
Abstract
The excitatory amino acid transporters (EAATs) are transmembrane proteins responsible for the uptake of (S)-glutamate from the synaptic cleft. To date, five subtypes EAAT1-5 have been identified for which selective inhibitors have been discovered for EAAT1 and EAAT2. By screening of a commercially available compound library consisting of 4,000 compounds, N-acyl-N-phenylpiperazine analog (±)- exo -1 was identified to be a non-selective inhibitor at EAAT1-3 displaying IC50 values in the mid-micromolar range (10 μ M, 40 μ M and 30 μ M at EAAT1, 2 and 3, respectively). Subsequently, we designed and synthesized a series of analogs to explore the structure-activity-relationship of this scaffold in the search for analogs characterized by increased inhibitory potency and/or EAAT subtype selectivity. Despite extensive efforts, all analogs of (±)- exo -1 proved to be either inactive or to have least 3-fold lower inhibitory potency than the lead, and furthermore none of the active analogs displayed selectivity for a particular subtype amongst the EAAT1-3. On the basis of our findings, we speculate that (±)- exo -1 binds to a recess (deepening) on the EAAT proteins than a well-defined pocket.
Similar content being viewed by others


Background
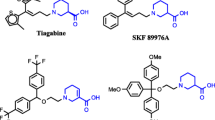
In the central nervous system (CNS), the excitatory amino acid transporters (EAATs) are transmembrane proteins responsible for the uptake of (S)-glutamate (Glu) from the synaptic cleft. Five subtypes have been identified, named EAAT1–EAAT5 in humans and GLAST, GLT-1, EAAC1, EAAT4 and EAAT5, respectively, in rodents. (Bunch et al. 2009) While EAAT5 is found exclusively in the retina, subtypes EAAT1–4 are expressed differentially within the CNS with respect to brain regions as well as at the cellular level: EAAT1 and EAAT2 are expressed primarily on astrocytes, but EAAT2 is also found in neurons, astrocytes and oligodendrocytes. (Lauriat et al. 2007) Subtype EAAT3 is distributed predominantly in postsynaptic neuronal sites, (Nieoullon et al. 2006) whereas EAAT4 is distributed in Purkinje cells as well as in the cerebral cortex. (Massie et al. 2001) Discovery of subtype selective ligands for the EAATs has attracted much attention over the past decade, (Jensen et al. 2009) the latest being the disclosure of UCPH-101 as first subtype selective EAAT1-inhibitor (Figure 1). (Jensen et al. 2009; Erichsen et al. 2010; Huynh et al. 2012,ab).

Results and discussion
From screening of a 4,000 compounds-library at HEK293 cells stably expressing human EAAT1-3 N-acyl-N-phenylpiperazine analog (±)- exo -1 was identified as a non-selective inhibitor at the transporter exhibiting IC50 values in the mid-micromolar range (10 μ M, 40 μ M and 30 μ M at EAAT1, -2 and -3, respectively, Figure 2).

Chemical structure of screening hit N -acyl- N -phenylpiperazine (±)-exo-1 displaying inhibitory activity at EAAT1-3 in the mid-micromolar range (IC 50 = 10, 40 and 30 μ M, respectively).
Although phenylpiperazines are promiscuous hits in high-throughput screenings (HTS) and a frequent core skeleton in marketed drugs, (Millan et al. 2001; Fragasso et al. 2006; Weisberg et al. 2007) we were motivated to explore the structure-activity-relationship (SAR) of this new class of EAAT inhibitors. A conventional medicinal chemistry analysis of (±)-exo-1 suggests that the amide functionality, the aniline nitrogen, the phenyl ring and the trifluoromethyl group may play key roles in binding of this class of EAAT inhibitors. Consequently, the chemical structure can be broken down into three fragments: a core skeleton being the acyl-phenylpiperazine scaffold and two substituents being the trifluoromethyl- and the bicyclo[2.2.1]heptanyl group (Figure 1). The SAR study was designed as to study the influence of one of the two substituents (Figure 1) individually including the stereochemical organization around the α-carbonyl carbon.
The SAR study commenced by investigation of the influence of bicycle[2.2.1]heptanyl group on the EAAT inhibitory activity. The stereochemical configuration of the α-cabonyl carbon was addressed by the synthesis of endo-conformer (±)-endo-1 (Table 1) from commercially available (±)-endo-carboxylic acid and N-(3-trifluoromethylphenyl)-piperazine 4, the latter prepared by a palladium-catalyzed amination of commercially available piperazine (Scheme 1). (Nishiyama et al. 1998) Furthermore, the racemic and diastereomeric mixture (±)-endo-exo-1 was prepared from the corresponding acid (±)-endo-exo-6 obtained from oxidation of commercially available (±)-endo-exo-bicyclo[2.2.1]heptanylmethyl alcohol ((±)-endo-exo-5) (Scheme 2) using KMnO4 and K2CO3 in H2O. Isolation of (±)-endo-exo-6 turned out to be difficult for which reason it was used directly in the next step. (Gudipati et al. 1993) To search for the optimal bulkiness of the lipophilic substituent, larger as well as smaller rigid hydrophobic ring-systems were introduced (2.1-2.5). Moreover, analogs 2.6-2.10 comprising alkyl group of varying length and bulkiness were designed to explore the effect of increased flexibility of this substituent on ligand binding. Furthermore, analogs 2.11–2.17 address if the bicyclo[2.2.1]heptane could be substituted for an aromatic moiety, whereas the analogs 2.18–2.21 were designed to explore the distinct substitution for a hydrophobic group. The synthesis of piperazine analogs 1 and 2.1-2.21 was carried out by amidation of 4 using the respective carboxylic acids, acid chlorides, benzenesulfonyl chloride and benzyl carbonochloridate afforded the corresponding amides in moderate to good yields (Scheme 1). The rationally designed 3-trifluoromethylphenylpiperazine analogs were supplemented by commercially available analogs 2.22–2.31, as a quick way of expanding the SAR into the chemical space beyond rational guidance. Finally, the importance of the amide functionality was explored by the synthesis of carbamate 2.32 by acylation of 4 with carbonochloridate, sulfonamide 2.33 by treatment of 4 with phenylsulfonyl chloride, amine 2.34 by reduction of (±)-endo-exo-1 with LiAlH4, (Cook et al. 1992) and N-benzyl analog 2.35. by alkylation of phenylpiperazine 4 (Burkhard et al. 2010) In addition, these analogs were supplemented by five commercially available structurally diverse analogs 2.36–2.40.

Synthesis of phenylpiperazine 4 and analogs 1, 2.1-2.21 and 2.33-2.35 a.
Synthesis of phenylpiperazine 4 and analogs 1, 2.1-2.21 and 2.33-2.35 a . aReagents and conditions: (a) 3-Trifluoromethylphenylbromide, Pd(OAc)2, P(tBu)3, NaOtBu, o-xylene, 120°C, 17 h, 77%. (b) Appropriate carboxylic acid, TBTU, DIPEA, dry DMF, rt, 20 h, 51-92%. (c) Appropriate acid chloride (except for the synthesis of 2.32: benzyl carbonochloridate), Et3N, dry dichloromethane, rt, 30 min, 71-78%. (d) Benzenesulfonyl chloride, Et3N, dry dichloromethane, rt, 30 min, 71%. (e) BnBr, Et3N, dry dichloromethane, rt, 24 h, 57%. (f) Starting from (±)-endo-exo-1: LiAlH4, THF, rt, 72 h, 71%.

Synthesis of piperazine analog (±)- endo-exo -3.2 a . a Reagents and conditions: (a) KMnO4, K2CO3, H2O, rt, 17 h. (b) Piperazine, DIPEA, TBTU, DMF, rt, 24 h, 49%. (c) 1-Bromo-4-(trifluoromethyl)benzene, Pd(OAc)2, P(tBu)3, NaOtBu, dry o-xylene, 120°C, 24 h, 56%.
We then turned to the design of analogs for investigation of the influence on EAAT inhibitory activity of the chemical nature of the trifluoromethyl group as well as its position on the phenyl ring. A series of 12 analogs were included in the SAR study, all wherein the (±)-endo-exo-bicyclic[2.2.1]-acyl group was conserved (analogs 3.1-3.12, Table 2): Simplification of the chemical structure by depletion of the trifluoromethyl group provides analog (±)-endo-exo-3.1, while shifting the 3-trifluoromethyl group to the 4- or 2-positions affords analogs (±)-endo-exo-3.2 and (±)-endo-exo-3.5, respectively (Table 2 and Scheme 2). The latter two analogs were supplemented by commercially available analogs (±)-endo-exo- 3.3, (±)-endo-exo-3.4 and (±)-endo-exo-3.6. Continuing the design stage, substitution of the 3-trifluoromethyl group for a chloride, hydroxyl-, cyano- and methoxy group, provides analogs (±)-endo-exo-3.7-3.10 respectively (Table 2), whereas 2,4-difluorophenyl analog (±)-endo-exo-3.11 was included due to readily available starting materials. Analog (±)-endo-exo-3.12 could be obtained from commercial suppliers and thus included with the notion that it comprises an N-diphenylmethyl group, which is indeed chemically distinct from the N-3-trifluoromethylphenyl group and furthermore the connecting nitrogen will be protonated at physiological pH=7.4. The analogs were synthesized starting from the appropriate phenylpiperazine and (±)-endo-exo-6 under standard coupling conditions (TBTU, DIPEA in DMF) for the amide formation to afford the target compounds in moderate yields (Scheme 3) (Balalaie et al. 2007).

Synthesis of piperazine analogs 3.1, 3.5, 3.8-3.11 by use of the coupling reagent TBTU a . a Reagents and conditions: (a) TBTU, DIPEA, dry DMF, rt, 20 h, 40-58%.
