can be observed in Figure 2b. Although, in our simulations, we do not include the applied electric field used to produce p-Si by electrochemical etching, it is possible to fabricate p-Si by chemical etching with HF without any electrical current. Figure 3 shows a porous sample obtained by electrochemical etching with 40 mA cm−2 current in only 60 s and then chemically etched by the sample in the HF solution during 68 h without any current. As it can be observed, chemical etching is a very slow process but capable of growing porous in a silicon substrate.

Crystallinity of the p-Si structure. TEM images of a p-Si sample prepared from a boron p-type silicon wafer with a resistivity of 0.001 to 0.005 Ω cm−1. The sample was placed on an amorphous carbon grid for measurement. From (a) the Fourier-filtered diffraction lattice image and (b) the transmission electron diffraction pattern, it can be observed that the p-Si final structure remains crystalline.

Porous silicon topology. Porous silicon produced by electrochemical etching on a silicon substrate in an electrolyte composed of ethanol, HF and glycerol. (a) Low electrical-resistivity substrate (10−3Ω cm−1), (b) high-resistivity substrate (10 Ω cm−1).

Electrochemical etching without an electrical current. Porous silicon produced first by electrochemical etching of a low-resistivity substrate (applying a 40-mA cm−2 current for only 60 s). The sample was chemically etched in HF solution for 68 h without an electrical current. The electrolyte is composed of ethanol, HF and glycerol (volume ratio 7:3:1).
Results and discussion
To analyze the PSiHB models from a topological point of view, we begin by studying their relaxed structures and the behavior of a B atom localized in the bulk (Figure 4b) and on the pore surface of the PSiHB models (Figure 4c). Considering that the length of the Si-B bond is 2.00 [12] or 2.05 Å [13] and that bond lengths Si-H and B-H are 1.66 and 1.20 Å [12], respectively, we can see from the figures that when the B impurity is in the bulk, it is bonded to three Si atoms at less than 2.05 Å, which is in agreement with [12] and [13]. However, when the boron atom is on the pore surface, it has only one bond with a Si atom at 2.05 Å. This suggests that under room temperature conditions, porous silicon will not have boron atoms on the pore surface.

Porous silicon models. (a) Boron positions (magenta) in the PSiHB non-relaxed models. (b) PSiHB-relaxed structure with the boron atom (green) in the bulk and three bonds with the Si atoms 1, 2 and 3. The impurity is no longer bonded with Si atom number 4. (c) PSiHB-relaxed structure with the boron atom (green) on the pore surface. The impurity has just one bond with Si atom number 2.
We also report the superposition of the total RDFs of each PSiHB model and the RDF of the PSiH model (Figure 5a). The RDFs of the PSiHB models present sharper peaks for the first and second silicon neighbors than the PSiH RDF, which indicates that the PSiHB structures are more ordered than the PSiH structure [14]. Between the first and second silicon neighbors of the PSiHB RDFs, a peak appears, unlike the classical three broad peaks of the RDF of the amorphous silicon (a-Si) network [15]. For the PSiHB models, the mean interatomic distance between first silicon neighbors is 2.35 Å, the second silicon neighbors are distributed in two peaks at 3.25 and 3.85 Å, and the third neighbors are at a distance of 4.85 Å. For the PSiH model, its first, second, and third silicon neighbors are at 2.45, 3.85, and 5.05 Å respectively. Now, remembering that the crystalline silicon interatomic distance is 2.35 Å and that its second and third silicon neighbors are at 3.85 and 4.50 Å, respectively, we conclude that the boron impurities of the PSiHB models attract their silicon neighbors within an approximate radius of 5.00 Å, causing a crystalline ordering between silicon first neighbors. This brings some of the second neighbors from 3.85 to 3.25 Å and moves the third neighbors distance from 5.05 to 4.85 Å.

