
Abstract
Hydroquinone-1,2-dioxygenase, an enzyme involved in the degradation of alkylphenols in Sphingomonas sp. strain TTNP3 was purified to apparent homogeneity. The extradiol dioxygenase catalyzed the ring fission of hydroquinone to 4-hydroxymuconic semialdehyde and the degradation of chlorinated and several alkylated hydroquinones. The activity of 1 mg of the purified enzyme with unsubstituted hydroquinone was 6.1 μmol per minute, the apparent Km 2.2 μM. ICP-MS analysis revealed an iron content of 1.4 moles per mole enzyme. The enzyme lost activity upon exposure to oxygen, but could be reactivated by Fe(II) in presence of ascorbate. SDS-PAGE analysis of the purified enzyme yielded two bands of an apparent size of 38 kDa and 19 kDa, respectively. Data from MALDI-TOF analyses of peptides of the respective bands matched with the deduced amino acid sequences of two neighboring open reading frames found in genomic DNA of Sphingomonas sp strain TTNP3. The deduced amino acid sequences showed 62% and 47% identity to the large and small subunit of hydroquinone dioxygenase from Pseudomonas fluorescens strain ACB, respectively. This heterotetrameric enzyme is the first of its kind found in a strain of the genus Sphingomonas sensu latu.
Similar content being viewed by others


Introduction
Both Sphingomonas sp. strain TTNP3 and Sphingobium xenophagum Bayram are able to degrade several branched isomers of nonylphenol and bisphenol A, well-known endocrine disruptors, by ipso substitution. i.e. ipso-hydroxylation and subsequent detachment of the side chain of the alkylphenol. In these pathways hydroquinone is formed as a key metabolite (Kolvenbach et al. 2007; Corvini et al. 2006; Gabriel et al. 2007a; Gabriel et al. 2007b; Gabriel et al. 2005). Hydroquinone (HQ) is also a key intermediate in the degradation of several other compounds of environmental importance, such as 4-nitrophenol (Spain and Gibson 1991), γ-hexachlorocyclohexane (Miyauchi et al. 1999), 4-hydroxyacetophenone (Moonen et al. 2008a) and 4-aminophenol (Takenaka et al. 2003).
There are two established pathways in the literature for the degradation of hydroquinone. One involves direct ring cleavage of hydroquinone by dioxygenases containing Fe(II) in their active center, resulting in the formation of 4-hydroxymuconic acid semialdehyde (HMSA) (Chauhan et al. 2000; Miyauchi et al. 1999; Moonen et al. 2008b). The second pathway requires the hydroxylation of hydroquinone to benzene-1,2,4-triol (Eppink et al. 2000) which is then cleaved to yield maleylacetic acid (Rieble et al. 1994; Jain et al. 1994) by dioxygenases containing Fe(III) in their active center (Latus et al. 1995; Travkin et al. 1997; Ferraroni et al. 2005).
The hydroquinone dioxygenases (HQDO) can be divided into two subtypes that have few similarities. Members of type I are phylogenetically related to the well-described extradiol catechol dioxygenases, (Eltis and Bolin 1996) and are monomeric (Xu et al. 1999). Moreover, they are involved in the degradation of HQ and chlorinated HQ formed during degradation of pentachlorophenol and γ-hexachlorocyclohexane by several members of the Sphingomonas genus (Cai and Xun 2002; Miyauchi et al. 1999; Lal et al. 2010). Supposedly, more homologs exist as DNA sequences with similarities of 99% and higher to the PcpA encoding sequence have been attributed to γ-hexachlorocyclohexane degradation in other sphingomonads, i.e. strains of the genus Sphingomonas sensu latu (Dogra et al. 2004; Manickam et al. 2008; Yamamoto et al. 2009; Lal et al. 2010) and nitrophenol degradation in Cupriavidus necator Jmp134(Yin and Zhou 2010). Type II dioxygenases consist of two different subunits forming an α2β2 heterotetramer. These enzymes are responsible for ring cleavage of HQ formed during degradation in the degradation pathway of hydroxyacetophenone (Moonen et al. 2008b) and in the degradation pathway of p-nitrophenol (Wei et al. 2010; Zhang et al. 2009; Shen et al. 2010). Interestingly, members of type II have not been found in sphingomonad strains yet.
Recently, PcpA, a type I HQDO from Sphingobium chlorophenolicum, has been subjected to homology based structural modeling in combination with site directed mutagenesis, yielding information on the native tertiary structure and the histidine residues responsible for chelating the Fe(II) in the active center (Machonkin et al. 2009). However little is known about HQDO in general, as until now only the HQDO from Pseudomonas fluorescens strain ACB has been purified and thoroughly characterized (Moonen et al. 2008b).
Here, we describe the purification and the properties of a novel type II heterotetrameric HQDO that we isolated from Sphingomonas sp. strain TTNP3.
Materials and methods
Materials
Tris, ammonium sulfate, ascorbic acid were purchased from Applichem (Axon Lab, Baden-Dättwil, Switzerland), hydroquinone and technical grade nonylphenol were purchased from Fluka (Buchs, Switzerland). Standard I Medium was purchased from Merck (Zug, Switzerland). Methylhydroquinone was obtained from Sigma (Buchs, Switzerland), ethylhydroquinone and t-butylhydroquinone were obtained from ACBR (Karlsruhe, Germany), propyl-, pentyl- and hexylhydroquinone were obtained from Labotest (Niederschöna, Germany). 2-(1-methyl-1-octyl)-hydroquinone was synthesized by Friedel-Crafts alkylation from hydroquinone with 2-nonanol obtained from Sigma (Buchs, Switzerland) according to the protocol of Corvini et al: (Corvini et al. 2004b). All other chemicals were of analytical grade. All columns used for protein purification were purchased from GE Healthcare (Uppsala, Sweden).