Pharmacological characterization
In total, 54 piperazine analogs 2.1-2.40 and 3.1-3.12 were characterized pharmacologically at stable HEK293 cells expressing human EAAT1-3 in a [3H]-D-aspartate uptake assay, (Jensen & Bräuner-Osborne 2004) and the results are summarized in Table 1 and Table 2. The endo-isomer 1 (endo-isomer) displayed inhibitory activities at EAAT1-3 comparable with those of the exo-isomer 1 (lead structure) (IC50 = 14 μ M, 32 μ M and 10 μ M vs. 10 μ M, 40 μ M and 30 μ M, respectively). In line with this, endo-exo 1, which is a 1:1 ratio of endo/exo moiety displayed IC50 values at EAAT1-3 of IC50 = 14 μ M, 42 μ M and 14 μ M, respectively. Usually, such findings would lead to the conclusion that the bicyclo-[2.2.1]-heptanyl group occupies a promiscuous hydrophobic pocket, which could be optimized for increased potency. However, upon increasing or decreasing the hydrophobic bulk and/or flexibility, a clear drop in potency was observed (analogs 2.1-2.17, Table 1). Except for analogs 2.3 and 2.11, which displayed only a 5-15 fold drop in inhibitory potency across the subtypes, all of these analogs would be characterized as inactive (IC50 >100 μ M or >300 μ M, Table 1). These findings could open up for the hypothesis that the pocket is indeed not hydrophobic but instead hydrophilic in nature. Upon binding of the hydrophobic alkane group, water molecules are forced out and ligand binding is entropically driven rather than enthalpically. Thus analogs 2.18-2.31, which comprise a hydrophobic group, could be potential inhibitors. However, none of these displayed any inhibitory activity at EAAT1-3 (IC50 >100, >300 or >1000 μ M, Table 1). Continuing the characterization, neither the carbamate 2.32 nor the sulfonamide 2.33 analog displayed inhibitory activity at EAAT1-3, and likewise all amines 2.34-2.40 were found to be inactive at all three subtypes.
The pharmacological results for the twelve analogs 3.1-3.12, which address the influence of substituent 2 (Figure 2) on inhibitory activity at EAAT1-3 are summarized in Table 2. While it was not surprising that removal or repositioning of the 3-trifluoromethyl group (analog 3.1, 3.2 and 3.5, respectively) resulted in loss of inhibitory activity, the further nine analogs 3.3, 3.4, 3.6-3.12 were also inactive or displayed at least a 3-fold lower inhibitory activity than (±)- exo -1.
Conclusion
In conclusion, screening of a compound library identified (±)-exo -1 as a broad range EAAT1-3 inhibitor exhibiting IC50 values at the three transporters in the mid-micromolar range. Subsequently, rational design and synthesis of 33 analogs of (±)-exo -1 was carried out, together with the purchase of 21 analogs. Thus, a total of 54 piperazine analogs were characterized pharmacologically as inhibitors at EAAT1-3 but only the endo diastereomer (±)-endo -1 displayed inhibitory potency in the mid-micromolar range comparable to that of the lead structure (±)-exo -1. The remaining analogs were inactive or at least three fold weaker inhibitors at EAAT1-3 than the lead, none of them displaying signs of subtype-selectivity. Given the structural diversity of the analogs characterized pharmacologically, we speculate if the lead structure (±)-exo -1 adheres to a recess (deepening) in the surface of the protein rather than binds in an organized way to a well-defined pocket.
Experimental section
All commercially available reagents were used without further purification. THF was distilled over sodium/benzophenone, Et2O was dried over neatly cut sodium and dichloromethane was dried over 3 Å molecular sieves. All solvents were tested for water content using a Karl Fisher apparatus. All reactions involving dry solvents or sensitive agents were performed under a nitrogen atmosphere, and glassware was dried prior to use. All reactions were monitored by analytical thin-layer chromatography (TLC, Merck silica gel 60 F254 aluminum sheets). Flash chromatography was carried out using Merck silica gel 60A (35-70 micron). 1H NMR spectra were recorded on a 300 MHz Varian Mercury 300BB or a 400 MHz Avance Bruker and 13C NMR spectra on a 75 MHz Varian Gemini 2000BB or a 100 MHz Avance Bruker. Preparative HPLC was done using Agilent Prep HPLC systems with Agilent 1100 series pump, Agilent 1200 series diode array, multiple wavelength detector (G1365B), and Agilent PrepHT High Performance Preparative Cartridge Column (Zorbax, 300 SB-C18 Prep HT, 21.2 × 250 mm, 7 μ m). LC-MS spectra were recorded using an Agilent 1200 series solvent delivery system equipped with an autoinjector coupled to an Agilent 6400 series triple quadrupole mass spectrometer equipped with an electrospray ionization source. Gradients of 5% aqueous MeCN + 0.05% HCOOH (eluent A), and 95% aqueous MeCN + 0.043% formic acid (eluent B) were employed. Melting points were measured using a MPA 100 Optimelt automatic melting point system and are stated uncorrected. Compounds were dry either under high vacuum or freeze dried using a Holm & Halby, Heto LyoPro 6000 freezedrier.
General procedure a: synthesis of amides using O-benzotriazole-1-yl-N,N,N',N'-tetramethyluronium tetrafluoroborate (TBTU) as coupling reagent
To a suspension of the appropriate phenylpiperazine analog (0.33 mmol), the carboxylic acid (0.40 mmol) and TBTU (0.43 mmol) in dry DMF (4 mL) under an N2 atmosphere, was added DIPEA (1.32 mmol) and reaction mixture was stirred for 20 h at rt. The reaction mixture was quenched with brine (5 mL) and extracted with dichloromethane (3 × 20 mL). The combined organic phases were washed with H2O (20 mL) and brine (20 mL) and dried over anhydrous Na2SO4. After concentration in vacuo, the crude product was purified by column chromatography on silica gel in accordance with details described for the analog.
General procedure B: synthesis of amides using acid chlorides
To a suspension of 1-(3-(trifluoromethyl)phenyl)piperazine (4) (0.33 mmol) in dry dichloromethane (5 mL) at 0°C under a N2 atmosphere was added Et3N (0.91 mmol). The reaction mixture was stirred for 10 min at 0°C, then the appropriate acid chloride (0.48 mmol) was added and stirring continued for 30 minutes at rt. The reaction mixture was quenched with sat. NH4Cl (5 mL) and extracted with dichloromethane (3 × 20 mL). The combined organic phases were washed with H2O (20 mL) and brine (20 mL) and dried over Na2SO4. After concentration in vacuo, the crude product was purified by column chromatography on silica gel in accordance with details described for the analog.
(±)-endo- Bicyclo[2.2.1]heptan-2-yl(4-(3-(trifluoromethyl)phenyl)piperazin-1-yl)methanone ((±)-endo- 1)
Obtained from 1-(3-(trifluoromethyl)phenyl)piperazine (4) and commercially available (±)-endo-bicyclo[2.2.1]heptane-2-carboxylic acid by general procedure A in 61% yield as a pale-yellow oil. R f 0.25 (heptane/EtOAc 3:1). 1H NMR (300 MHz, CDCl3) δ 7.36 (t, J = 8.4 Hz, 1H), 7.12-7.04 (m, 3H), 3.94-3.89 (m, 1H), 3.79-3.62 (m, 3H), 3.30-3.08 (m, 4H), 2.95 (dt, J = 10.8, 4.2 Hz, 1H), 2.40 (br s, 1H), 2.29 (br s, 1H), 1.95 (ddd, J = 12.0, 4.5, 2.4 Hz, 1H), 1.64-1.28 (m, 7H). 13C NMR (75 MHz, CDCl3) δ 172.3, 151.1, 131.5 (q, J = 31.5 Hz), 129.7, 124.2 (q, J = 270.7 Hz), 119.3, 116.5 (q, J = 3.8 Hz), 112.6 (q, J = 3.8 Hz), 49.5, 45.2, 43.7, 41.7, 40.8, 40.5, 40.2, 37.1, 32.2, 29.1, 21.1. LC-MS (m/z) calcd for C19H23F3N2O [M+H+], 353.2; found, 353.2. Anal. calcd for C19H23F3N2O × 1HCl: C 58.69, H 6.22, N 7.20 found C 59.28, H 6.24, N 7.18.
(±)-endo- exo-Bicyclo[2.2.1]heptan-2-yl(4-(3-(trifluoromethyl)phenyl)piperazin-1-yl)methanone ((±)-endo-exo- 1)
Prepared from 1-(3-(trifluoromethyl)phenyl)piperazine (4) and (±)-endo-exo- bicyclo[2.2.1]heptane-2-carboxylic acid ((±)-endo-exo-(6)) by general procedure A in 36% yield as a pale-yellow oil. R f 0.50 (heptane/EtOAc 1:1). 1H NMR (300 MHz, CDCl3) δ 7.38-7.33 (m, 1H), 7.12-7.02 (m, 3H), 3.98-3.96 (m, 1H), 3.75 (br s, 2H), 3.67 (br s, 2H), 3.21 (br s, 4H), 2.98-2.92 (m, 0.6H), 2.40 (br s, 1.4H), 2.29 (br s, 1H), 1.95-1.92 (m, 1H), 1.62-1.25 (m, 6H). 13C NMR (75 MHz, CDCl3) δ 173.1, 151.3, 131.5 (q, J = 31.5 Hz), 129.7, 124.2 (q, J = 270.7 Hz), 119.2, 116.5 (q, J = 3.8 Hz), 112.7 (q, J = 3.8 Hz), 50.2, 49.8, 45.2, 43.7, 41.7, 40.8, 40.4, 36.7, 32.3, 28.9, 21.9. LC-MS (m/z) calcd for C19H23F3N2O [M+H+], 353.2; found, 353.4. HPLC: purity254 > 99%.
Bicyclo[2.2.2]octan-2-yl(4-(3-(trifluoromethyl)phenyl)piperazin-1-yl)methanone (2.1)
Obtained from 1-(3-(trifluoromethyl)phenyl)piperazine (4) and racemic (±)-endo-exo-bicyclo[2.2.2]octane-2-carboxylic acid ((±)-endo-exo-(6)) by general procedure A in 59% yield as a pale-yellow oil. R f 0.95 (1:1 heptane/EtOAc). 1H NMR (400 MHz, CDCl3) δ 7.12 (t, J = 6.0 Hz, 1H), 6.50 (dd, J = 6.0, 1.0 Hz, 1H), 6.42-6.35 (m, 2H), 3.82 (br s, 2H), 3.64 (br s, 2H), 3.32 (br s, 4H), 2.79-2.76 (m, 1H), 2.19-2.16 (m, 1H), 1.79-1.73 (m, 1H), 1.69-1.18 (m, 10H). 13C NMR (100 MHz, CDCl3) δ 174.0, 151.2, 131.5 (q, J = 31.5 Hz), 129.7, 125.6 (q, J = 270.7 Hz), 119.3, 116.5 (q, J = 3.8 Hz), 112.6 (q, J = 3.8 Hz), 49.2, 45.2, 41.6, 38.8, 28.4, 27.6, 26.5, 25.3, 25.2, 23.9, 21.5. LC-MS (m/z) calcd for C20H25F3N2O [M+H+], 366.2; found, 366.2. HPLC: purity254 > 98%.