Radial distribution functions. (a) Radial distribution functions of the PSiHB models (red line) compared with the RDF of the hydrogenated porous silicon model (blue line). (b) Boron radii of influence of the PSiHB models (in blue) compared with the atomic coordinate change produced in a p-type silicon crystal structure due to the presence of a boron impurity (red line).
In Figure 5b, we present the superposition of the PSiHB boron radii of influence (rB) defined in the previous section, and we compare it with the effect of one B atom in a p-type silicon crystal. The value of the boron radii of influence is bigger than the 10% for the atoms that are at less than 5 Å from the B impurities. The boron radii of influence coincide with the approximate boron effect observed in the PSiHB RDFs and with the contractions of the first and second Si nearest neighbors toward the substitutional impurity calculated by [13, 16] and observed by [17].
Besides the RDFs, we have obtained the plane angle distributions of the PSiH and PSiHB models with the purpose of understanding the topological B effect in the p-type p-Si structure. The plain angle distribution of the PSiH model is showed in Figure 6a, and the distribution of one of the PSiHB models is presented in Figure 6b. Both distributions are broad and exhibit two peaks around 40° and 110°. The median of the PSiHB and PSiH bond angle distributions is 107.62° and 106.93° respectively. Recalling that the crystalline bond angle is 109.47°, we conclude that for both structures, most of the tetrahedral bonds are a bit distorted from the crystalline value, but their distributions are different from the a-Si ones [18], thus remain crystalline. Because the PSiH and PSiHB are similar, we are not able to distinguish the effect of the B atom in the bond angle distortion.

Plane angle distributions. (a) Plane angle distribution of the PSiH and (b) plane angle distribution of one of the PSiHB models. The plane angle value for the silicon crystal structure is in black. The median and mean of the distributions are in red and magenta, respectively.
We have calculated the PSiHB RelD(E)s and PSiH D(E) on the planes where the B atom and its Si neighbors lie. The PSiH D(E) is presented in Figure 7a. The electronic densities of the silicon atoms are represented as little hills and, as we can notice, there are open spaces because of the pore. The RelD(E) of one of the PSiHB models is presented in Figure 7b; a huge electronic charge appears around the B impurity, and the hills surrounding the B atom represent the redistribution of the electronic density, caused by the impurity present in the plane, beyond their silicon neighbors. The ten PSiHB RelD(E)s are similar; their maximum, minimum, and mean heights are 1.90, 1.61 and 1.75 electrons/Bohr2, respectively. The height of the PSiHB RelD(E) model with the B atom on the pore surface is 1.77 electrons/Bohr2 near the mean value of the RelD(E) height distribution; we have not observed any peculiarity in this RelD(E)s with respect to the RelD(E)s of the rest of the PSiHB models with impurity in the bulk. Considering the plane of the impurity of first neighbors, the presence of the B atom modifies the silicon electronic charge distribution in a range less than two atomic layers; this result agrees with [16].