Bacterial strains and culture conditions
Sphingomonas sp. strain TTNP3 was obtained from Professor Willy Verstraete (LabMet, University Ghent, Belgium). The strain was grown on Standard I Medium as described previously (Corvini et al. 2004c). Enzymatic activity was induced by the addition of 0.5 mM technical grade nonylphenol 16 hours prior to harvesting the cells at an OD550 of about 3.0. Cultures were then centrifuged at 4,500 * g for 15 minutes, resuspended in 50 mM Tris, pH 7.5 at 4°C. This washing procedure was repeated twice. In the last step, the cells were resuspended to an OD550 of 60 and stored at -20°C.
Sequence data
DNA analysis of Sphingomonas sp. strain TTNP3 was performed with data obtained from genome shotgun sequencing.
Nucleotide sequence accession number
The nucleotide and amino acid sequence data reported in this paper have been deposited in the GenBank sequence database under accession number JF440299.
Purification of HQDO from strain TTNP3
Purification steps were performed on a Pharmacia FPLC liquid chromatography system. All steps were performed at 4°C, unless stated otherwise. Buffers for purification were stored under argon (Messer AG, Switzerland). Thawed cells were diluted to an OD550 of 20 in 16 mL 50 mM Tris, pH 7.5, 4-hydroxybenzoic acid (HBA, 1 M in Ethanol) and ascorbic acid (0.5 M dissolved in equimolar NaOH) were added to final concentration of 0.5 mM and 2.5 mM, respectively. Cells were disrupted by sonication on ice (20 minutes at 100% intensity, 0.6 s/s duty cycle using a Labsonic M sonicator by B. Braun Biotech, equipped with a 3 mm probe). After centrifugation (21,500 * g for 15 min), five preparations of cell extract were pooled to a volume of 65 mL and subjected to ammonium sulfate precipitation, by adding ammonium sulfate to 40% saturation with subsequent centrifugation at 21,500 * g for 30 min. The supernatant was diluted to 20% ammonium sulfate saturation with 50 mM Tris, pH 7.5, containing 0.5 mM HBA (buffer A) and loaded onto two coupled Phenyl Sepharose High Performance columns with a total volume of 10 mL, previously equilibrated with buffer A containing 20% ammonium sulfate (buffer B). After washing with 40 mL of buffer B, HQDO activity was eluted by applying a linear gradient from 100% buffer B to 100% buffer A in 100 mL. Active fractions were pooled and desalted over 4 coupled Hi Trap Desalting columns (total volume of 20 mL), equilibrated with buffer A, and then applied to a 20 mL DEAE column. After washing with 40 mL buffer A, proteins were eluted with a linear gradient from 0 to 400 mM NaCl in 200 mL buffer A. Active fractions were desalted as described above and loaded onto a Mono Q column. After washing with 10 mL buffer A, activity was eluted with a linear gradient from 0 to 1 M NaCl in 40 mL buffer A and stored at -20°C under argon. Size exclusion chromatography of the native enzyme was carried out on a HP Agilent Series 1050 HPLC system (Agilent Technologies, Basel, Switzerland) equipped with a Superose 6 column equilibrated with phosphate buffer (10 mM, pH 7.0) containing 137 mM NaCl. The system was calibrated with a standard mixture of thyroglobulin, myosin, ovalbumin, RNAse A and aprotinin (Sigma, Switzerland) and detection was carried out at 280 nm
Enzyme activity
Enzyme activity was routinely measured at 25°C by measuring the formation of HMSA at 320 nm (ε320 = 11000 M-1 * cm-1 (Spain and Gibson 1991)) on a Synergy 2 multi-mode microplate reader (Biotek, Luzern, Switzerland). The assay mixture (250 μL) typically contained ca. 50 nM enzyme solution in 250 μL air saturated 50 mM Tris, pH 7.0, reactions were started by the addition of 100 μL freshly prepared solution of 350 μM HQ in 50 mM Tris buffer, pH 7.0, resulting in a final substrate concentration of 100 μM. Activity of HQDO on substituted hydroquinones was determined by measuring oxygen consumption with a Clarke type oxygen electrode (Oxytherm system, Hansatech, Reutlingen, Germany). To a total volume of 800 μL, about 100 nM of enzyme was added before the addition of 8 μL of an ethanolic solution of 20 mM substrate to reach a final substrate concentration of 200 μM.
As the enzyme was subject to suicide deactivation upon incubation with HQ, only initial rates recorded within 20 seconds after the addition of substrate were used for determination of kinetics. kM was determined by Prism version 5.02(GraphPad).
Enzyme stability
The stability of HQDO at 30°C was studied by incubating the purified enzyme in 50 mM Tris buffer, pH 7.0 at 30°C in absence of 4-HBA under argon and, in the presence and absence of 0.5 mM 4-HBA under normal atmosphere, respectively.
Enzyme inactivation by iron chelators
The inactivation of HQDO was determined by incubation of the purified enzyme at 30°C in the presence of 0.1 mM and 1 mM 2,2'-bipyridyl, 0.1 mM and 1 mM o-phenanthroline, respectively, before testing for remaining activity after 15 minutes. The purified enzyme was also incubated at 30°C in the presence of 0.1 mM hydrogen peroxide, before assaying for remaining activity after one minute.
Protein content/SDS-PAGE
Protein content was determined using the Bio-Rad Protein Assay (Biorad) using lysozyme as a standard. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was carried out with 15% Tris-glycine minigels according to a standard protocol (Laemmli 1970) in a Mini-PROTEAN Tetra Cell (BioRad).
