
Abstract
In this study, we used the yeast carotenogenic producer Pichia pastoris Pp-EBIL strain, which has been metabolically engineered, by heterologously expressing β-carotene-pathway enzymes to produce β-carotene, as a vessel for recombinant astaxanthin expression. For this purpose, we designed new P. pastoris recombinant-strains harboring astaxanthin-encoding genes from carotenogenic microorganism, and thus capable of producing xanthophyllic compounds. We designed and constructed a plasmid (pGAPZA-WZ) containing both the β-carotene ketolase (crtW) and β-carotene hydroxylase (crtZ) genes from Agrobacterium aurantiacum, under the control of the GAP promoter and containing an AOX-1 terminator. The plasmid was then integrated into the P. pastoris Pp-EBIL strain genomic DNA, producing clone Pp-EBILWZ. The recombinant P. pastoris (Pp-EBILWZ) cells exhibited a strong reddish carotenoid coloration and were confirmed, by HPLC, to produce not only the previous described carotenoids lycopene and β-carotene, but also de novo synthesized astaxanthin.
Similar content being viewed by others

Introduction
Carotenoids are natural lipid-soluble pigments produced primarily by bacteria, algae and plants. These pigments are in part responsible for the wide variety of colors seen in nature. In some organisms, carotenoids such as β-carotene are modified with oxygen-containing functional groups, thus creating xanthophylls such as astaxanthin.
Astaxanthin is an abundant carotenoid found in marine animals, including salmonids and crustaceans (Miki et al. [1982]; Wade et al. [2005]) and is a commonly encountered keto-carotenoid in certain algae, many invertebrates and fish. The use of astaxanthin as colorant in aquaculture, especially as feed supplement in farmed trout, salmon and prawns, is necessary to obtain the red–pink coloration present in their wild counterparts, since neither fish nor prawns are capable of de novo carotenoid synthesis. Incorporation of astaxanthin into the fish and prawn feed not only increases their nutritional value, but also considerably enhances their appeal to customers and hence their commercial value.
Astaxanthin has attracted commercial interest not only in its role as a pigment, but also as a potent antioxidant capable of delaying aging and the onset of degenerative diseases in animals (Hix et al. [2004]; Kurihara et al. [2002]; Neuman et al. [2000]). Furthermore, epidemiological and experimental studies have suggested that astaxanthin also possesses anticarcinogenic and antitumor activities (Neuman et al. [2000]; Bertram & Vine [2005]; Kozuki et al. [2000]), hence astaxanthin’s relevance is progressively increasing in the pharmaceutical and cosmetic industries.
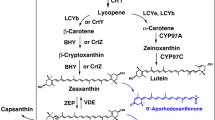
The cluster genes responsible for the synthesis of xanthophylls have been isolated from the marine bacterium A. aurantiacum. Analysis of its nucleotide sequence revealed five open reading frames, designated as genes crtW crtZ crtY crtI, and crtB, respectively (Misawa et al. [1995]) and functionally analyzed in E. coli (Misawa et al. [1995]). β-carotene ketolase (crtW genes) converts β-carotene to canthaxanthin, with echinenone as an intermediary step; whereas β-carotene hydroxylase (crtZ genes) mediates the conversion of β-carotene to zeaxanthin, via β-cryptoxanthin. As seen in Figure 1, the crtW and crtZ gene products, in combination, catalyze all the necessary steps for the conversion of β-carotene into astaxanthin (Figure 1).

Schematic diagram of astaxanthin biosynthetic pathways and possible intermediates in Agrobacterium. aurantiacum , modified from Misawa et al.[1995.]
A variety of carotenoid biosynthesis genes that produce astaxanthin have been isolated from various sources, including the yeast Xanthophyllomyces dendrorhous (Johnson et al. [1980]), the green alga Haematococcus pluvialis (Bubrick [1991]), the gram-positive bacterium Brevibacterium linens (Krubasik & Sandmann [2000]), and the marine bacterium Paracoccus haeundaensis (Lee et al. [2004]), and the function of their gene products has been determined (Kurihara et al. [2002]; Krubasik & Sandmann [2000]; Armstrong et al. [1989]; Harker & Hirschberg [1998]; Harker & Hirschberg [1997]; Misawa et al. [1990]; Verdoes et al. [1999]).