Electronic densities. (a) PSiH D(E). The hills are the Si atoms electronic densities. (b) RelD(E) of a PSiHB model. The boron impurity is in the bulk and at the center of the plane showed.
Conclusions
We conclude that the presence of boron atoms in the porous silicon samples favors crystallinity since it reduces the interatomic distance among its first silicon neighbors around 10% with respect to pure silicon increasing the short range order, which is observed through the sharp peaks in the RDFs. Also, the plane angle distribution around the impurity is close to the crystalline value. Another interesting result of our simulations is that boron atoms do not remain on the pore surface because of weak bonding to the silicon network. We find that boron atoms present a bigger electronic density than silicon atoms which could indicate that during chemical etching, the HF ions will tend to avoid the impurities at a distance of at least 5 Å (two atomic layers). This might be the reason why p and p+ porous silicon samples present different topologies. When more boron atoms are present, there are not many possible trajectories for the HF and a more regular structure is obtained, while when a small number of impurities are present, the etching could be in many directions, generating a sponge-like structure.
Methods
We generated ten PSiHB models with 255 Si atoms, 154 hydrogen atoms, and one B atom. For nine of the ten models, the B atom is placed on different bulk locations, and for one of the models, B is on the pore surface. The PSiHB models have one central and regular pore along the z-axis of the supercell. The optimized structures were calculated using ABINIT that is an ab initio code based on total energy pseudopotential methods [19]. This code is based on the density functional theory [20, 21] and uses plane waves to expand the electronic wave functions.
To construct the PSiHB models, we followed the next steps:
-
1.
The diamond structure Si cell was replicated four times on each axis to obtain a crystalline supercell with 512 Si atoms, whose edge length is 21.72 Å, with a density of 2.33 g/cm3.
-
2.
The central pore was constructed eliminating 50% of the silicon atoms and with a regular transverse section.
-
3.
The silicon dangling bonds were passivated with hydrogen atoms.
-
4.
For each PSiHB model, one Si atom was substituted by a B one. The position of the impurity is different for each model. Just one of the PSiHB structures has a B atom bonded on the inner pore surface, while the other structures have their impurities in the bulk (Figure 4a). In this step, the B atom is bonded to four atoms, but this is an artificial state because B has just three valence electrons.
-
5.
Once the PSiHB models have been constructed, an optimization of their geometries is implemented in the ABINIT code to obtain the lowest energy configuration. The VMD code was used to visualize the resulting optimized structures.
-
6.
The Si and H atoms were simulated with a local type Troullier-Martins pseudopotential; the B atoms, with Troullier-Martins-Fermi. The exchange-correlation energy functional used for the local density approximation was the one proposed by Goedecker et al. [22].
-
7.
For geometry optimization, a cutoff energy radius of 10 Ha and a tolerance force of 5×10−5 Ha/Bohr were used.
-
8.
To reproduce the doping level of the experimental p-type silicon wafers, the PSiHB resistivity (ρ) was set as ρ=1×10−3 Ω cm−1 corresponding to a 1×1020 carriers/cm3 concentration [23].
We are interested in the changes on the topology and electronic densities due to the presence of a B atom impurity in the PSiHB models. We have obtained their RDFs with the VMD code and plane angles analyzing the ABINIT files. We define a boron radius of influence (rB) as follows:
where rPSiH and rPSiHB are the distance vectors of each Si atom from the B impurity, at the origin of the coordinate system, on a pure porous silicon structure passivated with hydrogen (PSiH) and on a PSiHB structure, respectively. To study the electronic behavior around the B impurity, we calculate the relative electronic densities (RelD(E)) of the PSiHB structures as follows:
where D(E)PSiHB is the electronic density of a PSiHB model, and D(E)PSiH is the electronic density of a pure porous silicon model passivated with hydrogen. We have obtained the RelD(E) of each PSiHB model on a plane determined by a B atom and two of its first Si atom neighbors. In order to visualize the local changes around the boron, we consider rB > 1% and RelD(E) > 0.01 electrons/Bohr2.
References
Lugo JE, del Río JA, Martínez JT: Influence of surface coverage on the effective optical properties of porous silicon modeled as a Si-wire array. J. Appl. Phys 1997, 81: 1923–1928. 10.1063/1.364047