ICP-MS
Iron concentrations in fractions eluting from the MonoQ columns were determined using an inductively coupled plasma-mass spectrometry (ICP-MS) system (Agilent 7500cx) equipped with an Octopole Reaction System. Water and hydrochloric acid were added to 750 μL of each fraction to a total volume of 2 mL and a HCl concentration of 1.5%, before measuring the samples on the inductively coupled plasma-mass spectrometry system. The measurements were performed using a radio frequency power of 1500W, a carrier gas flow of 0.79 L/min, a make-up gas flow of 0.30 L/min at a sample depth of 8 mm. Fe was quantified on m/z = 56 whereas m/z = 57 served as control to verify quantification results. Other elements assayed were Mg (m/z = 24), Mn (m/z = 55), Ni (m/z = 60). All measurements were carried out in collision mode with an optimized helium flow of 5 mL/min. Indium served as internal standard.
GC-MS
Samples for GC-MS analysis were acidified with a drop of 6 M HCl and extracted with two volumes of ethyl acetate three times; the organic phase was dried over Na2SO4 before evaporation under a gentle nitrogen stream. Extracts were redissolved in acetonitrile/BSTFA (90:10 v/v) for derivatization at 75°C for 15 minutes. Samples were analyzed in an Agilent 7890A series gas chromatograph (Agilent Technologies, Basel, Switzerland) equipped with a Zebron ZB-5MS column, (30 m by 0.25 mm, 0.25 μm film thickness, Phenomenex) coupled to an Agilent 5975C series mass spectrometer. The mass selective detector (EI) was operated in the scan mode (mass range m/z 50-600) with an electron energy of 70 eV. The temperature program was 70°C for 3 min, 8°C per minute to 250°C; the injector temperature was 90°C; the interface temperature 280°C. The injection volume was 1 μL (split 1:30). The carrier gas was helium (1 mL/min).
Protein identification
Briefly, protein bands were picked from the SDS gel. The proteins were digested tryptically in gel and identified by nanoHPLC-nanoESI-MS/MS. Fully automated online pre-concentration and separation of the tryptically digested samples was performed using a set of capillary- and nanoHPLC instruments of the 1100 Series (Agilent, Waldbronn, Germany) operated in series. Mass spectrometric detection was carried out by online coupling nanoHPLC with a QSTAR XL (QqTOF) mass spectrometer (Applied Biosystems/MDS/Sciex, Darmstadt, Germany) operated in MS and MS/MS mode. The instrument was equipped with an online nano-electrospray ion source (NanoSpray II Source) and upgraded with a heated interface (Vester et al. 2009).
A first data interpretation of acquired product-ion spectra of the nanoHPLC-nanoESI-MS/MS analysis, was performed by an automatic database search with MASCOT™ (version 2.2, Matrix Science, London, UK) (Perkins et al. 1999). For all searches, the MASCOT peptide fragmentation mass fingerprint algorithm screening against all species of the actual NCBI non-redundant database (2010-04-20) was used to identify the corresponding peptides. A detailed description of this procedure was previously reported (Vester et al. 2009). Additionally, most abundant peptides were selected and manually de novo sequenced using an in-house software tool.
Phylogenetic analysis of HqdA and HqdB
A phylogenetic tree of HqdA and HqbB found in Sphingomonas sp. strain TTNP3 and respectively corresponding sequences from 21 other bacterial strains that were found to be similar by BLAST analysis was constructed by rendering a ClustalX 2 alignment and using Treeview 1.6.6
Results
Purification of HQDO from Sphingomonas sp. strain TTNP3
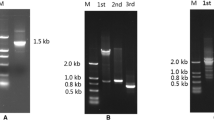
Even though strain TTNP3 appears to express the HQ cleaving enzyme constitutively (Corvini et al. 2006), higher amounts of enzyme activity could be achieved by inducing the cells with technical nonylphenol mixture prior to harvesting them. Without the addition of a reversible inhibitor, HQDO lost activity rapidly, impeding success of early purification attempts. Table 1 presents the result of a typical preparation of purified enzyme from 8 g of cells. Purification in four steps typically resulted in a yield of 30%, a purification factor of 42 and a specific activity of 6.1 U mg-1. SDS-PAGE analysis showed the presence of two major protein bands, corresponding to masses of 38 kDa and 19 kDa, respectively (Figure 1). The purified enzyme eluted from the Superose 6 column in one symmetrical peak with an apparent molecular mass of 120 kDa (data not shown).

SDS-PAGE of HQDO from Sphingomonas sp. strain TTNP3. Lane A, marker proteins: lane B, crude cell extract; lane C, ammonium sulfate fractionation supernatant; lane D, phenyl-Sepharose pool; lane E, DEAE pool; lane F; MonoQ pool.
Physico-chemical properties of the enzyme
ICP-MS analysis of the fractions eluting from the final purification step, i.e. MonoQ column, revealed a clear correlation between the enzyme activity in the fraction and its respective iron content. Based on the apparent molecular mass of 120 kDa, 1 μmol of enzyme contained 1.4 μmol of iron and 0.04 μmol of manganese. Other metal species could not be attributed to fractions containing enzyme activity. HQDO showed an absorption maximum at 279 nm, slight absorption between 300 nm and 400 nm, yet none longer wavelengths.
Catalytic properties
HQDO from Sphingomonas sp. strain TTNP3 catalyzed the ring cleavage of hydroquinone to HMSA under consumption of an equimolar amount of molecular oxygen (data not shown). Maximal enzyme activity was observed between pH 7 and pH 8. The apparent Km for HQ was determined to be 2.2 μM with a standard error of. 0.2. kcat was determined to be 811 min-1 with a standard error of 15 for the heterotetrameric enzyme and k¬cat/kM was determined to be 369 min-1.
HQDO was shown to readily lose activity upon incubation with its substrate, HQ. Inactivation of the enzyme appeared to be irreversible, as enzyme activity could not be restored by incubation with Fe(II) ions (compare Enzyme stability). Nevertheless, fresh enzyme added to a spent reaction mixture transformed the substrate at the normal rate.
Besides acting on hydroquinone as a substrate, HQDO catalyzed the conversion of several other substituted hydroquinones (Table 2). Phenol, catechol, resorcinol and 4-mercaptophenol were not used as substrate by the enzyme (data not shown).