Adamantan-1-yl(4-(3-(trifluoromethyl)phenyl)piperazin-1-yl)methanone (2.2)
Prepared from 1-(3-(trifluoromethyl)phenyl)piperazine (4) and 1-adamantanecarboxylic acid by general procedure A in 73% yield as a pale-yellow oil. R f 0.68 (100% EtOAc). 1H NMR (300 MHz, CDCl3) δ 7.39-7.32 (m, 1H), 7.12-7.04 (m, 3H), 3.87 (dd, J = 6.0, 3.0 Hz, 4H), 3.21 (dd, J = 6.0, 3.0 Hz, 4H), 2.06-2.03 (m, 9H), 1.70-1.77 (m, 6H). 13C NMR (75 MHz, CDCl3) δ 176.2, 151.5, 131.1 (q, J = 31.5 Hz), 130.1, 127.2 (q, J = 270.8 Hz), 119.4, 116.9 (q, J = 3.8 Hz), 112.8 (q, J = 3.8 Hz), 55.2, 49.7, 49.6, 45.4, 43.9, 42.1, 39.5, 37.1, 28.9, 19.1. LC-MS (m/z) calcd for C22H27F3N2O [M+H+], 393.2; found, 393.2. Anal. calcd for C22H27F3N2O × 1HCl: C 61.61, H 6.58, N 6.53 found C 61.38, H 6.38, N 6.38.
Cyclohexyl(4-(3-(trifluoromethyl)phenyl)piperazin-1-yl)methanone (2.3)
Prepared from 1-(3-(trifluoromethyl)phenyl)piperazine (4) and cyclohexanecarboxylic acid by general procedure A in 59% yield as a yellow oil. R f 0.15 (heptane/EtOAc 3:1). 1H NMR (300 MHz, CDCl3) δ 7.35 (t, J = 7.8 Hz, 1H), 7.12-7.03 (m, 3H), 3.78 (br s, 2H), 3.67 (br s, 2H), 3.21 (br s, 4H), 2.50 (tt, J = 11.4, 3.3 Hz, 1H), 1.83-1.69 (m, 5H), 1.55 (dq, J = 11.4, 3.9 Hz, 2H), 1.35-1.23 (m, 3H). 13C NMR (75 MHz, CDCl3) δ 175.0, 151.5, 131.9 (q, J = 31.5 Hz), 130.1, 124.6 (q, J = 270.8 Hz) 119.7 (q, J = 1.5 Hz), 117.0 (q, J = 3.8 Hz), 113.0 (q, J = 3.8 Hz), 49.8, 49.5, 45.1, 40.9, 29.8, 26.3. LC-MS (m/z) calcd for C18H23F3N2O [M+H+], 341.1; found, 341.1. Anal. calcd for C18H23F3N3O × 1HCl: C 57.37, H 6.42, N 7.43 found C 57.38, H 6.20, N 7.38.
Cyclopentyl(4-(3-(trifluoromethyl)phenyl)piperazin-1-yl)methanone (2.4)
Prepared from 1-(3-(trifluoromethyl)phenyl)piperazine (4) and cyclopentanecarbonyl chloride by general procedure B in 85% yield as a yellow oil. R f 0.50 (heptane/EtOAc 1:1). 1H NMR (400 MHz, CDCl3) δ 7.37 (t, J = 8.0 Hz, 1H), 7.13-7.06 (m, 3H), 3.80 (dd, J = 8.0, 4.0 Hz, 2H), 3.70 (dd, J = 8.0, 4.0 Hz, 2H), 3.24 (dd, J = 8.0, 4.0 Hz, 2H ), 3.19 (dd, J = 8.0, 4.0 Hz, 2H), 1.92-1.54 (m, 9H). 13C NMR (100 MHz, CDCl3) δ 176.1, 147.0, 134.2 (q, J = 31.5 Hz), 132.4, 125.0 (q, J = 270.7 Hz), 124.9, 118.1 (q, J = 3.8 Hz), 117.6 (q, J = 3.8 Hz), 54.7, 43.1, 42.4, 31.6, 27.5. LC-MS (m/z) calcd for C17H21F3N2O [M+H+], 327.2; found, 327.2. HPLC: purity254 > 97%.
Cyclopropyl(4-(3-(trifluoromethyl)phenyl)piperazin-1-yl)methanone (2.5)
Prepared from 1-(3-(trifluoromethyl)phenyl)piperazine (4) and cyclopropanecarboxylic acid by general procedure A in 51% yield as a yellow oil. R f 0.55 (100% EtOAc). 1H NMR (300 MHz, CDCl3) δ 7.39-7.32 (m, 1H), 7.12-7.05 (m, 3H), 3.83 (br s, 4H), 3.26 (br s, 2H), 3.24 (br s, 2H), 1.78 (m, 1H), 1.05-1.00 (m, 2H), 0.84-0.78 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 172.5, 151.5, 131.9 (q, J = 31.5 Hz), 130.1, 127.5 (q, J = 270.8 Hz), 119.6 (q, J = 0.8 Hz), 116.9 (q, J = 3.8 Hz), 113.0 (q, J = 3.8 Hz), 49.5, 49.3, 45.6, 42.2, 11.4, 8.0. LC-MS (m/z) calcd for C15H17F3N2O [M+H+], 299.1; found, 299.1. Anal. calcd for C15H17F3N2O × 1HCl: C 53.82, H 5.42, N 8.37 found C 56.15, H 5.15, N 8.17.
2,2-Dimethyl-1-(4-(3-(trifluoromethyl)phenyl)piperazin-1-yl)propan-1-one (2.6)
Prepared from 1-(3-(trifluoromethyl)phenyl)piperazine (4) and pivaloyl chloride by general procedure B in 78% yield as a clear oil. R f 0.28 (heptane/EtOAc 3:1). 1H NMR (300 MHz, CDCl3) δ 7.35 (dt, J = 8.1, 0.9 Hz, 1H), 7.11-7.03 (m, 3H), 3.81 (dd, J = 5.1, 5.1 Hz, 4H), 3.21 (dd, J = 5.1, 5.1 Hz, 4H), 1.32 (s, 9H). 13C NMR (75 MHz, CDCl3) δ 176.4, 151.1, 131.5 (q, J = 31.5 Hz), 129.7, 124.2 (q, J = 270.8 Hz), 119.1 (q, J = 1.5 Hz), 116.5 (q, J = 3.8 Hz), 112.4 (q, J = 3.8 Hz), 49.0, 44.8, 38.7, 28.4. LC-MS (m/z) calcd for C16H21F3N2O [M+H+], 315.2; found, 315.2. Anal. calcd for C16H21F3N2O × 1HCl: C 55.11, H 6.21, N 7.96 found C 54.78, H 6.32, N 7.99.
1-(4-(3-(Trifluoromethyl)phenyl)piperazin-1-yl)ethanone (2.7)
Obtained from 1-(3-(trifluoromethyl)phenyl)piperazine (4) and acetic acid by general procedure A in 64% yield as a yellow oil. R f 0.22 (100% EtOAc). 1H NMR (300 MHz, CDCl3) δ 7.36 (t, J = 9.0 Hz, 1H), 7.13-7.04 (m, 3H), 3.78 (dd, J = 6.0, 3.0 Hz, 2H), 3.64 (dd, J = 6.0, 3.0 Hz, 2H), 3.24 (dd, J = 6.0, 3.0 Hz, 2H), 3.20(dd, J = 6.0, 3.0 Hz, 2H), 2.15 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 169.4, 151.4, 131.9 (q, J = 31.5 Hz), 130.1, 124.6 (q, J = 270.8 Hz), 119.7 (q, J = 1.5 Hz), 117.1 (q, J = 3.8 Hz), 113.1 (q, J = 3.8 Hz), 49.5, 49.3, 46.4, 41.6, 21.8. LC-MS (m/z) calcd for C13H15F3N2O [M+H+], 273.1; found, 273.1. Anal. calcd for C13H15F3N2O × 1HCl: C 50.58, H 5.22, N 9.07 found C 50.73, H 5.02, N 8.74.
3-Methyl-1-(4-(3-(trifluoromethyl)phenyl)piperazin-1-yl)butan-1-one (2.8)
Prepared from 1-(3-(trifluoromethyl)phenyl)piperazine (4) and isovaleryl chloride by general procedure B in 59% yield as a pale-yellow oil. R f 0.40 (heptane/EtOAc 1:1). 1H NMR (400 MHz, CDCl3) δ 7.39 (t, J = 6.9 Hz, 1H), 7.16-7.10 (m, 3H), 3.82 (dd, J = 6.0, 3.0 Hz, 2H), 3.68 (dd, J = 6.0, 4.0 Hz, 2H), 3.23 (br s, 4H), 2.27 (d, J = 4.0 Hz, 2H), 2.17 (septet, J = 6.0 Hz, 1H), 1.00 (d, J = 6.0 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 173.7, 147.8, 133.4 (q, J = 31.5 Hz), 132.1, 123.9, 123.6 (q, J = 270.8 Hz), 117.1 (q, J = 3.8 Hz), 113.4 (q, J = 3.8 Hz), 47.8, 42.6, 41.3, 27.0, 22.9. LC-MS (m/z) calcd for C16H21F3N2O [M+H+], 315.2; found, 315.2. HPLC: purity254 > 99%.