Recombinant carotenoid biosynthesis was successful, by introduction and modification of heterologous carotenogenic genes, in originally non-carotenogenic yeasts, such as Saccharomyces cerevisiae (Lange & Steinbüchel [2011]; Ukibe et al. [2009]; Verwaal et al. [2007]; Yamamo et al. [1994]), both S. cerevisiae and Candida utilis (Misawa & Shimada [1998]), C. utilis (Miura et al. [1998]; Misawa & Shimada [1998]), P. pastoris (Araya-Garay et al. [2012]; Bhataya et al. [2009]), and the filamentous fungus Mucor circinelloides (Papp et al. [2006]).
In the present work, we successfully modified the carotenoid production of P. pastoris Pp-EBIL strain by incorporating in its genome the crtW and crtZ genes from the marine bacterium A. aurantiacum. This resulted in a recombinant P. pastoris which synthesized astaxanthin as well as pathway intermediates such as lycopene, β-carotene and canthaxanthin.
Materials and methods
Strains, plasmid and culture conditions
Plasmid pGAPZαA was purchased from Invitrogen Corporation (Carlsbad, CA, USA), whereas the β-carotene producer Pp-EBIL strain of P. pastoris was constructed as previously described (Araya-Garay et al. [2012]).
P. pastoris cells were grown in YPD medium supplemented with Zeocin (100 μg/mL; Invitrogen) and incubated at 30°C, in a rotary shaker at 200 rpm for 72 h. Escherichia coli TOP10 cells were grown in low salt LB medium at 37°C for 12 h, and clones containing plasmid pGAPZαA were selected by their Zeocin (25 μg/mL) resistance. pGAPZαA* (a mutant pGAPZαA missing an Avr II site) was generated by site-directed mutagenesis (Araya-Garay et al. [2012]). Genes crtW and crtZ were amplified from the plasmid pAK96K (Misawa et al. [1995]), which harbors both the A. aurantiacum crtW (β-carotene ketolase) and crtZ (β-carotene hydroxylase) genes, and was shown to mediate the conversion of β-carotene into astaxanthin in recombinant E. coli cells. This plasmid was a gift from Prof. Misawa (Research Institute for Bioresources and Biotechnology, Ishikawa Prefectural University, Japan). Amplification of the above mentioned genes was carried out using 5′ primers that contained a restriction Sfu I site, followed by an optimized Kozak consensus sequence (ATGG), as well as a start codon, and a 3′ primer containing an EcoR I restriction site (Table 1). All DNA ligations were carried out with T4 DNA ligase (New England BioLabs, Beverly, MA, USA), as recommended by the manufacturer. After DNA ligation, the plasmids were transformed into chemically-competent E. coli Top 10 “One shot” (Invitrogen), and grown on low salt Luria–Bertani media (0.5% Yeast extract, 1% Tryptone, 0.5% NaCl) plates containing 25 μg/mL Zeocin. The plates were then incubated overnight at 37°C and recombinant colonies selected and grown overnight in low salt LB media containing 25 μg/mL Zeocin.
The Graphical Codon Usage Analyser 2.0 (Fuhrmann et al. [2004]) was used for differential codon usage analysis.
Construction of carotenoid expression vectors
The DNA coding for crtW was inserted into the Sfu I and EcoR I restriction sites of plasmid pGAPZαA, and the same restriction sites were used for inserting crtZ DNA into pGAPZαA*. The resulting expression vectors were denominated pGAPZA-W and pGAPZA*-Z, respectively and both plasmids lacked the alpha factor (Figure 2). The BamH I–Bgl II DNA fragment from pGAPZA*-Z was subcloned into the BamH I site of plasmid pGAPZA-W to generate the pGAPZA-WZ expression vector (Figure 2). All plasmids constructed in this study were subjected to DNA sequencing before use and are shown in Table 2.