Enzyme activity was inhibited by the substrate analog 4-HBA. Inhibition was shown to be reversible, as samples showed normal reaction rates after removal of 4-HBA by gel filtration (data not shown). A number of other phenolic compounds inhibited the degradation reaction as well. The strongest inhibitions were observed with 4-hydroxybenzonitrile, 4-mercaptophenol, benzoquinone and vanillin (Table 3).
Product identification
GC-MS analysis of the trimethylsilylated HQ ring cleavage products resulted in a chromatogram with five peaks that showed similar mass spectra (Figure 2A, peak 1b: m/z 286 (M+., 1.2%); 271 (M+. - .CH3, 16.4%); 257 (M+. - .CHO, 23.4%); 243 (2.4%); 196 (M+. - .OSi(CH3)3, 2.1%); 169 (M+. - . Si(CH3)3 - CO2, 17.5%); 147 ([(Si(CH3)3)2 + H]+, 48.1%); 143 (M+. - . Si(CH3)3 - CO2 - HC≡CH, 33, 33.3%); 93 (5.1%); 77 (30.1%); 75 (56.2%); 73 +Si(CH3)3, 100%, compare Table 4). Based on mass spectral analysis and published data (Miyauchi et al. 1999; Kohler et al. 1993), we identified the corresponding products as stereoisomers (cis-trans-isomers and conformers) of 4-hydroxmuconic acid semialdehyde (4-hydroxy-6-oxohexa-2,4-dienoic acid).

A, GC-MS total ion chromatogram of the trimethylsilylated ring cleavage product of hydroquinone; B, mass spectrum of peak 1b from Figure 2A; C, GC-MS total ion chromatogram of the trimethylsilylated ring cleavage product of 2-methylhydroquinone; D, mass spectrum of the product peak 2c from Figure 2C.
Similarly, work-up and analysis of the cleavage products of methylhydroquinone showed a chromatogram with six peaks. The spectra corresponding to the two by far most intensive peaks were very similar, showing signals at m/z 300 (M+.), 285 (M+. - .CH3), 271 (M+. - .CHO), 257 (M+. - 43), 210, 183 (M+. - . Si(CH3)3 - CO2), 147 ([(Si(CH3)3)2 + H]+), 143 (M+. - . Si(CH3)3 - CO2 - HC≡CCH3) (compare Figure 2C, peak 2c, Table 5 range above m/z 140). Loss of a neutral mass of 29 amu is indicative of the presence of an aldehyde group. Combining this conclusion with a general mass spectral analysis and biochemical reasoning, we propose that the two major chromatographic peaks correspond to stereo or position isomers of trimethylsilylated methyl-4-hydroxymuconic acid semialdhyde (4-hydroxy-6-oxohexa-2,4-dienoic acid with a methyl substituent at positions 2, 3 or 5). Hence, ring cleavage proceeded between a C-OH and a neighboring C-H group of the methylhydroquinone substrate (and not between the neighboring C-OH and C-CH3 groups). Assuming that m/z 143 ions were produced by loss of H-C≡C-CH3 (R1-C≡C-R2, see additional file 1) from m/z 183 ions (M+. - . Si(CH3)3 - CO2) further restricts the possible cleavage sites to the ring bonds C(1) - C(6) and C(4)-C(5) in methylhydroquinone (cleavage of the C(3) -C(4) bond would have led to a loss of HC≡CH from the m/z 183 ions).
Enzyme stability
After incubation of the desalted enzyme at 30°C under normal atmosphere and without inhibitor for two hours, more than 20% of the initial activity was lost, while no loss of activity was observed when stored under argon or with the inhibitor 4-HBA, respectively. Incubation of 2.7 μM purified enzyme in presence of 0.5 mM inhibitor under argon atmosphere for 15 days, resulted in 16% and 9% loss of activity when kept at 0°C and 20°C, respectively. In the former case, incubation of the enzyme with 0.1 mM Fe2SO4 and 0.1 mM ascorbate on ice for 30 minutes prior to the assay could partially restore the activity, leaving a loss of 5% relative to the initial activity.
Part of the activity could be recovered by incubating the enzyme with 0.1 mM Fe2SO4 and 0.1 mM ascorbate on ice for 30 minutes prior to the assay.
ortho-Phenanthroline and 2,2'-dipyridyl, inactivated HQDO (Table 6). Rapid and complete inactivation also occurred upon incubation of the purified enzyme with the oxidizing agent hydrogen peroxide at 100 μM (Table 6).
Sequence data
nanoHPLC-nanoESI-MS/MS-analysis of bands resulting from SDS-PAGE of the purified enzyme and subsequent de novo sequencing yielded four peptides for the 19 kDa band, and six peptides for the 38 kDa band, respectively. These matched with amino acid sequences deduced from open reading frames that had been identified in genomic DNA from Sphingomonas sp. strain TTNP3, tentatively named HqdA and HqdB (Figure 3). A MASCOT search against a user database containing the sequences of HqdA and HqdB confirmed the identification for HqdA (Mowse Score: 435, sequence coverage: 30%) and HqdB (Mowse Score: 318, sequence coverage: 44%). HqdA and HqdB showed a sequence identity of 61% and 47% compared to the small and large subunit of HQDO from Pseudomonas fluorescens strain ACB, respectively.

Amino acid sequences deduced from open reading frames found in a part of genomic DNA from Sphingomonas sp. strain TTNP3. The amino acids marked with asterisks were identified by nanoHPLC-nanoESI-MS/MS (from residues 175 to 194, two peptides were matched, one ranging from 175 to 184, the other one from 185 to 194).