3,3-Dimethyl-1-(4-(3-(trifluoromethyl)phenyl)piperazin-1-yl)butan-1-one (2.9)
Prepared from 1-(3-(trifluoromethyl)phenyl)piperazine (4) and 3,3-dimethylbutanoyl chloride by general procedure B in 73% yield as a pale-orange oil. R f 0.50 (heptane/EtOAc 1:1). 1H NMR (400 MHz, CDCl3) δ 7.39-7.35 (m, 1H), 7.14-7.11 (m, 2H), 7.08-7.06 (m, 1H), 3.81 (dd, J = 8.0, 4.0 Hz, 2H), 3.69 (dd, J = 8.0, 4.0 Hz, 2H), 3.22 (dd, J = 8.0, 4.0 Hz, 2H), 3.21 (dd, J = 8.0, 4.0 Hz, 2H), 2.31 (s, 2H), 1.08 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 171.9, 147.3, 134.2 (q, J = 31.5 Hz), 132.3, 126.2, 124.7 (q, J = 270.8 Hz), 123.4 (q, J = 3.8 Hz), 117.4 (q, J = 3.8 Hz), 54.5, 46.1, 33.0, 31.5, 21.1. LC-MS (m/z) calcd for C17H23F3N2O [M+H+], 329.2; found, 329.2. HPLC: purity254 > 96%.
4-Methyl-1-(4-(3-(trifluoromethyl)phenyl)piperazin-1-yl)pentan-1-one (2.10)
Prepared from 1-(3-(trifluoromethyl)phenyl)piperazine (4) and 4-methylvaleryl chloride by general procedure B in 87% yield as a clear oil. R f 0.35 (heptane/EtOAc 1:1). 1H NMR (400 MHz, CDCl3) δ 7.37 (t, J = 6.0 Hz, 1H), 7.14-7.07 (m, 3H), 3.79 (dd, J = 8.0, 4.0 Hz, 2H), 3.65 (dd, J = 8.0, 4.0 Hz, 2H), 3.24 (dd, J = 8.0, 4.0 Hz, 2H), 3.22 (dd, J = 8.0, 4.0 Hz, 2H), 2.35 (dd, J = 6.0, 6.0 Hz, 2H), 1.63-1.52 (m, 3H), 0.93 (d, J = 3.0 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 172.1, 151.1, 131.6 (q, J = 31.5 Hz), 129.7, 124.2 (q, J = 271.0 Hz), 119.3, 116.6 (q, J = 4.0 Hz), 112.7 (q, J = 4.0 Hz), 49.2, 49.0, 45.4, 41.3, 34.2, 31.3, 27.9, 22.7, 22.4. LC-MS (m/z) calcd for C17H23F3N2O [M+H+], 329.2; found, 329.2. HPLC: purity254 > 99%.
Phenyl(4-(3-(trifluoromethyl)phenyl)piperazin-1-yl)methanone (2.11)
Prepared from 1-(3-(trifluoromethyl)phenyl)piperazine (4) and benzoic acid by general procedure A in 58% yield as a pale-yellow oil. R f 0.33 (heptane/EtOAc 2:3). 1H NMR (300 MHz, CDCl3) δ 7.47-7.39 (m, 6H), 7.26-7.20 (m, 2H), 7.10 (d, J = 7.5 Hz, 1H), 3.75 (br s, 2H), 3.48 (br s, 2H), 3.28 (br s, 4H). 13C NMR (75 MHz, CDCl3) δ 170.8, 151.5, 135.8, 132.1 (q, J = 31.5 Hz), 130.3 (d, J = 1.5 Hz), 130.1 (d, J = 1.5 Hz), 129.0, 127.5, 124.6 (q, J = 267.0 Hz), 119.9, 117.2, 113.3, 49.7, 47.8. LC-MS (m/z) calcd for C18H17F3N2O [M+H+], 335.1; found, 335.1. Anal. calcd for C18H17F3N2O × 1HCl: C 58.30, H 4.89, N 7.55 found C 58.20, H 4.78, N 7.43.
Benzo[d][1,3]dioxol-5-yl(4-(3-(trifluoromethyl)phenyl)piperazin-1-yl)methanone (2.12)
Obtained from 1-(3-(trifluoromethyl)phenyl)piperazine (4) and benzo[d][1,3]dioxole-5-carboxylic acid by general procedure A in 60% yield as a white solid. R f 0.18 (heptane/EtOAc 3:1). 1H NMR (300 MHz, CDCl3) δ 7.36 (t, J = 9.0 Hz, 1H), 7.13-7.05 (m, 3H), 6.97-6.82 (m, 3H), 6.00 (s, 2H), 3.77 (br s, 4H), 3.24 (br s, 4H). 13C NMR (75 MHz, CDCl3) δ 169.9, 151.0, 149.0, 147.7, 131.5 (q, J = 31.5 Hz), 129.7, 128.9, 124.1 (q, J = 270.8 Hz), 121.7, 119.4, 116.8 (q, J = 3.8 Hz), 112.8 (q, J = 3.8 Hz), 108.2, 108.1, 101.5, 49.2, 42.8; mp 87-89°C. LC-MS (m/z) calcd for C19H17F3N2O3 [M+H+], 379.1; found, 379.1. Anal. calcd for C19H17F3N2O3: C 60.32, H 4.53, N 7.40 found C 59.95, H 4.40, N 7.02.
(3-Phenoxyphenyl)(4-(3-(trifluoromethyl)phenyl)piperazin-1-yl)methanone (2.13)
Prepared from 1-(3-(trifluoromethyl)phenyl)piperazine (4) and 3-phenoxybenzoic acid by general procedure A in 62 % yield as a pale-yellow oil. R f 0.20 (heptane/EtOAc 3:1). 1H NMR (300 MHz, CDCl3) δ 7.40-7.22 (m, 5H), 7.16-6.99 (m, 8H), 3.90 (br s, 2H), 3.60 (br s, 2H), 3.25 (br s, 2H), 3.16 (br s, 2H). 13C NMR (75 MHz, CDCl3) δ 170.1, 158.1, 156.7, 151.4, 137.4, 132.0 (q, J = 31.5 Hz), 130.6, 130.4, 130.1, 124.6 (q, J = 270.8 Hz), 124.4, 121.9, 120.2, 119.9, 119.6, 117.3 (q, J = 3.8 Hz), 113.3 (q, J = 3.8 Hz), 49.7, 42.9. LC-MS (m/z) calcd for C24H21F3N2O2 [M+H+], 427.1; found, 427.1. Anal. calcd for C24H21F3N3O2 × 1HCl: C 62.27, H 4.79, N 6.05 found C 63.52, H 4.64, N 5.90.
1-Phenylcyclopentyl-(4-(3-(trifluoromethyl)phenyl)piperazin-1-yl)methanone (2.14)
Prepared from 1-(3-(trifluoromethyl)phenyl)piperazine (4) and 1-phenylcyclopentanecarboxylic acid by general procedure A in 92 % yield as an off-white solid. R f 0.37 (heptane/EtOAc 3:1). 1H NMR (300 MHz, CDCl3) δ 7.40-7.17 (m, 8H), 7.05 (d, J = 6.0 Hz, 1H), 3,78 (br s, 4H), 3.25 (br s, 2H), 3.13 (br s, 2H), 2.70-2.41 (m, 4H), 2.06-1.87 (m, 4H). 13C NMR (75 MHz, CDCl3) δ 175.2, 151.4, 145.8, 143.2, 131.9 (q, J = 31.5 Hz), 130.0, 128.7, 127.5, 126.8, 124.6 (q, J = 270.8 Hz) 119.4 (q, J = 1.5 Hz), 116.8 (q, J = 3.8 Hz), 112.9 (q, J = 3.8 Hz), 59.3, 58.9, 48.7, 38.8, 36.4, 25.7, 24.0; mp 77-79°C. LC-MS (m/z) calcd for C23H25F3N2O [M+H+], 403.2; found, 403.2. Anal. calcd for C23H25F3N2O: C 68.64, H 6.26, N 6.96 found C 70.90, H 6.37, N 6.73.
Naphthalen-1-yl(4-(3-(trifluoromethyl)phenyl)piperazin-1-yl)methanone (2.15)
Prepared from 1-(3-(trifluoromethyl)phenyl)piperazine (4) and 1-naphthoic acid by general procedure A in 83% yield as a colorless oil. R f 0.19 (heptane/EtOAc 3:1). 1H NMR (300 MHz, CDCl3) δ 7.90-7.83 (m, 3H), 7.55-7.32 (m, 5H), 7.12-7.02 (m, 3H), 4.21-4.01 (m, 2H), 3.42-3.35 (m, 4H), 3.11-2.99 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 169.9, 151.4, 134.1, 133.9, 132.0 (q, J = 31.5 Hz), 130.1, 129.8, 128.9, 127.6, 127.0, 125.6, 125.1, 124.5 (q, J = 270.8 Hz), 124.3, 119.4 (q, J = 1.5 Hz), 117.3 (q, J = 3.8 Hz), 113.3 (q, J = 3.8 Hz), 50.1, 49.7, 47.3, 42.0. LC-MS (m/z) calcd for C22H19F3N2O [M+H+], 385.1; found, 385.1. Anal. calcd for C22H19F3N2O × 1HCl: C 62.79, H 4.79, N 6.66 found C 64.82, H 4.72, N 6.26.
Thiophen-2-yl(4-(3-(trifluoromethyl)phenyl)piperazin-1-yl)methanone (2.16)
Prepared from 1-(3-(trifluoromethyl)phenyl)piperazine (4) and thiophene-2-carboxylic acid by general procedure A in 69% yield as a pale-yellow oil. R f 0.68 (100% EtOAc). 1H NMR (300 MHz, CDCl3) δ 7.46 (dd, J = 6.0, 3.0 Hz, 1H), 7.39-7.32 (m, 2H), 7.13-7.04 (m, 4H), 3.92 (dd, J = 6.0, 3.0 Hz, 4H), 3.28 (dd, J = 6.0, 3.0 Hz, 4H). 13C NMR (75 MHz, CDCl3) δ 164.1, 151.4, 137.1, 131.9 (q, J = 31.5 Hz), 130.1, 129.5, 129.3, 127.2, 124.6 (q, J = 270.8 Hz), 119.7 (q, J = 1.5 Hz), 117.2 (q, J = 3.8 Hz), 113.1 (q, J = 3.8 Hz), 49.5, 45.7. LC-MS (m/z) calcd for C16H15F3N2OS [M+H+], 341.0; found, 341.0. Anal. calcd for C16H15F3N2OS ×1HCl: C 51.00, H 4.28, N 7.43 found C 51.41, H 4.28, N 7.33.