Construction of plasmids pGAPZA-W and pGAPZA*-Z, containing ctrW and ctrZ genes, respectively. The expression plasmid pGAPZA-WZ, coding for both ctrW and ctrZ, was used to transform P. pastoris Pp-EBIL cells. Plasmid pGAPZA* represents a plasmid pGAPZA in which the Avr II restriction site has been eliminated by mutation.
Plasmid transformation
Plasmid pGAPZA-WZ was linearized with the restriction enzyme Avr II (New England BioLabs) and transformed into electrocompetent P. pastoris Pp-EBIL cells by electroporation, using a Bio-Rad Micropulser (Bio-Rad Laboratories, Inc Hercules, CA) as described previously (Araya-Garay et al. [2012]). Recombinant P. pastoris cells were then selected on YPDS (1% yeast extract, 2% peptone, 2% glucose, and 1 M sorbitol) plates, supplemented with Zeocin (200 μg/mL). The plates were incubated at 30°C until colonies were visible (48–72 h), transferred to room temperature, and incubated for a further 48–72 h. Gene integration into the P. pastoris genome was analyzed by PCR, using P. pastoris genomic DNA extracted with the Master Pure Yeast DNA Purification Kit (Epicentre Biotechnologies, Madison, WI, USA).
Yeast culture and harvest
Red (Pp-EBILWZ) P. pastoris colonies, obtained in YPDS agar plates, were selected and grown, for 72 h at 30°C, with shaking at 200 rpm, in 100 to 500 mL of YPD (yeast extract 1%, peptone 2%, and glucose 2%) media containing 200 μg/mL Zeocin. The cell culture was then harvested, washed with distilled water, centrifuged and lyophilized for 48 h at 0.1 mbar in a Telstar Cryodos Lyophilizer.
Carotenoid extraction
Prior to carotenoid extraction, 50 mg of lyophilized yeast cells were incubated in 3 mL of DMSO, pre-warmed at 55°C for 30 min, with strong shaking for 1 min, and then maintained for an extra 30 min without shaking (Dos Santos et al. [2011]). Residual cell-suspension, from each of the above treatments, was extracted with 10 mL of acetone and vortexed for 5 min at 4°C. Extracts were then combined with 5 mL of hexane and 1 mL of 0.1 M phosphate buffer, followed by vortexing for 30 s and centrifugation at 3000 g for 10 min. This extraction procedure was repeated until both the supernatant and residual cell pellet were colorless. The crude solvent extract thus obtained was then evaporated, under a stream of N2 flow, and kept at −80°C until high performance liquid chromatography (HPLC) analysis. All above treatments were carried out on ice and under dim light conditions, to prevent photo-degradation, isomerization and structural carotenoid changes.
HPLC analysis of carotenoids
Carotenoid samples were prepared for HPLC by dissolving their cryo-preserved dry extracts in 2 mL of chlorophorm:metanol:acetone (3:2:1, v:v:v) and filtering them through polycarbonate 0.22 μm membranes. HPLC was carried out on a C30 carotenoid column (250 mm x 4,6 mm, 5 μm; YMC Europe), as previously described (Araya-Garay et al. [2012]). Carotenoids were identified by comparing their HPLC retention time and color with commercial standards. The β-carotene and astaxanthin standards were obtained from Sigma-Aldrich (Madrid, Spain). For each elution, a Maxplot chromatogram was obtained, displaying the carotenoid elution profile and its corresponding maximum absorbance wavelength. Qualitative analyses were carried out by comparing the carotenoid profiles obtained with the retention times for the β-carotene and astaxanthin standards.