A dendrographic tree of HqdA and HqbC found in S. sp. strain TTNP3 and respectively corresponding sequences from 21 other bacterial strains that were found to be similar by BLAST analysis was constructed by amino acid sequence alignment via Clustal × version 2.0.11 (Larkin et al. 2007) and drawn by Treeview version 1.6.6 http://taxonomy.zoology.gla.ac.uk/rod/treeview.html (Figure 4). For complete multiple sequence alignments refer to the additional files 2 and 3.

Phylogenetic trees of the sequences of HqdA (A) and HqdB (B) and the respective homolog sequences from Burkholderia sp. 383 (gi 78063587 and gi 78063586), Burkholderia sp. CCGE1002 (gi 295680998 and gi 295680997), Burkholderia sp. H160 (gi 209517843 and gi 209517844), Burkholderia ambifaria AMMD (gi 115359956 and gi 115359957), Burkholderia ambifaria IOP40-10 (gi 170700037 and gi 170700038), Burkholderia ambifaria MC40-6 (gi 172062406 and gi 172062407), Burkholderia ambifaria MEX-5 (gi 171319707 and gi 171319708), Burkholderia cenocepacia HI2424 (gi 116691528 and gi 116691529), Burkholderia cenocepacia J2315 (gi 206562327 and gi 206562328), Burkholderia multivorans ATCC 17616 (gi 161523095 and gi 161523094) Burkholderia multivorans CGD1 (gi 221212137 and gi 221212136), Burkholderia multivorans CGD2M (gi 221199017 and gi 221199016), Burkholderia phymatum STM815 (gi 186470422 and gi 186470423), Photorhabdus luminescens subsp. laumondii TTO1 (gi 37524165 and gi 37524166), Pseudomonas aeruginosa PA7 (gi 152988009 and gi 152987326), Pseudomonas fluorescens ACB (gi 182374631 and gi 182374632), Pseudomonas putida (gi 224460045 and gi 260103908), Pseudomonas sp. 1-7 (gi 284176971 and gi 284176972), Pseudomonas sp. NyZ402 (gi 269854714 and gi 269854713), Pseudomonas sp. WBC-3 (gi 156129389 and gi 156129388) and Variovorax paradoxus S110(gi 239820773 and gi 239820774). Sequences were retrieved from NCBI via BLAST search, subsequently aligned using ClustalX 2 and rendered using Treeview 1.6.6.
Discussion
We were able to isolate and characterize a protein from Sphingomonas sp. strain TTNP3 that catalyzes the Fe2+- and O2-dependent conversion of HQ to 4-hydroxymuconic semialdehyde. Like nonylphenol ipso-hydroxylases, i.e. the first enzyme in the degradation pathway of nonylphenol and bisphenol A (Kolvenbach et al. 2007; Gabriel et al. 2007a; Gabriel et al. 2007b; Gabriel et al. 2005; Corvini et al. 2006), the HQDO represents an interesting class of enzymes that has been little studied.
The enzyme readily lost activity upon exposure to its substrate HQ, which distinguishes it from HQDO from Pseudomonas fluorescens ACB (Moonen et al. 2008b). This characteristic has previously also been reported for a HQDO from a Moraxella strain(Spain and Gibson 1991), and for other extradiol type dioxygenases, such as catechol dioxygenases (Cerdan et al. 1994; Bartels et al. 1984) and protocatechuate dioxygenases (Ono et al. 1970). This is possibly due to oxidation of ferrous iron in the active centre to ferric iron, a process that is reversed in vivo by redox-dependent reactions catalyzed by ferredoxins (Tropel et al. 2002).
The hypothesis of a ferrous iron in the active center of the enzyme is strongly supported by the results from experiments with hydrogen peroxide or with chelators of ferrous iron, in which enzyme activity was significantly reduced, Hydrogen peroxide is likely to inactivate the enzyme by oxidizing the ferrous iron to its ferric form (Spain and Gibson 1991; Lendenmann and Spain 1996; Moonen et al. 2008b).
The molecular mass determined by size exclusion chromatography in combination with the molecular masses determined by SDS-PAGE and the similarities to HQDO of Pseudomonas fluorescens ACB (Moonen et al. 2008b) indicate that the enzyme may be a tetramer in its native form. The iron content determined by ICP-MS suggests the presence of 1.4 atoms of iron per tetrameric unit. Taking into account that the loss of activity during the purification process may partially have been caused by the removal of iron from the enzyme, it can be assumed that the actual enzyme contains two iron atoms per tetrameric unit. This would be in agreement with data reported for other heteromeric extradiol dioxygenases, namely the protocatechuate 4,5-dioxygenases from Pseudomonas pseudoalcaligenes JS45 (Lendenmann and Spain 1996) and Sphingomonas paucimobilis SYK-6 (Sugimoto et al. 1999).
Our findings support reports that the substituent in para to the hydroxyl group adjacently to the cleaving site is an important discriminator for substrate binding to HQDO (Moonen et al. 2008b). Phenolic compounds possessing functional groups in para to the hydroxyl group, i.e. 4-mercaptophenol, 4-hydroxybenzonitrile and 4-nitrophenol, exhibited a strong inhibitory effect, whereas those lacking substituents in para position, such as phenol, catechol and resorcinol, led to enzyme inhibition of less than 10%. Furthermore, less than 5% inhibition was observed with caffeic acid and p-coumaric acid, which might indicate that the propenyl side chain in para position is sterically preventing the inhibitor from accessing the active site of the enzyme. The strong inhibitory effect on HQDO observed benzoquinone can be of relevance in vivo, as benzoquinone may be formed in the cell by oxidation of hydroquinone.
The degradation of technical nonylphenol mixtures in Sphingomonas sp. TTNP3 and Sphingobium xenophagum Bayram leads to the formation of minor amounts of 2-alkylated hydroquinones (Corvini et al. 2004a; Gabriel et al. 2005), potentially toxic metabolites that may pose oxidative stress on the organism. Even though Sphingomonas sp. TTNP3 has shown to lack the ability to degrade both 2(3',5'-dimethyl-3'-heptyl)-1,4-benzenediol and 2(2',6'-dimethyl-2'-heptyl)-1,4-benzenediol (Corvini et al. 2006) we wanted to investigate if other alkylated hydroquinones could be degraded by HQDO. Interestingly, degradation of 2-methylhydroquinone appeared to proceed at a higher rate than that of HQ, whereas with increasing length of the alkyl chain degradation (e.g. 2-hexylhydroquinone) rates decreased to less than two per cent of the degradation rate of HQ.