1-(4-(3-(Trifluoromethyl)phenyl)piperazin-1-yl)-2-phenylethanone (2.17)
Prepared from 1-(3-(trifluoromethyl)phenyl)piperazine (4) and 2-phenylacetyl chloride by general procedure B in 77% yield as a clear oil. R f 0.28 (heptane/EtOAc 1:1). 1H NMR (300 MHz, CDCl3) δ 7.34-7.20 (m, 6H), 7.09-6.96 (m, 3H), 3.79 (dd, J = 6.0, 3.0 Hz, 2H), 3.78 (s, 2H), 3.58 (dd, J = 6.0, 3.0 Hz, 2H), 3.16 (dd, J = 6.0, 3.0 Hz, 2H), 2.99 (dd, J = 6.0, 3.0 Hz, 2H). 13C NMR (75 MHz, CDCl3) δ 169.8, 151.3, 137.8, 131.3 (q, J = 31.5 Hz), 129.4, 129.1, 128.3, 127.1, 124.3 (q, J = 270.8 Hz), 118.6 (d, J = 0.8 Hz), 115.6 (q, J = 3.8 Hz), 112.0 (q, J = 3.8 Hz), 62.9, 52.8, 48.6. LC-MS (m/z) calcd for C19H19F3N2O [M+H+], 349.1; found, 349.1. Anal. Calcd for C19H19F3N2O × 1HCl: C 60.20, H 5.16, N 7.12 found C 59.30, H 5.24, N 7.28.
4-Aminophenyl-(4-(3-(trifluoromethyl)phenyl)piperazin-1-yl)methanone (2.18)
Prepared from 1-(3-(trifluoromethyl)phenyl)piperazine (4) (103 mg, 0.37 mmol) and 4-((tert-butoxycarbonyl)amino)benzoic acid in accordance with general procedure A to provide N-Boc-2.18 in 48% yield as a brown oil. R f 0.44 (heptane/EtOAc 1:1). 1H NMR (400 MHz, MeOD) δ 7.37-7.27 (m, 5H), 7.07-6.99 (m, 3H), 3.81-3.61 (m, 4H), 3.27-3.07 (m, 4H), 1.46 (s, 9H). 13C NMR (100 MHz, MeOD) δ 170.3, 152.5, 151.1, 140.1, 131.8, 131.5, 129.7, 128.5, 127.5, 125.6, 119.4, 118.1, 116.8, 114.1, 112.9 (d, J = 4.0 Hz), 81.1, 49.2, 31.9, 29.0, 28.3, 22.7. LC-MS (m/z) calcd for C23H26F3N3O3 [M+H+], 450.1, found, 450.1. The intermediate product N-Boc-2.18 was dissolved in DCM (2 mL) and TFA (2 mL) and stirred for 1h at rt. The reaction mixture was evaporated and the title compound 2.18 was obtained in 69% yield as TFA-salt. 1H NMR (400 MHz, MeOD) δ 7.43-7.39 (m, 1H), 7.35-7.32 (m, 2H), 7.23-7.20 (m, 2H), 7.12-7.10 (m, 1H), 6.90-6.87 (m, 2H), 3.79 (s, 4H), 3.30-3.26 (m, 4H). 13C NMR (100 MHz, MeOD) δ 173.1, 152.1, 148.3, 132.8, 132.5 (q, J = 8.0 Hz), 131.0, 130.4, 127.2, 126.7, 124.5, 120.8, 118.6, 117.2 (q, J = 1.0 Hz), 117.0, 115.0, 113.6 (q, J = 1.0 Hz), 50.1, 49.7. LC-MS (m/z) calcd for C18H18F3N3O [M+H+], 350.1; found, 350.1. HPLC: purity254 >97%.
3-Aminophenyl-(4-(3-(trifluoromethyl)phenyl)piperazin-1-yl)methanone (2.19)
Prepared from 3-trifluoromethylphenylpiperazine (4) (101 mg, 0.37 mmol) and 3-((tert-butoxycarbonyl)amino)benzoic acid by general procedure A in 85% yield as a reddish oil. R f 0.42 (heptane/EtOAc 1:1). 1H NMR (400 MHz, MeOD) δ 7.45 (s, 1H), 7.36-7.23 (p, J = 8.0 Hz, 3H), 7.06-6.98 (m, 4 H), 6.78 (s, 1H), 3.95-3.75 (m, 2H), 3.66-3.46 (m, 2H), 3.27-3.01 (m, 4H), 1.45 (s, 9H). 13C NMR (100 MHz, MeOD) δ 172.4, 155.2, 152.9, 141.1 (q, J = 2.0 Hz), 137.2, 132.6 (q, J = 4.0 Hz), 131.0, 130.3, 129.8, 127.2, 124.8, 122.0, 120.9 (q, J = 2.0 Hz), 118.0, 117.3, 113.7, 81.2, 79.5, 54.8, 30.7, 28.7, 24.3, 23.8. LC-MS (m/z) calcd for C23H26F3N3O3 [M+H+], 450.1, found, 450.1. The intermediate product N-Boc-2.19 was dissolved in DCM (2 mL) and TFA (2 mL) and stirred for 1h at rt. The reaction mixture was evaporated to afford the title compound in 99% yield as TFA-salt. 1H NMR (400 MHz, MeOD) δ 7.67-7.62 (m, 2H), 7.57-7.49 (m, 3H), 7.44-7.40 (m, 1H), 7.24-7.22 (m, 2H), 7.14-7.12 (m, 1H), 4.01-3.80 (m, 2H), 3.74-3.50 (m, 2H), 3.45-3.18 (m, 4H). 13C NMR (100 MHz, MeOD) δ 170.8, 159.0 (q, J = 10.5 Hz), 152.7, 138.6, 134.6, 134.24, 133.5, 132.5 (q, J = 8.0 Hz), 131.7, 131.5, 131.01, 130.6, 128.5, 127.5, 127.2, 125.0, 124.9, 124.5, 122.3, 120.9 (d, J = 1.0 Hz), 117.4 (q, J = 1.0 Hz), 114.7, 113.7 (q, J = 1.0 Hz), 54.5, 54.2. LC-MS (m/z) calcd for C18H18F3N3O [M+H+], 350.1; found, 350.1. HPLC: purity254 >96%.
2-Amino-1-(4-(3-(trifluoromethyl)phenyl)piperazin-1-yl)ethanone (2.20)
Prepared from (4) (104 mg, 0.37 mmol) and N-Boc-glycine-OH (105 mg, 0.36 mmol) by general procedure A to provide N-Boc-2.20 in 86% yield as a colorless oil. R f 0.25 (heptane/EtOAc 1:1). 1H NMR (400 MHz, MeOD) δ 7.41 (t, J = 8.0 Hz, 1H), 7.23-7.20 (m, 2H), 7.11 (d, J = 8.0 Hz, 1H), 3.97 (s, 2H), 3.75 (s, 2H), 3.66 (s, 2H), 3.28-3.23 (m, 4H), 1.45 (s, 9H). The intermediate N-Boc-2.20 was dissolved in DCM (2 mL) and TFA (2 mL) and stirred for 30 min at rt. The reaction mixture was evaporated to afford the title compound as the TFA-salt in quantitative yield. 1H NMR (400 MHz, MeOD) δ 7.45-7.40 (m, 1H), 7.24-7.21 (m, 2H), 7.14-7.12 (m, 1H), 4.00 (s, 2H), 3.80 (t, J = 4.0 Hz, 2H), 3.62 (t, J = 4.0 Hz, 2H), 3.30-3.26 (m, 2H). 13C NMR (100 MHz, MeOD) δ 165.7, 159.9 (q, J = 41.0 Hz), 152.7, 132.40 (t, 32.0 Hz), 131.0, 127.2, 124.5, 120.9, 117.5, 114.7, 113.6 (q, J = 4.0 Hz), 49.9, 49.7, 45.5, 43.1, 41.0, 27.7. LC-MS (m/z) calcd for C13H16F3N3O [M+H+], 288.1; found, 288.1. HPLC: purity254 >99%.
2-Amino-1-(4-(3-(trifluoromethyl)phenyl)piperazin-1-yl)ethanone (2.21)
Prepared from (16) (103 mg, 0.37 mmol) and N-Boc-glycine-OH (69 mg, 0.36 mmol) by general procedure A to provide N-Boc-2.21 in 51% yield as a colorless oil. R f 0.18 (heptane/EtOAc 1:1). The intermediate N-Boc-2.20 was dissolved in DCM (2 mL) and TFA (2 mL) and stirred for 45 min at rt. The reaction mixture was evaporated to afford the title compound as the TFA-salt in quantitative yield. 1H NMR (400 MHz, MeOD) δ 7.44-7.40 (m, 1H), 7.24-7.20 (m, 2H), 7.12 (dt, J = 8.0 Hz, 1H), 3.78 (t, J = 4.0 Hz, 2H), 3.68 (t, J = 4.0, 2H), 3.26-3.21 (m, 6H), 2.83 (t, J = 4.0 Hz, 2H). 13C NMR (100 MHz, MeOD) δ 170.4, 152.8, 132.7, 132.4, 131.0, 127.2, 120.8, 117.5, 117.3 (q, J = 4.0 Hz), 113.5 (q, J = 4.0 Hz), 54.4, 50.0, 46.16, 42.65. LC-MS (m/z) calcd for C14H18F3N3O [M+H+], 302.1; found, 302.1. HPLC: purity254 >98%.
Benzyl 4-(3-(trifluoromethyl)phenyl)piperazine-1-carboxylate (2.32)
Prepared from 1-(3-(trifluoromethyl)phenyl)piperazine (4) and benzyl carbonochloridate by general procedure B in 73% yield as a clear oil. R f 0.43 (heptane/EtOAc 3:1). 1H NMR (300 MHz, CDCl3) δ 7.36-7.29 (m, 6H), 7.10-7.01 (m, 3H), 5.15 (s, 2H), 3.66 (dd, J = 6.0, 3.0 Hz, 4H), 3.17 (br s, 4H). 13C NMR (75 MHz, CDCl3) δ 155.1, 151.2, 136.5, 131.3 (q, J = 31.5 Hz), 129.6, 128.5, 128.1, 127.9, 124.2 (q, J = 270.8 Hz), 119.4 (q, J = 1.5 Hz), 116.5 (q, J = 3.8 Hz), 112.4 (q, J = 3.8 Hz), 67.3, 48.9, 43.5. LC-MS (m/z) calcd for C19H19F3N2O2 [M+H+], 365.1; found, 365.1. Anal. Calcd for C19H19F3N2O2 × 1HCl: C 57.35, H 5.03, N 6.91 found C 56.93, H 5.03, N 6.99.