Results
Construction of expression plasmids
The coding regions for genes crtW and crtZ, from A. aurantiacum, were PCR amplified from the plasmid pAK96K (Misawa et al. [1995]). The PCR products were then subcloned into the Sfu I and EcoR I sites of pGAPZαA, an expression vector containing a constitutive GAP promoter and an AOX-1 terminator, generating plasmids pGAPZA-W and pGAPZA*-Z (without Avr II site) (Figure 2). The BamH I–Bgl II fragment from plasmid pGAPZA*-Z was then subcloned into the BamH I restriction site of plasmid pGAPZA-W and the resulting construct integrated into P. pastoris Pp-EBIL DNA genome by recombination events. Finally, plasmid pGAPZA-WZ (5046 bp) was designed, constructed and introduced into the yeast P. pastoris to produce astaxanthin (Figure 2).
Characterization of the Pichia pastoris recombinant clones
The wild type P. pastoris X-33 yeast cells, shown in Figure 3A, display the typical white color characteristic of this strain. On the other hand, the Pp-EBIL recombinant strain, we used as the base for our transformation, shows an orange color (Figure 3B) typical of a strain producing lycopene and β-carotene (Araya-Garay et al. [2012]). Finally, integration of the plasmid pGAPZA-WZ into the Pp-EBIL genome resulted in yet another visible change in the color of the recombinant cells. The red cultures thus obtained (Figure 3C) are what will be expected from cells capable of de novo production of the carotenoid astaxanthin. The Pp-EBILWZ recombinant cells were confirmed, by PCR analyses, to contain the six recombinant genes we transformed integrated in their genomic DNA.

Photographs of P. pastoris wet cell pellets (left) and agar plates (right). (A) Non-transgenic culture; (B) Recombinant Pp-EBIL cells producing lycopene and β-carotene; (C) Recombinant Pp-EBIL strain harboring the plasmid pGAPZA-WZ and producing lycopene, β-carotene and astaxanthin. The reddish color corresponds to the carotenoids produced by the transgenic cultures.
HPLC analyses of carotenoids
To further confirm the nature and composition of the carotenoids produced by the red recombinant Pp-EBILWZ cultures, the photochromic compounds were extracted from the lyophilized cells and analyzed by high resolution liquid chromatography, coupled to a photodiode array detector (HPLC-PDA). These analyses revealed that the Pp-EBIL strain, carrying the plasmid pGAPZA-WZ, did indeed synthesize astaxanthin and this was accompanied by the accumulation of biosynthesis precursors, such as lycopene, β-carotene and a small amount of canthaxanthin, but no zeaxanthin was detected (Figure 4). The astaxanthin concentration produced by the cultures was 3.7 μg per g of cells (dry weight).

MaxPlot chromatograms of cell extracts from P. pastoris : (A) Pp-EBIL strain, producing lycopene (peak 1) and β-carotene (peak 2); (B) Pp-EBIL strain harboring plasmid pGAPZA-WZ and producing lycopene (peak 1), β-carotene (peak 2) and astaxanthin (peak 3).
It has now been known for some time (Komar et al. [1999]) that synonymous codon substitutions may not always be silent, they can change protein structure and function and can be responsible for low expression of heterologous proteins (recently reviewed by (Angov [2011])). To investigate whether the low astaxanthin production by our recombinant Pp-EBIL strain could be attributed to differences in synonymous codon usage between expression and natural hosts, we used the Graphical Codon Usage Analyser 2.0 (Fuhrmann et al. [2004]) to compare codon usage by the expression host (P. pastoris), the two natural hosts (E. uredovora and A. aurantiacum) and the fig tree (F. carica). As shown in Figure 5, the codon usage by E. uredovora is markedly different (differences ranging from 33.72 to 35.66%) from that of P. pastoris. The difference is even more marked (~51%) with A. aurantiacum, whereas the fig tree appears to be more closely related to our expression host (only 19.31% differences).

Differences in synonymous codon usage in Pichia pastoris genome, as compared to Erwinia uredovora ctrE , ctrB and ctrI genes, Agrobacterium aurantiacum ctrW and ctrZ genes, and Ficus carica crtL gene.