2-methylhydroquinone seemed to be preferentially cleaved adjacently to a ring-hydrogen, and not the electron donating methyl substituent. In contrast, type I HQDO in Sphingomonas paucimobilis (LinE) and type II HQDO in Pseudomonas fluorescens ACB appear to cleave 2-chlorohydroquinone and 2-fluorohydroquinone, respectively, both between two carbon atoms (C-1 and C-2) substituted by a hydroxyl and the electron withdrawing halogen group (Miyauchi et al. 1999; Moonen et al. 2008b). Future degradation experiments with a halogen monosubstituted hydroquinone derivative will determine whether steric or electronic constraints are predominant in determining the cleavage site.
Interestingly, 2-t-butylhydroquinone appeared to be degraded slower than linear 2-propyl- and 2-pentylhydroquinone, respectively, although a degradation rate in the same range as that of the latter derivatives would have been expected. A direct comparison to 2-butylhydroquinone, which bears a linear alkyl, was not possible as it could not be commercially obtained. Concluding from our data, an involvement of HQDO in the degradation of 2-nonylhydroquinones appears improbable, as the apparent degradation of 2-(1-methyl-1-octyl)hydroquinone was not unequivocally distinguishable from the oxygen consumption caused by chemical oxidation of the substrate. But considering the apparent negative effects of both alkyl chain length and the occurrence of branched chains it can be reasoned that other 2-nonylhydroquinone isomers, branched or not, will not be degraded by HQDO.
Quinonoide compounds, derived from hydroquinones are agents of oxidative stress and have a high toxic potential (Monks et al. 1992; Kappus 1987), which is why a rapid further metabolization of this intermediate is necessary to minimize exposure time and thus to avoid damage to the cell. Therefore, the elucidation of the nature of HQDO contributes substantially to the understanding of the mechanisms to prevent oxidative stress present in Sphingomonas sp. strain TTNP3.
According to our results the deduced amino acid sequences of the subunits of HQDO of Sphingomonas sp. TTNP3 show similarities to sequences of HQDO and putative proteins found in other strains, namely Photorhabdus, Pseudomonas, Burkholderia, and Variovorax (Figure 4). No sequence similarities to known sequences of HQDO reported for other sphingomonads could be found. Surprisingly, both HqdA and HqdB were found to be most similar to homologous proteins from Photorhabdus luminescens subsp. laumondii TTO1, a bacterium that can be found in the gut of entomopathogenic nematodes (Bowen et al. 1998).
Our data show that the HQDO of strain TTNP3, can be attributed to the type II HQDO. It represents the first enzyme of this type that has been identified in a Sphingomonas strain.
References
Bartels I, Knackmuss H-J, Reineke W: Suicide Inactivation of Catechol 2,3-Dioxygenase from Pseudomonas putida mt-2 by 3-Halocatechols. Appl Environ Microbiol 1984,47(3):500–505.
Bowen D, Rocheleau TA, Blackburn M, Andreev O, Golubeva E, Bhartia R, ffrench-Constant RH: Insecticidal Toxins from the Bacterium Photorhabdus luminescens. Science 1998,280(5372):2129–2132. 10.1126/science.280.5372.2129
Cai M, Xun L: Organization and Regulation of Pentachlorophenol-Degrading Genes in Sphingobium chlorophenolicum ATCC 39723. J Bacteriol 2002,184(17):4672–4680. 10.1128/JB.184.17.4672-4680.2002
Cerdan P, Wasserfallen A, Rekik M, Timmis KN, Harayama S: substrate-specificity of catechol 2,3-dioxygenase encoded by tol plasmid pwwo of pseudomonas-putida and its relationship to cell-growth. Journal of Bacteriology 1994,176(19):6074–6081.
Chauhan A, Samanta SK, Jain RK: Degradation of 4-nitrocatechol by Burkholderia cepacia: a plasmid-encoded novel pathway. Journal of Applied Microbiology 2000,88(5):764–772. 10.1046/j.1365-2672.2000.01018.x
Corvini PFX, Hollender J, Ji R, Schumacher S, Prell J, Hommes G, Priefer U, Vinken R, Schaffer A: The degradation of alpha-quaternary nonylphenol isomers by Sphingomonas sp strain TTNP3 involves a type II ipso-substitution mechanism. Applied Microbiology and Biotechnology 2006,70(1):114–122. 10.1007/s00253-005-0080-0
Corvini PFX, Meesters RJW, Schaffer A, Schroder HF, Vinken R, Hollender J: Degradation of a nonylphenol single isomer by Sphingomonas sp strain TTNP3 leads to a hydroxylation-induced migration product. Applied and Environmental Microbiology 2004,70(11):6897–6900. 10.1128/AEM.70.11.6897-6900.2004
Corvini PFX, Meesters RJW, Schäffer A, Schröder HF, Vinken R, Hollender J: Degradation of a nonylphenol single isomer by Sphingomonas sp. strain TTNP3 leads to a hydroxylation-induced migration product. Applied and Environmental Microbiology 2004,70(11):6897–6900. 10.1128/AEM.70.11.6897-6900.2004
Corvini PFX, Vinken R, Hommes G, Schmidt B, Dohmann M: Degradation of the radioactive and non-labelled branched 4(3 ',5 '-dimethyl 3 '-heptyl)-phenol nonylphenol isomer by Sphingomonas TTNP3. Biodegradation 2004,15(1):9–18.