1-(Phenylsulfonyl)-4-(3-(trifluoromethyl)phenyl)piperazine (2.33)
Prepared from 1-(3-(trifluoromethyl)phenyl)piperazine (4) and benzenesulfonyl chloride by general procedure B in 71% yield as a white solid. R f 0.39 (heptane/EtOAc 3:1). 1H NMR (300 MHz, CDCl3) δ 7.80-7.76 (m, 2H), 7.65-7.51 (m, 3H), 7.32 (t, J = 7.5 Hz, 1H), 7.11-6.97 (m, 3H), 3.30-3.26 (m, 4H), 3.19-3.16 (m, 4H). 13C NMR (75 MHz, CDCl3) δ 151.1, 135.6, 133.5, 131.9 (q, J = 31.5 Hz), 130.2, 129.6, 128.2, 124.5 (q, J = 270.8 Hz), 119.9 (q, J = 1.5 Hz), 117.4 (q, J = 3.8 Hz), 113.5 (q, J = 3.8 Hz), 49.1, 46.4; mp 109-111°C (decomposed). LC-MS (m/z) calcd for C17H17F3N2O2S [M+H+], 371.1; found, 371.1. Anal. Calcd for C17H17F3N2O2S: C 55.13, H 4.63, N 7.56 found C 54.60, H 4.44, N 7.41.
(±)-endo-exo-Bicyclo[2.2.1]heptan-2-ylmethyl)-4-(3-(trifluoromethyl)phenyl)piperazine (2.34)
A solution of (±)-endo-exo-bicyclo[2.2.1]heptan-2-yl(4-(3-(trifluoromethyl)phenyl)piperazin-1-yl)methanone ((±)-endo-exo 2.3) (116.5 mg, 0.30 mmol) in dry THF (3 mL) was added dropwise to a solution of LiAlH4 (13 mg, 0.33 mmol) in THF (5 mL) at 0°C under a N2 atmosphere. The reaction mixture was stirred for 3 days at rt, quenched with 2N NaOH (2 mL) and extracted with dichloromethane (3 × 25 mL). The combined organic phases were washed with H2O (20 mL) and brine (20 mL). The organic phase was dried over anhydrous Na2SO4. After concentration in vacuo, the crude product was purified by column chromatography on silica gel to afford the titled compound as a pale-yellow oil (72 mg, 0.21 mmol, 71% yield): R f 0.42 (heptane/EtOAc 2:1). 1H NMR (400 MHz, CDCl3) δ 7.37 (t, J = 8.0 Hz, 1H), 7.13-7.00 (m, 3H), 3.91-3.66 (m, 4H), 3.25-3.16 (m, 4H), 2.97-2.41 (m, 2H), 2.32-2.28 (m, 1H), 1.98-1.92 (m, 1H), 1.62-1.19 (m, 9H). 13C NMR (100 MHz, CDCl3) δ 151.2, 130.1 (q, J = 31.5 Hz), 130.2, 124.5 (q, J = 270.7 Hz), 119.3 (d, J = 3.8 Hz), 117.3 (q, J = 3.8 Hz), 113.2 (q, J = 3.8 Hz), 49.0, 44.3, 40.9, 40.4, 37.2, 36.7, 36.0, 34.9, 32.2, 29.5, 28.9, 24.5. LC-MS (m/z) calcd for C19H25F3N2 [M+H+], 339.2; found, 339.2. HPLC: purity254 > 99%.
1-Benzyl-4-(3-(trifluoromethyl)phenyl)piperazine (2.35)
BnBr (54 μ L, 0.46 mmol) was added dropwise to a solution of 1-(3-(trifluoromethyl)phenyl)piperazine (4) (100 mg, 0.43 mmol) and Et3N (126 μ L, 0.46 mmol) in dichloromethane (5 mL) at rt under a N2 atmosphere. The reaction mixture was stirred for 24 hours, quenched with saturated NH4Cl (5 mL) and extracted with dichloromethane (3 × 20 mL). The combined organic phases were washed with H2O (20 mL) and brine (20 mL). The organic phase was dried over anhydrous Na2SO4. After concentration in vacuo, the crude product was purified by column chromatography on silica gel to afford the titled compound as a clear oil (79 mg, 0.25 mmol, 57% yield): R f 0.41 (heptane/EtOAc 2:1). 1H NMR (300 MHz, CDCl3) δ 7.34-7.19 (m, 6H), 7.08-6.99 (m, 3H), 3.55 (br s, 2H), 3.21 (dd, J = 6.0, 6.0 Hz, 4H), 2.59 (dd, J = 6.0, 6.0 Hz, 4H). 13C NMR (75 MHz, CDCl3) δ 151.3, 135.2, 131.7 (q, J = 31.5 Hz), 130.1, 129.3, 128.9, 127.4 124.6 (q, J = 270.8 Hz), 119.6 (d, J = 3.8 Hz), 117.0 (q, J = 3.8 Hz), 113.0 (q, J = 3.8 Hz), 49.3, 49.1, 46.2, 41.9, 41.5. LC-MS (m/z) calcd for C18H19F3N2 [M+H+], 321.2; found, 321.2. Anal. Calcd for C18H19F3N2 × 1HCl: C 60.59, H 5.65, N 7.85 found C 55.74, H 5.23, N 7.12.
Bicyclo[2.2.1]heptan-2-yl(4-phenylpiperazin-1-yl)methanone (3.1)
Prepared in accordance with general procedure A. The crude product was purified by flash chromatography to afford 3.1 as a clear oil (240 mg, 43%). R f 0.20 (heptane/EtOAc 3:1). 1H NMR (400 MHz, CDCl3 ) δ 7.31-7.25 (m, 2H), 6.95-6.85 (m, 3H), 3.99-3.61 (m, 4H), 3.30-3.03 (m, 4H), 2.98-2.91 (m, 0.5H), 2.44-2.39 (m, 1.5H), 2.34-2.26 (m, 1H), 1.99-1.90 (m, 1H), 1.64-1.16 (m, 7H). 13C NMR (100 MHz, CDCl3) δ 173.9, 172.2, 151.0, 129.2, 120.5, 120.5, 116.6, 50.2, 49.8, 49.7, 49.6, 45.4, 45.3, 44.2, 43.7, 41.9, 41.7, 40.8, 40.6, 40.4, 37.2, 36.7, 36.0, 34.9, 32.2, 29. 5, 28.9, 28.9, 24.5. LC-MS (m/z) calcd for C18H24N2O [M+H+], 285.1; found, 285.1. HPLC: purity254 >95%.
(±)-endo-exo-Bicyclo[2.2.1]heptan-2-yl(4-(4-(trifluoromethyl)phenyl)piperazin-1-yl)methanone (3.2)
Pd(OAc)2 (1.0 mg, 4.6 μ mol) and P(tBu)3 (1.0 M in toluene, 16 μ L, 0.016 mmol) were added to a solution of 4-(bicyclo[2.2.1]heptane-2-carbonyl)piperazin-1-ium chloride (±)-endo-exo- 7 (101 mg, 0.41 mmol), 1-bromo-4-(trifluoromethyl)benzene (63 μ L, 0.45 mmol) and NaOtBu (86 mg, 0.90 mmol) in dry o-xylene (1.3 mL) at rt under a N2 atmosphere. The reaction mixture was stirred at 120°C for 24 hours, quenched with H2O (10 mL) and extracted with EtOAc (3 × 20 mL). The combined organic phases were washed with H2O (20 mL) and brine (20 mL). The organic phase was dried over anhydrous Na2SO4. After concentration in vacuo, the crude product was purified by column chromatography on silica gel to afford the titled compound as a white solid (80 mg, 0.23 mmol, 56%): R f 0.25 (heptane/EtOAc 3:1). 1H NMR (400 MHz, CDCl3) δ 7.42 (d, J = 8.6 Hz, 2H), 6.86 (d, J = 8.8 Hz, 2H), 3.90-3.80 (m, 0.5H), 3.75-3.62 (m, 4H), 3.31-3.08 (m, 4H), 2.91-2.80 (m, 0.5H), 2.40-2.30 (m, 1H), 2.28-2.19 (m, 1H), 1.91-1.82 (m, 1H), 1.60-1.10 (m, 7H). 13C NMR (100 MHz, CDCl3) δ 174.0, 172.3, 153.0, 126.6, 126.5, 126.4, 126.4, 125.9, 123.2, 121.4, 121.1, 114.9, 48.8, 48.4, 48.3, 48.2, 45.1, 44.9, 44.2, 43.7, 41.6, 41.4, 40.8, 40.3, 37.1, 36.7, 36.0, 34.9, 32.2, 29.5, 28.9, 28.9, 24.5; mp: 126-127°C (decomposed). LC-MS (m/z) calcd for C19H23F3N2O [M+H+], 353.2; found, 353.2. HPLC: purity254 > 95%.
(±)-endo-exo-Bicyclo[2.2.1]heptan-2-yl(4-(2-(trifluoromethyl)phenyl)piperazin-1- yl)methanone ((±)-endo-exo-3.5)
Prepared in accordance with general procedure A. The crude product was purified twice by flash chromatography to afford (±)-endo-exo -3.5 as a clear oil (48 mg, 14%). R f 0.32 (heptane/EtOAc 3:1). 1H NMR (400 MHz, CDCl3 ) δ 7.64 (d, J = 7.86 Hz, 1H), 7.52 (t, J = 7.69 Hz, 1H), 7.31 (d, J = 8.02 Hz, 1H), 7.28-7.22 (m, 1H), 3.72 (s, 4H), 2.99-2.85 (m, 4.5H), 2.45-2.36 (m, 1.5H) 2.33-2.25 (m, 1H), 2.00-1.90 (m, 1H), 1.65-1.15 (m, 7H). 13C NMR (100 MHz, CDCl3) δ 173.9, 172.3, 151.8, 132.8, 127.6, 127.3, 127.2, 125.2, 124.0, 122.6, 54.1, 53.8, 53.3, 53.2, 46.0, 45.9, 44.2, 43.7, 42.4, 42.2, 40.8, 40.6, 40.3, 37.2, 36.7, 36.0, 34.9, 32.2, 29.4, 28.9, 28.9, 24.5. LC-MS (m/z) calcd for C19H23F3N2O [M+H+], 353.1; found, 353.1. HPLC: purity254 > 95%.