From the Figure 5 results, it appears that the low astaxanthin production by our recombinant Pp-EBIL strain could indeed be due to differences in synonymous codon usage between P. pastoris and the recombinant genes natural hosts, and this is an area we are currently investigating.
Discussion
Although S. cerevisiae and P. pastoris share considerable genetic similarity that has enabled expression of similar genes and compatibility between vectors, P. pastoris has a strong preference for respiratory metabolism. This means that the latter can grow at high cell densities without the accumulation of ethanol, an event that usually occurs in S. cerevisiae (Cereghino et al. [2002]) and hinders culture growth and hence protein production. Other advantages of using P. pastoris for heterologous protein expression reside on the simplicity of this system, the availability of strong promoters to drive gene expression, and the ability of this system to perform eukaryotic post-translational modifications at low cost (Cregg et al. [2002]; Lin Cereghino & Cregg [2000]).
On the other hand, yeasts have several cellular organelles which are physically separated from other cellular components by membrane structures (Karpichev & Small [2000]). The heterologously expressed six enzymes were designed to be randomly distributed in P. pastoris, and both cellular and cytoplasmic membranes can be putative locations for membrane-bound enzymes to settle in (Bhataya et al. [2009]). Therefore, since other yeasts such as S. cerevisiae and X. dendrorhous have similarity on the structural constrains of the cells and they have higher levels of astaxanthin production, we believe that the structural constrains of P. pastoris is it not a limit factor for astaxanthin production.
In the present work, we have succeeded in constructing genetically-stable astaxanthin-producing P. pastoris strains (Pp-EBILWZ). We achieved this by introducing the carotenogenic genes crtW (β-carotene ketolase) and crtZ (β-carotene hidroxylase) into a β-carotene-producing P. pastoris strain (Pp-EBIL) we previously engineered (Araya-Garay et al. [2012]) under the control of a GAP promoter.
DNA integration into a GAP locus requires linearization of the expression vectors with Avr II, and there is a recognition site for this restriction enzyme within the coding region of the GAP promoter. To avoid this complication, we removed, by site-directed mutagenesis, the Avr II restriction site within the pGAPZαA plasmid thus generating the silent-mutated plasmid pGAPZαA*. This plasmid was further modified by addition of the two crt genes required for the synthesis of astaxanthin from β-carotene (Figure 2), giving rise to the integrative plasmid we named pGAPZA-WZ. Recombinant plasmid pGAPZA-WZ was then integrated into Pp-EBIL genomic DNA, resulting in the production of yeast cells with a red coloration (Figure 3).
To determine the composition of the carotenoids produced by Pp-EBILWZ, this strain was grown for 3 days in liquid culture containing Zeocin (200 μg/mL), and the carotenoid content in the yeast cells analyzed by HPLC.As shown in Figure 4, our recombinant P. pastoris strain was indeed capable of synthesizing new xanthophylls, but its astaxanthin production level was below its β-carotene production. Additionally, the accumulation of astaxanthin metabolic intermediates indicates that the flux through the carotenogenic pathway was not fully efficient. The astaxanthin yield we obtained from our recombinant yeast is lower than those previously reported for heterologous astaxanthin production in C. utilis (Miura et al. [1998]) with the amounts of 400 μg per g of cells (dry weight) and S. cerevisiae (Ukibe et al. [2009]) with 29 μg per g of cells (dry weight); although it is very close to the yield obtained in M. circinelloides (Papp et al. [2006]) with 3 μg per g of cells (dry weight). Whereas in other microorganisms such as X. dendrorhous and H. pluvialis a significantly higher level of production are observed (120 μg and 114 μg per g of cells [dry weight], respectively). It should be noted that Pp-EBIL cells accumulated more β-carotene (339 μg per g [dry weight] of cells) than the total amounts of astaxanthin and β-carotene in the wild-type cells of X. dendrorhous (270 μg per g [dry weight] of cells). The Pp-EBILWZ had an additional drawback, as its growth was slower than that of the Pp-EBIL strain it originated from.