Dogra C, Raina V, Pal R, Suar M, Lal S, Gartemann K-H, Holliger C, van der Meer JR, Lal R: Organization of lin Genes and IS6100 among Different Strains of Hexachlorocyclohexane-Degrading Sphingomonas paucimobilis: Evidence for Horizontal Gene Transfer. J Bacteriol 2004,186(8):2225–2235. 10.1128/JB.186.8.2225-2235.2004
Eltis LD, Bolin JT: Evolutionary relationships among extradiol dioxygenases. Journal of Bacteriology 1996,178(20):5930–5937.
Eppink MHM, Cammaart E, van Wassenaar D, Middelhoven WJ, van Berkel WJH: Purification and properties of hydroquinone hydroxylase, a FAD-dependent monooxygenase involved in the catabolism of 4-hydroxybenzoate in Candida parapsilosis CBS604. European Journal of Biochemistry 2000,267(23):6832–6840. 10.1046/j.1432-1033.2000.01783.x
Ferraroni M, Seifert J, Travkin VM, Thiel M, Kaschabek S, Scozzafava A, Golovleva L, Schlomann M, Briganti F: Crystal structure of the hydroxyquinol 1,2-dioxygenase from Nocardioides simplex 3E, a key enzyme involved in polychlorinated aromatics biodegradation. Journal of Biological Chemistry 2005,280(22):21144–21154. 10.1074/jbc.M500666200
Gabriel FLP, Cyris M, Giger W, Kohler HPE: ipso-substitution: A general biochemical and biodegradation mechanism to cleave alpha-quaternary alkylphenols and bisphenol A. Chemistry & Biodiversity 2007,4(9):2123–2137. 10.1002/cbdv.200790170
Gabriel FLP, Cyris M, Jonkers N, Giger W, Guenther K, Kohler HPE: Elucidation of the ipso-substitution mechanism for side-chain cleavage of alpha-quaternary 4-nonylphenols and 4-t-butoxyphenol in Sphingobium xenophagum Bayram. Applied and Environmental Microbiology 2007,73(10):3320–3326. 10.1128/AEM.02994-06
Gabriel FLP, Heidlberger A, Rentsch D, Giger W, Guenther K, Kohler HPE: A novel metabolic pathway for degradation of 4-nonylphenol environmental contaminants by Sphingomonas xenophaga Bayram-ipso-hydroxylation and intramolecular rearrangement*. Journal of Biological Chemistry 2005,280(16):15526–15533. 10.1074/jbc.M413446200
Jain RK, Dreisbach JH, Spain JC: Biodegradation of p-nitrophenol via 1, 2, 4-benzenetriol by an Arthrobacter sp. Applied and Environmental Microbiology 1994,60(8):3030–3032.
Kappus H: Oxidative stress in chemical toxicity. Archives of Toxicology 1987,60(1):144–149. 10.1007/BF00296968
Kohler HPE, Schmid A, van der Maarel M: Metabolism of 2, 2'-dihydroxybiphenyl by Pseudomonas sp. strain HBP1: production and consumption of 2, 2', 3-trihydroxybiphenyl. Journal of Bacteriology 1993,175(6):1621–1628.
Kolvenbach B, Schlaich N, Raoui Z, Prell J, Zuhlke S, Schaffer A, Guengerich FP, Corvini PFX: Degradation Pathway of Bisphenol A: Does ipso Substitution Apply to Phenols Containing a Quaternary {alpha}-Carbon Structure in the para Position? Appl Environ Microbiol 2007,73(15):4776–4784. 10.1128/AEM.00329-07
Laemmli UK: Cleavage Of Structural Proteins During Assembly Of Head Of Bacteriophage-T4. Nature 1970,227(5259):680.. 10.1038/227680a0
Lal R, Pandey G, Sharma P, Kumari K, Malhotra S, Pandey R, Raina V, Kohler HPE, Holliger C, Jackson C, Oakeshott JG: Biochemistry of microbial degradation of hexachlorocyclohexane and prospects for bioremediation. Microbiology and Molecular Biology Reviews 2010,74(1):58–80. 10.1128/MMBR.00029-09
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG: Clustal W and Clustal × version 2.0. Bioinformatics 2007,23(21):2947–2948. 10.1093/bioinformatics/btm404
Latus M, Seitz HJ, Eberspacher J, Lingens F: Purification and characterization of hydroxyquinol 1, 2-dioxygenase from Azotobacter sp. strain GP1. Applied and Environmental Microbiology 1995,61(7):2453–2460.
Lendenmann U, Spain J: 2-aminophenol 1,6-dioxygenase: a novel aromatic ring cleavage enzyme purified from Pseudomonas pseudoalcaligenes JS45. J Bacteriol 1996,178(21):6227–6232.
Machonkin T, Holland P, Smith K, Liberman J, Dinescu A, Cundari T, Rocks S: Determination of the active site of Sphingobium chlorophenolicum 2,6-dichlorohydroquinone dioxygenase (PcpA). Journal of Biological Inorganic Chemistry 2009,15(3):291–301.
Manickam N, Reddy MK, Saini HS, Shanker R: Isolation of hexachlorocyclohexane-degrading Sphingomonas sp. by dehalogenase assay and characterization of genes involved in Y-HCH degradation. Journal of Applied Microbiology 2008,104(4):952–960. 10.1111/j.1365-2672.2007.03610.x
Miyauchi K, Adachi Y, Nagata Y, Takagi M: Cloning and sequencing of a novel meta-cleavage dioxygenase gene whose product is involved in degradation of gamma-hexachlorocyclohexane in Sphingomonas paucimobilis. Journal of Bacteriology 1999,181(21):6712–6719.