(±)-endo-exo-Bicyclo[2.2.1]heptan-2-yl(4-(3-chlorophenyl)piperazin-1-yl)methanone ((±)-endo-exo-3.7)
Prepared in accordance with general procedure A. The crude product was purified by flash chromatography to afford 3.7 as a clear oil (90 mg, 46%). R f 0.31 (heptane/EtOAc 3:1). 1H NMR (400 MHz, CDCl3 ) δ 7.17 (t, J = 8.09 Hz, 1H), 6.87 (t, J = 2.04 Hz, 1H), 6.85 (d, J = 7.84 Hz, 1H) 6.78 (dd, J = 8.35, 2.15 Hz, 1H), 3.95-3.60 (m, 4H), 3.25-3.05(m, 4H), 2.97-2.90 (m, 0.5H), 2.45-2.37 (m, 1.5H), 2.33-2.25 (m, 1H) 1.98-1.88 (m, 1H), 1.64-1.06 (m, 7H). 13C NMR (100 MHz, CDCl3) δ 173.8, 172.1, 151.9, 134.9, 130.0, 119.9, 116.2, 114.3, 49.4, 49.1, 48.9, 45.1, 44.9, 44.9, 43.6, 41.5, 41.3, 40.7, 40.5, 40.2, 37.0, 36.6, 35.9, 34.8, 32.0, 29.3, 28.7, 24.4. LC-MS (m/z) calcd for C18H23ClN2O [M+H+], 319.1; found, 319.1. HPLC: purity254 > 97%.
(±)-endo-exo- Bicyclo[2.2.1]heptan-2-yl(4-(3-hydroxyphenyl)piperazin-1-yl)methanone ((±)-endo-exo-3.8)
Prepared from (±)-endo-exo-bicyclo[2.2.1]heptane-2-carboxylic acid ((±)-endo-exo -6) and 3-(piperazin-1-yl)phenol by general procedure A in 40% yield as a white solid. R f 0.45 (heptane/EtOAc 2:1). 1H NMR (400 MHz, CDCl3) δ 7.13 (t, J = 8.0 Hz, 1H), 6.52-6.36 (m, 3H), 3.93-3.64 (m, 4H), 3.22-2.92 (m, 4H), 2.45-2.41 (m, 1H), 2.31-2.28 (m, 1H), 1.97-1.88 (m, 1H), 1.63-1.18 (m, 8H). 13C NMR (100 MHz, CDCl3) δ 173.5, 157.4, 152.3, 130.1, 108.7, 107.7, 103.6, 50.2, 49.8, 49.1, 45.3, 44.3, 43.7, 41.8, 40.6, 40.5, 37.2, 36.8, 36.0, 35.1, 32.2, 29.5, 28.9, 28.9, 24.5; mp: 187-189°C (decomposed). LC-MS (m/z) calcd for C18H24N2O2 [M+H+], 301.2; found, 301.2. HPLC: purity254 > 98%.
3-(4-(bicyclo[2.2.1]heptane-2-carbonyl)piperazin-1-yl)benzonitrile (3.9)
Prepared in accordance with general procedure A. The crude product was purified by flash chromatography to afford 3.9 as a clear oil (63 mg, 63%). R f 0.15 (heptane/EtOAc 3:1). 1H NMR (400 MHz, CDCl3 ) δ 7.37-7.31(m, 1H), 7.15-7.10 (m, 3H), 3.98-3.62 (m, 4H), 3.30-3.10 (m, 4H), 2.98-2.90 (m, 0.5H), 2.46-2.36 (m, 1.5H), 2.34-2.26 (m, 1H), 1.98-1.88 (m, 1H), 1.64-1.10 (m, 7H). 13C NMR (100 MHz, CDCl3) δ 173.9, 172.3, 151.0, 130.0, 123.3, 120.4, 119.1, 118.9, 113.2, 49.1, 48.8, 48.6, 45.0, 44.9, 44.2, 43.7, 41.5, 41.3, 40.8, 40.6, 40.3, 37.1, 36.7, 36.0, 34.9, 32.2, 29.4, 28.9, 28.8, 24.5. LC-MS (m/z) calcd for C19H23N3O [M+H+], 310.1; found, 310.1. HPLC: purity254 > 99%.
(±)-endo-exo-Bicyclo[2.2.1]heptan-2-yl(4-(3-methoxyphenyl)piperazin-1-yl)methanone ((±)-endo-exo-3.10)
Prepared from (±)-endo-exo- bicyclo[2.2.1]heptane-2-carboxylic acid ((±)-endo-exo -6) and 1-(3-methoxyphenyl)piperazine by general procedure A in 45% yield as a clear oil. R f 0.35 (heptane/EtOAc 2:1). 1H NMR (400 MHz, CDCl3) δ 7.19 (t, J = 8.0 Hz, 1H), 6.55-6.44 (m, 3H), 3.92-3.61 (m, 4H), 3.48 (s, 3H), 3.24-2.92 (m, 4H), 2.44-2.40 (m, 1H), 2.31-2.27 (m, 1H), 1.97-1.89 (m, 1H), 1.61-1.17 (m, 8H). 13C NMR (100 MHz, CDCl3) δ 173.2, 160.7, 152.4, 129.9, 109.3, 105.1, 103.1, 55.2, 50.8, 50.0, 49.6, 49.6, 49.4, 45.4, 45.3, 44.3, 43.8, 41.9, 41.7, 40.8, 40.6, 40.4, 37.2, 36.8, 36.0, 34.9, 32.2, 29.5, 29.0, 28.9, 27.0, 24.5. LC-MS (m/z) calcd for C19H26N2O2 [M+H+], 315.2; found, 315.2. HPLC: purity254 > 98%.
(±)-endo-exo-Bicyclo[2.2.1]heptan-2-yl(4-(2,4-difluorophenyl)piperazin-1-yl)methanone ((±)-endo-exo-3.11)
Prepared from (±)-endo-exo-bicyclo[2.2.1]heptane-2-carboxylic acid ((±)-endo-exo -6) and 1-(2,4-difluorophenyl)piperazine by general procedure A. After chromatography (R f = 0.43, heptane/EtOAc 2:1). The title compound was isolated in 58% yield as a clear oil. 1H NMR (400 MHz, CDCl3) δ 6.92-6.77 (m, 3H), 3.93-3.64 (m, 4H), 3.07-2.90 (m, 4H), 2.44-2.27 (m, 2H), 1.98-1.90 (m, 1H), 1.62-1.17 (m, 8H). 13C NMR (100 MHz, CDCl3) δ 173.9, 172.3, 159.5 (d, J = 12.0 Hz), 157.0 (d, J = 12.0 Hz), 154.6 (d, J = 12.0 Hz), 136.4 (d, J = 4.0 Hz), 136.2 (d, J = 4.0 Hz), 129.0, 128.4, 126.9, 119.9 (q, J = 4.0 Hz), 110.9 (d, J = 2.0 Hz), 110.7 (d, J = 2.0 Hz), 105.1, 104.9 (d, 2.0 Hz), 104.6, 53.4, 51.8, 51.4, 51.0, 45.7, 45.6, 44.2, 43.2, 42.0, 41.8, 40.8, 40.6, 40.4, 37.2, 36.8, 36.0, 29.0, 28.9, 24.6. LC-MS (m/z) calcd for C18H22F2N2O [M+H+], 321.2; found, 321.2. HPLC: purity254 > 97%.
1-(3-(Trifluoromethyl)phenyl)piperazine (4)
Piperazine (5.74 g, 66.6 mmol), 3-bromobenzotrifluoride (5.0 g, 22.2 mmol), Pd(OAc)2 (50 mg, 220 μ mol), P(tBu)3 (216 μ L, 880 μ mol) and NaOtBu (3 g, 31.1 mmol) were stirred in dry o-xylene (50 mL) at 120ºC under a N2 atmosphere for 17 h. H2O (25 mL) was added and the crude reaction was extracted with EtOAc (3 × 50 mL) and the combined organic phases were washed with H2O (30 mL) and brine (30 mL). The organic phase was dried over anhydrous Na2SO4. After concentration in vacuo, the crude product was purified by column chromatography on silica gel. This afforded the titled compound (3.91 g, 17.0 mmol, 77%) as a yellow oil: R f 0.23 (Et2O/MeCN/MeOH/Et3N 10:1:1:0.5). 1H NMR (300 MHz, CDCl3) δ 7.33 (t, J = 7.95 Hz, 1H), 7.10-7.03 (m, 3H), 3.18 (dd, J = 9.0, 5.4 Hz, 4H), 3.04 (dd, J = 6.9, 5.1 Hz, 4H), 2.0 (s, 1H). 13C NMR (75 MHz, CDCl3) δ 152.2, 131.9 (q, J = 30.8 Hz), 129.9, 124.7 (q, J = 271.5 Hz), 119.1 (q, J = 1.5 Hz), 116.2 (q, J = 3.8 Hz), 112.5 (q, J = 4.5 Hz), 50.1, 46.3. LC-MS (m/z) calcd for C11H13F3N2 [M+H+], 231.1; found, 231.1. HPLC: purity254 > 95%.
(±)-endo-exo-Bicyclo[2.2.1]heptane-2-carboxylic acid ((±)-endo-exo-6)
A solution of KMnO4 (53.2 g, 336.5 mmol) in H2O (190 mL) was added dropwise to a solution of bicyclo[2.2.1]heptan-2-ylmethanol (17.0 g, 134.6 mmol) and K2CO3 (7.4 g, 53.8 mmol) in H2O (380 mL) at 0°C and stirred at room temperature for 24 hours. The crude reaction was quenched with 4N HCl (pH ≈ 2) and extracted with EtOAc (3 × 1000 mL). The combined organic phases were washed with H2O (500 mL), brine (250 mL) and dried over anhydrous MgSO4. After concentration in vacuo, the crude product was directly used for the next step without purification.