From the results shown in Figure 5, it appears that the low astaxanthin production by our recombinant Pp-EBILWZ strain could be due to differences in synonymous codon usage between P. pastoris and the recombinant genes natural hosts. This codon usage appears to be related to the intracellular availability of each tRNA, whose concentration is relatively proportional to the frequency of its complementary codon coding sequences population. This suggests that the speed of translation and, therefore, carotenoid protein production, may be limited and our recombinant strain cannot achieve high protein expression level for all of the six foreign genes the cells host. It must also be taken into account that the six recombinant genes are all members of the same pathway and are under the same GAP promoter. This could cause metabolic stress in the yeast cells, by limiting the availability of transcription factors required for proper expression of all the pathway proteins. Metabolic overload could be the cause of the slowing down of the cell growth observed in Pp-EBILWZ, as compared with the two strains (Pp-EBIL and P. pastoris X-33) it originates from.
However, based on the published strategies for improvements in the production of carotenoids described for other organisms, either by over-expression of genes, codon usage optimization or modification of gene members of the pathway, we believe that it is possible to increase our current astaxanthin production levels in P. pastoris to an industrially-relevant yield. One approach worth considering is that reported by Verwaal et al. (Verwaal et al. [2007]) and Yuan et al. (Yuan et al. [2006]), using mutated cultures and special fermentation conditions in large volumes. This strategy has worked well for X. dendrorhous, resulting in a marked increase in astaxanthin production (An et al. [1989]).
In conclusion, the results shown here indicate that it is indeed feasible to biosynthesize astaxanthin using the β-carotene-producing P. pastoris strain (Pp-EBIL) here described, although further investigation is required in order to improve the protein yield. This represents a further step in recombinant carotenoid production, and carotenoids, astaxanthin in particular, play an important role in the aquaculture industry and their addition into the fish and prawn feed not only increases their nutritional value, but also considerably enhances their appeal to customers and hence their commercial value. Additionally, there is increasing concern about food security, in particular fish and sea food, and aquaculture is progressively replacing shortages in fish catches, caused by overfishing, pollution, climate change and other insults to the marine habitats.
References
An GH, Shuman DB, Johnson EA: Isolation of Phaffia rhodozyma mutants with increased astaxanthin content. Appl Environ Microbiol 1989, 55: 116–124.
Angov E: Codon usage: nature’s roadmap to expression and folding of proteins. Biotech J 2011, 6: 650–659. 10.1002/biot.201000332
Araya-Garay JM, Feijoo-Siota L, Rosa-dos-Santos F, Veiga-Crespo P, Villa TG: Construction of new Pichia pastoris X-33 strains for production of lycopene and β-carotene. Appl Microbiol Biotechnol 2012, 93: 2483–2492. 10.1007/s00253-011-3764-7
Armstrong GA, Alberti M, Leach F, Hearst JE: Nucleotide sequence, organization, and nature of the protein products of the carotenoid biosynthesis gene cluster of Rhodobacter capsulatus. Mol Gen Genet 1989, 216: 254–268. 10.1007/BF00334364
Bertram JS, Vine AL: Biochim Biophys Acta. 2005, 1740: 170–178. 10.1016/j.bbadis.2005.01.003
Bhataya A, Schmidt-Dannert C, Lee PC: Metabolic engineering of Pichia pastoris X-33 for lycopene production. Process Biochem 2009, 44: 1095–1102. 10.1016/j.procbio.2009.05.012
Bubrick P: Production of astaxanthin from Haematococcus. Bioresour Technol 1991, 38: 237–239. 10.1016/0960-8524(91)90161-C
Cereghino GP, Cereghino JL, Ilgen C, Cregg JM: Production of recombinant proteins in fermenter cultures of the yeast Pichia pastoris: heterologous protein expression in the methylotrophic yeast Pichia pastoris. Curr Opin Biotechnol 2002, 13: 329–332. 10.1016/S0958-1669(02)00330-0
Cregg JM, Cereghino JL, Shi J, Higgins DR: Recombinant protein expression in Pichia pastoris. Mol Biotechnol 2002, 16: 23–52.