Tagüeña-Martínez J, Rubo YG, Cruz M, Beltran MR Wang: Tight-binding description of disordered nanostructures: an application to porous silicon. Appl. Surf. Sci 1998, 142: 564–568.
Cruz M, Yuri R, Rubo G, CWJTnM Beltrán: PRB: Supercell approach to the optical properties of porous silicon. Phys. Rev. B 1999, 59: 1–15387. 10.1103/PhysRevB.59.1
Agarwal V, Malpuech G, Zamfirescu M, Kavokin A, Coquillat D, Scalbert D, Vladimirova M, Gil: Photon bloch oscillations in porous silicon optical superlattices. Phys Rev. Lett 2004, 92: 097401–097406.
Agarwal V, del Río J: Filters, mirrors and microcavities from porous silicon. Int. J. Mod. Phys. B 2006, 20: 99–110. 10.1142/S0217979206033115
Palestino AG, del Río JA, Gergely C, de la Mora MB, Pérez E: Three-dimensional spatial resolution of the nonlinear photoemission from biofunctionalized porous silicon microcavity. Appl. Phys. Lett 2007, 91: 091–1219093.
Nava R: Assisted electrochemical etching of photonic luminescent multilayers of porous silicon. J. Phys. D: Appl. Phys 2010, 43: 455102–455107. 10.1088/0022-3727/43/45/455102
de la Mora MB, Nava R, Tagüeña-Martínez J, del Río JA, Jaramillo OA: Viability study of porous silicon photonic mirrors as secondary reflectors for solar concentration systems. Sol. Energy Mater. Sol. Cells 2009, 93: 1218–1224. 10.1016/j.solmat.2009.01.007
Loustau ERL, del Río JA, Tagüeña-Martínez J, Sansores LE, Nava R, de la Mora M B: Ab initio simulation of p-type silicon crystals. Solid State Commun 2012, 152: 1619–1624. 10.1016/j.ssc.2012.04.072
Lehmann V, Föll H: Formation Mechanism and Properties of Electrochemically Etched Trenches in n-Type Silicon. J. Electrochem. Soc 1990, 137: 653–659. 10.1149/1.2086525
Polisski G, Kovalev D, GDTSFK: Boron in mesoporous Si — Where have all the carriers gone? Physica B 1999, 273–274: 951–954.
Gilbert J, Mains Bock C W, Trachtman M: Theoretical structures for boron-silicon fluorohydrides. J. Phys. Chem 1989, 93: 1745–1752. 10.1021/j100342a016
Fritsch J, Scmidt KE, Page JB, Adams GB: First-principles local-orbital study of the boron-induced reconstruction of Si(001). Phys. Rev. B 1998, 57: 9745–9756. 10.1103/PhysRevB.57.9745
Loustau ERL: Imulación ab initio del silicio amorfo poroso Saarbrücken, Germany. Española: Editoral Académica; 2012.
Alvarez F, Diaz CC, Valladares AA: Radial distribution functions of ab initio generated amorphous covalent networks. Phys. Rev. B 2002, 65: 08–113112.
Chang J, Stott MJ: Si(001/B) surface reconstruction. Phys Rev. B 1996, 53: 0–13704.
Wang Y, Hammers RJ, Kaxiras E: Atomic structure and bonding of boron-induced reconstructions on Si(001). Phys. Rev. Lett 1995,74(3):403–406. 10.1103/PhysRevLett.74.403
Alvarez F, Valladares AA: Ab initio generation of amorphous semiconducting structures. The case of a-Si. Boron in mesoporous 2002, 299: 259–264.
Payne MC, Teter MP, Joannopoulos JD, DCATAA: Iterative minimization techniques for ab initio total-energy calculations: molecular dynamics and conjugate gradients. Rev. Mod. Phys 1992, 64: 1045–1096. 10.1103/RevModPhys.64.1045
Hohenberg P, Kohn W: Inhomogeneous electron gas. Phys. Rev. B 1964, 136: 864–871. 10.1103/PhysRev.136.B864
Kohn W, Sham LJ: Self-consistent equations including exchange and correlation effects. Phys. Rev. A 1965, 140: 1133–1138.
Goedecker S, Teter JHM: Separable dual-space Gaussian pseudopotentials. Phys. Rev. B 1996, 54: 1703–1710. 10.1103/PhysRevB.54.1703
Thurber WR, Mattis RL, Filliben J, YML: Resistivity-dopant density relationship for phosphorus-doped silicon Solid State Science and Technology. J. Electrochem. Soc 1980, 127: 2291–2294. 10.1149/1.2129394
Acknowledgements
The authors thank Dr. Maria Beatriz de la Mora and Orlando Hernández Cristobal for the SEM and TEM images. Support from the National University of México (UNAM) through a Posdoctoral Grant to E. R. L. Loustau is acknowledged. We thank the Computing and Information Technology Division of UNAM and the National Supercomputing Center (CNS) for the computer resources. This work was supported in part by DGAPA-UNAM under grant PAPIIT IN109812.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
ERLL carried out the computational simulations and result analysis and participated in the manuscript elaboration. JAdR and JT coordinated the project, discussed the results, and helped to draft the manuscript. LES examined the results and computational simulation methodology and helped to draft the manuscript. RN carried out the experiments, obtained most of the experimental images, and participated to draft the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Landa Loustau, E.R., del Río, J.A., Tagüeña-Martínez, J. et al. Ab initio simulations of p-type porous silicon nanostructures. J Nanostruct Chem 3, 21 (2013). https://doi.org/10.1186/2193-8865-3-21
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2193-8865-3-21