(±)-endo-exo-Bicyclo[2.2.1]heptan-2-yl(piperazin-1-yl)methanone ((±)-endo-exo- 7)
DIPEA (2.8 g, 21.4 mmol) was added dropwise to a stirred solution of (±)-endo-exo-bicyclo[2.2.1]heptane-2-carboxylic acid ((±)-endo-exo -7) (1 g, 7.1 mmol) and piperazine (1.85 g, 21.4 mmol) in dry DMF (60 mL) at 0°C under a N2 atmosphere. A solution of TBTU (2.98 g, 9.27 mmol) in DMF (26 mL) was added to the reaction mixture and stirred at 0°C for 30 min. After 20 hours at rt, the reaction mixture was quenched with brine (30 mL) and extracted with dichloromethane (3 × 100mL). The combined organic phases were washed with H2O (3 × 200 mL) and brine (100 mL). The organic phase was dried over anhydrous Na2SO4. After concentration in vacuo, the crude product was purified by column chromatography on silica gel (Et2O/MeOH/MeCN/Et3N 8:2:2:0.5). This afforded the title compound as a pale-yellow oil (0.73 mg, 3.5 mmol, 49% yield). The pure product was dissolved in dichloromethane (40 mL) and 4N HCl in dioxane (0.88 mL, 3.5 mmol) was added. The solution was evaporated to afford the corresponding HCl salt. 1H NMR (300 MHz, CDCl3) δ 10.10 (br s, 2H), 4.10-3.80 (m, 4H), 3.22 (br s, 4H), 2.95-2.83 (m, 0.5H), 2.40-2.27 (m, 2.5H), 1.97-1.80 (m, 1H), 1.70-1.16 (m, 6H). 13C NMR (75 MHz, CDCl3) δ 173.8, 172.2, 45.9, 45.4, 45.2, 45.0, 44.9, 44.8, 44.0, 43.5, 41.2, 41.1, 40.7, 40.4, 40.1, 37.0, 36.6, 35.8, 34.7, 32.0, 29.3, 28.7, 24.4, 8.7.
Analogs 2.22-2.31, 2.36-2.40, 3.3, 3.4, 3.6 and 3.12 were obtained as 5 mg portions pre-dissolved in DMSO from ChemBridge corporation and used directly in the [3H]-Asp-uptake assay.
Abbreviations
- CNS:
-
Central nervous system
- EAAT:
-
Excitatory amino acid transporter
- SAR:
-
Structure-activity-relationship.
References
Balalaie S, Mahdidoust M, Eshaghi-Najafabadi R: 2-(1H-Benzotriazole-1-yl)-1,1,3,3-tetramethyluronium tetrafluoroborate as an efficient coupling reagent for the amidation and phenylhydrazation of carboxylic acids at room temperature. J Iran Chem Soc 2007, 4: 364-369. 10.1007/BF03245987
Bunch L, Erichsen MN, Jensen AA: Excitatory amino acid transporters as potential drug targets. Expert Opin Ther Targets 2009, 13: 719-731. 10.1517/14728220902926127
Burkhard JA, Wagner B, Fischer H, Schuler F, Mueller K, Carreira EM: Synthesis of azaspirocycles and their evaluation in drug discovery. Angew Chem-Int Edit 2010, 49: 3524-3527. 10.1002/anie.200907108
Cook G, Barta N, Stille J: Lewis acid-promoted 3-Aza-cope rearrangement of N-alkyl-N-allylenamines. J Org Chem 1992, 57: 461-467. 10.1021/jo00028a016
Dunlop J, McIlvain HB, Carrick TA, Jow B, Lu Q, Kowal D, Lin S, Greenfield A, Grosanu C, Fan K, Petroski R, Williams J, Foster A, Butera J: Characterization of novel aryl-ether, biaryl, and fluorene aspartic acid and diaminopropionic acid analogs as potent inhibitors of the high-affinity glutamate transporter EAAT2. Mol Pharmacol 2005, 68: 974-982. 10.1124/mol.105.012005
Erichsen MN, Huynh THV, Abrahamsen B, Bastlund JF, Bundgaard C, Monrad O, Bekker-Jensen A, Nielsen CW, Frydenvang K, Jensen AA, Bunch L: Structure-activity relationship study of first selective inhibitor of excitatory amino acid transporter subtype 1: 2-amino-4-(4-methoxyphenyl)-7-(naphthalen-1-yl)-5-oxo-5,6,7,8-tetrahydro-4 H -chromene-3-carbonitrile (UCPH-101). J Med Chem 2010, 53: 7180-7191. 10.1021/jm1009154
Fragasso G, Palloshi A, Puccetti P, Silipigni C, Rossodivita A, Pala M, Calori G, Alfieri O, Margonato A: A randomized clinical trial of trimetazidine, a partial free fatty acid oxidation inhibitor, in patients with heart failure. J Am Coll Cardiol 2006, 48: 992-998. 10.1016/j.jacc.2006.03.060
Gudipati M, Radziszewski J, Kaszynski P, Michl J: Bicyclo[3.2.2]non-1-Ene - matrix-isolation and spectroscopic characterization of a moderately strained bridgehead olefin. J Org Chem 1993, 58: 3668-3674. 10.1021/jo00066a018
Huynh THV, Shim I, Bohr H, Abrahamsen B, Nielsen B, Jensen AA, Bunch L: Structure–activity relationship study of selective excitatory amino acid transporter subtype 1 (EAAT1) inhibitor 2-amino-4-(4-methoxyphenyl)-7-(naphthalen-1-yl)-5-oxo-5,6,7,8-tetrahydro-4 H -chromene-3-carbonitrile (UCPH-101) and absolute configurational assignment using infrared and vibrational circular dichroism spectroscopy in combination with ab initio hartree–fock calculations. J Med Chem 2012, 55: 5403-5412. 10.1021/jm300345z
Huynh THV, Abrahamsen B, Madsen KK, Gonzalez-Franquesa A, Jensen AA, Bunch L: Design, synthesis and pharmacological characterization of coumarin-based fluorescent analogs of excitatory amino acid transporter subtype 1 selective inhibitors, UCPH-101 and UCPH-102. Bioorg Med Chem 2012, 20: 6831-6839. 10.1016/j.bmc.2012.09.049
Jensen AA, Bräuner-Osborne H: Pharmacological characterization of human excitatory amino acid transporters EAAT1, EAAT2 and EAAT3 in a fluorescence-based membrane potential assay. Biochem Pharmacol 2004, 67: 2115-2127. 10.1016/j.bcp.2004.02.013
Jensen AA, Erichsen MN, Nielsen CW, Stensbøl TB, Kehler J, Bunch L: Discovery of the first selective inhibitor of excitatory amino acid transporter subtype 1. J Med Chem 2009, 52: 912-915. 10.1021/jm8013458
Lauriat TL, Richler E, McInnes LA: A quantitative regional expression profile of EAAT2 known and novel splice variants reopens the question of aberrant EAAT2 splicing in disease. Neurochem Int 2007, 50: 271-280. 10.1016/j.neuint.2006.08.014
Massie A, Vandesande F, Arckens L: Expression of the high-affinity glutamate transporter EAAT4 in mammalian cerebral cortex. Neuroreport 2001, 12: 393-397. 10.1097/00001756-200102120-00041
Millan MJ, Cussac D, Milligan G, Carr C, Audinot V, Gobert A, Lejeune F, Rivet JM, Brocco M, Duqueyroix D, Nicolas JP, Boutin JA, Newman-Tancredi A: Antiparkinsonian agent piribedil displays antagonist properties at native, rat, and cloned, human alpha(2)-adrenoceptors: cellular and functional characterization. J Pharmacol Exp Ther 2001, 297: 876-887.
Nieoullon A, Canolle B, Masmejean F, Guillet B, Pisano P, Lortet S: The neuronal excitatory amino acid transporter EAAC1/EAAT3: does it represent a major actor at the brain excitatory synapse? J Neurochem 2006, 98: 1007-1018. 10.1111/j.1471-4159.2006.03978.x
Nishiyama M, Yamamoto T, Koie Y: Synthesis of N-arylpiperazines from aryl halides and piperazine under a palladium tri-tert-butylphosphine catalyst. Tetrahedron Lett 1998, 39: 617-620. 10.1016/S0040-4039(97)10659-1
Sagot E, Jensen AA, Pickering DS, Pu X, Umberti M, Stensbøl TB, Nielsen B, Assaf Z, Aboab B, Bolte J, Gefflaut T, Bunch L: Chemo-enzymatic synthesis of (2S,4R)-2-amino-4-(3-(2,2-diphenylethylamino)-3-oxopropyl)pentanedioic acid: a novel selective inhibitor of human excitatory amino acid transporter subtype 2. J Med Chem 2008, 51: 4085-4092. 10.1021/jm800091e
Weisberg E, Manley PW, Cowan-Jacob SW, Hochhaus A, Griffin JD: Second generation inhibitors of BCR-ABL for the treatment of imatinib-resistant chronic myeloid leukaemia. Nat Rev Cancer 2007, 7: 345-356. 10.1038/nrc2126
Acknowledgements
We would like to thank the Lundbeck Foundation, the Carlsberg Foundation, the Novo Nordisk Foundation, and the Danish Medical Research Council for financial support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
All authors declare no financial competing interests.
Authors’ contributions
THVH, molecular design, synthesis and manuscript preparation. CSD, molecular design, synthesis and manuscript preparation. BA, molecular design, pharmacological characterization and manuscript preparation. EM, synthesis. MF, synthesis. AAJ, molecular design, pharmacological characterization and manuscript preparation. LB, molecular design and manuscript preparation. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Huynh, T.H., Demmer, C.S., Abrahamsen, B. et al. Structure-activity-relationship study of N-acyl-N-phenylpiperazines as potential inhibitors of the Excitatory Amino Acid Transporters (EAATs): improving the potency of a micromolar screening Hit is not truism. SpringerPlus 2, 112 (2013). https://doi.org/10.1186/2193-1801-2-112
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2193-1801-2-112