Dos Santos RA, da Silva R, Kalil S, Veiga A, de Fernandes J: Different cell disruption methods for astaxanthin recovery by Phaffia rhodozyma. Afr J Biotechnol 2011, 10: 1165–1171.
Fuhrmann M, Hausherr A, Ferbitz L, Schödl T, Heitzer M, Hegemann P: Monitoring dynamic expression of nuclear genes in Clamydomonas reinhardtii by using a synthetic luciferace reporter gene. Plant Mol Biol 2004, 55: 869–881.
Harker M, Hirschberg J: Biosynthesis of ketocarotenoids in transgenic cyanobacteria expressing the algal gene for beta-C-4-oxygenase, crtO. FEBS Lett 1997, 404: 129–134. 10.1016/S0014-5793(97)00110-5
Harker M, Hirschberg J, Oren A: Paracoccus marcusii sp. nov., an orange gram-negative coccus. Int J Syst Bacteriol 1998, 48: 543–548. 10.1099/00207713-48-2-543
Hix LM, Lockwood SF, Bertram JS: Bioactive carotenoids: potent antioxidants and regulators of gene expression. Redox Rep 2004, 9: 181–191. 10.1179/135100004225005967
Johnson EA, Villa TG, Lewis MJ: Phaffia rhodozyma as an astaxanthin sources in salmonid diet. Aquaculture 1980, 20: 123–134. 10.1016/0044-8486(80)90041-1
Karpichev IV, Small GM: Evidence for a novel pathway for the targeting of a Saccharomyces cerevisiae peroxisomal protein belonging to the isomerase/hydratase family. J Cell Sci 2000, 113: 533–544.
Komar AA, Lesnik T, Reiss C: Synonymous codon substitutions affect ribosome traffic and protein folding during in vitro translation. FEBS Lett 1999, 462: 387–391. 10.1016/S0014-5793(99)01566-5
Kozuki Y, Miura Y, Yagasaki K: Inhibitory effects of carotenoids on the invasion of rat ascites hepatoma cells in culture. Cancer Lett 2000, 151: 111–115. 10.1016/S0304-3835(99)00418-8
Krubasik P, Sandmann G: A carotenogenic gene cluster from Brevibacterium linens with novel lycopene cyclase genes involved in the synthesis of aromatic carotenoids. Mol Gen Genet 2000, 263: 423–432. 10.1007/s004380051186
Kurihara H, Koda H, Asami S, Kiso Y, Tanaka T: Contribution of the antioxidative property of astaxanthin to its protective effect on the promotion of cancer metastasis in mice treated with restraint stress. Life Sci 2002, 70: 2509–2520. 10.1016/S0024-3205(02)01522-9
Lange N, Steinbüchel A: β-carotene production by Saccharomyces cerevisiae with regard to plasmid stability and culture media. Appl Microbiol Biotechnol 2011, 91: 1611–1622. 10.1007/s00253-011-3315-2
Lee JH, Kim YS, Choi TJ, Lee WJ, Kim YT: Paracoccus haeundaensis sp. nov., a gram-negative, halophilic, astaxanthin-producing bacterium. Int J Syst Evol Microbiol 2004, 54: 1699–1702. 10.1099/ijs.0.63146-0
Lin Cereghino J, Cregg JM: Heterologous protein expression in the methylotrophic yeast Pichia pastoris. FEMS Microbiol Rev 2000, 24: 45–66. 10.1111/j.1574-6976.2000.tb00532.x
Miki W, Yamaguchi K, Konosu S: Comparison of carotenoids in the ovaries of marine fish and shellfish. Comp Biochem Physiol 1982, 71: 7–11.
Misawa N, Shimada H: Metabolic engineering for the production of carotenoids in non-carotenogenic bacteria and yeasts. J Biotechnol 1998, 59: 169–181. 10.1016/S0168-1656(97)00154-5
Misawa N, Nakagawa M, Kobayashi K, Yamano S, Izawa Y, Nakamura K, Harashima K: Elucidation of the Erwinia uredovora carotenoid biosynthetic pathway by functional analysis of gene products expressed in Escherichia coli. J Bacteriol 1990, 172: 6704–6712.
Misawa N, Satomi Y, Kondo K, Yokoyama A, Kajiwara S, Saito T, Ohtani T, Miki W: Structure and functional analysis of a marine bacterial carotenoid biosynthesis gene cluster and astaxanthin biosynthetic pathway proposed at the gene level. J Bacteriol 1995, 177: 6575–6584.
Miura Y, Kondo K, Saito T, Shimada H, Fraser PD, Misawa N: Production of the carotenoid lycopene, beta-carotene, and astaxanthin in the food yeast Candida utililis. Appl Environ Microbiol 1998, 64: 1226–1229.
Neuman I, Nahum H, Ben-Amotz A: Reduction of exercise-induced asthma oxidative stress by lycopene, a natural antioxidant. Allergy 2000, 55: 1184–1189. 10.1034/j.1398-9995.2000.00748.x
Papp T, Velayos A, Bartók T, Eslava AP, Vágvölgyi C, Iturriaga E: Heterologous expression of astaxanthin biosynthesis genes in Mucor circinelloides. Appl Microbiol Biotechnol 2006, 69: 526–531. 10.1007/s00253-005-0026-6
Ukibe K, Hashida K, Yoshida N, Takagi H: Metabolic engineering of Saccharomyces cerevisiae for astaxanthin production and oxidative stress tolerance. Appl Environ Microbiol 2009, 75: 7205–7211. 10.1128/AEM.01249-09
Verdoes JC, Krubasik P, Sandmann G, van Ooyen AJ: Isolation and functional characterisation of a novel type of carotenoid biosynthetic gene from Xanthophyllonyces dendrorhous. Mol Gen Genet 1999, 262: 453–461. 10.1007/s004380051105
Verwaal R, Wang J, Meijnen J-P, Visser H, Sandmann G, van den Berg JA, van Ooyen A: High-level production of beta-carotene in Saccharomyces cereviseae by successive transformation with carotenogenic genes from Xanthophyllomyces dendrorhous. Appl Environ Microbiol 2007,73(13):4342–4350. 10.1128/AEM.02759-06
Wade N, Goulter KC, Wilson KJ, Hall MR, Degnan BM: Esterified astaxanthin levels in lobster epithelia correlate with shell colour intensity: potential role in crustacean shell colour formation. Comp Biochem Physiol Biochem Mol Biol 2005, 141: 307–313. 10.1016/j.cbpc.2005.04.004
Yamamo S, Ishii T, Nakagawa M, Ikenaga H, Misawa N: Metabolic engineering for production of beta-carotene and lycopene in Saccharomyces cerevisiae. Biosci Biotechnol Biochem 1994, 58: 1112–1114. 10.1271/bbb.58.1112
Yuan LZ, Rouviere PE, LaRossa RA, Suh W: Chromosomal promoter replacement of the isoprenoid pathway for enhancing carotenoid production in E. coli. Metab Eng 2006,8(1):79–90. 10.1016/j.ymben.2005.08.005
Acknowledgements
J. M. A-G. is the recipient of an AECID scholarship from the Spanish Foreign Affairs Ministry. The authors thank Dr. Norihiko Misawa (Research Institute for Bioresources and Biotechnology, Ishikawa Prefectural University) for the gift of plasmid pAK96K, and both the Faculty of Pharmacy and School of Biotechnology for their support throughout this project.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Araya-Garay, J.M., Ageitos, J.M., Vallejo, J.A. et al. Construction of a novel Pichia pastoris strain for production of xanthophylls. AMB Expr 2, 24 (2012). https://doi.org/10.1186/2191-0855-2-24
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2191-0855-2-24