Monks TJ, Hanzlik RP, Cohen GM, Ross D, Graham DG: Quinone chemistry and toxicity. Toxicology and Applied Pharmacology 1992,112(1):2–16. 10.1016/0041-008X(92)90273-U
Moonen MJH, Kamerbeek NM, Westphal AH, Boeren SA, Janssen DB, Fraaije MW, van Berkel WJH: Elucidation of the 4-hydroxyacetophenone catabolic pathway in Pseudomonas fluorescens ACB. Journal of Bacteriology 2008,190(15):5190–5198. 10.1128/JB.01944-07
Moonen MJH, Synowsky SA, van den Berg WAM, Westphal AH, Heck AJR, van den Heuvel RHH, Fraaije MW, van Berkel WJH: Hydroquinone dioxygenase from Pseudomonas fluorescens ACB: a novel member of the family of nonheme-iron(II)-dependent dioxygenases. Journal of Bacteriology 2008,190(15):5199–5209. 10.1128/JB.01945-07
Ono K, Nozaki M, Hayaishi O: Purification and some properties of protocatechuate 4,5-dioxygenase. Biochimica et Biophysica Acta (BBA)-Enzymology 1970,220(2):224–238. 10.1016/0005-2744(70)90008-2
Perkins DN, Pappin DJC, Creasy DM, Cottrell JS: Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 1999,20(18):3551–3567. 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2
Rieble S, Joshi DK, Gold MH: Purification and characterization of a 1, 2, 4-trihydroxybenzene 1, 2-dioxygenase from the basidiomycete Phanerochaete chrysosporium. Journal of Bacteriology 1994,176(16):4838–4844.
Shen W, Liu W, Zhang J, Tao J, Deng H, Cao H, Cui Z: Cloning and characterization of a gene cluster involved in the catabolism of p-nitrophenol from Pseudomonas putida DLL-E4. Bioresource Technology 2010, in press.
Spain JC, Gibson DT: Pathway For Biodegradation Of Para-Nitrophenol In A Moraxella Sp. Applied and Environmental Microbiology 1991,57(3):812–819.
Sugimoto K, Senda T, Aoshima H, Masai E, Fukuda M, Mitsui Y: Crystal structure of an aromatic ring opening dioxygenase LigAB, a protocatechuate 4,5-dioxygenase, under aerobic conditions. Structure 1999,7(8):953–965. 10.1016/S0969-2126(99)80122-1
Takenaka S, Okugawa S, Kadowaki M, Murakami S, Aoki K: The metabolic pathway of 4-aminophenol in Burkholderia sp strain AK-5 differs from that of aniline and aniline with C-4 substituents. Applied and Environmental Microbiology 2003,69(9):5410–5413. 10.1128/AEM.69.9.5410-5413.2003
Travkin VM, Jadan AP, Briganti F, Scozzafava A, Golovleva LA: Characterization of an intradiol dioxygenase involved in the biodegradation of the chlorophenoxy herbicides 2,4-D and 2,4,5-T. FEBS Lett 1997,407(1):69–72. 10.1016/S0014-5793(97)00297-4
Tropel D, Meyer C, Armengaud J, Jouanneau Y: Ferredoxin-mediated reactivation of the chlorocatechol 2,3-dioxygenase from Pseudomonas putida GJ31. Archives of Microbiology 2002,177(4):345–351. 10.1007/s00203-002-0399-1
Vester D, Rapp E, Gade D, Genzel Y, Reichl U: Quantitative analysis of cellular proteome alterations in human influenza A virus-infected mammalian cell lines. Proteomics 2009,9(12):3316–3327. 10.1002/pmic.200800893
Wei Q, Liu H, Zhang J-J, Wang S-H, Xiao Y, Zhou N-Y: Characterization of a para-nitrophenol catabolic cluster in Pseudomonas sp. strain NyZ402 and construction of an engineered strain capable of simultaneously mineralizing both para- and ortho-nitrophenols. Biodegradation 2010,21(6):915–921. 10.1007/s10532-010-9351-2
Xu L, Resing K, Lawson SL, Babbitt PC, Copley SD: Evidence That pcpA Encodes 2,6-Dichlorohydroquinone Dioxygenase, the Ring Cleavage Enzyme Required for Pentachlorophenol Degradation in Sphingomonas chlorophenolica Strain ATCC 39723. Biochemistry 1999,38(24):7659–7669. 10.1021/bi990103y
Yamamoto S, Otsuka S, Murakami Y, Nishiyama M, Senoo K: Genetic diversity of gamma-hexachlorocyclohexane-degrading sphingomonads isolated from a single experimental field. Letters in Applied Microbiology 2009,49(4):472–477. 10.1111/j.1472-765X.2009.02691.x
Yin Y, Zhou N-Y: Characterization of MnpC, a Hydroquinone Dioxygenase Likely Involved in the meta-Nitrophenol Degradation by Cupriavidus necator JMP134. Current Microbiology 2010,61(5):471–476. 10.1007/s00284-010-9640-3
Zhang J-J, Liu H, Xiao Y, Zhang X-E, Zhou N-Y: Identification and Characterization of Catabolic para-Nitrophenol 4-Monooxygenase and para-Benzoquinone Reductase from Pseudomonas sp. Strain WBC-3. J Bacteriol 2009,191(8):2703–2710. 10.1128/JB.01566-08
Acknowledgements
This work has been funded by Swiss National Science Foundation grant No. NF 200021-120574, the Czech Ministry for Education grant No. M0520 and the European Union within the Seventh Framework Programme under grant agreement n° 265946 (MINOTAURUS project).
Sphingomonas sp. strain TTNP3 was a generous gift from Prof. Willy Verstraete (LabMet, University Ghent, Belgium). We thank Robert Heyer and Markus Pioch for programming an in-house software tool that facilitates manual de novo sequencing of peptides. We gratefully acknowledge Daniela Tobler (Institute for Chemistry and Bioanalytics, University of Applied Sciences, Northwestern Switzerland) for liquid chromatography SEC analysis of HQDO.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
BAK carried out the enzyme purification and biochemical experiments and drafted the manuscript. ML performed IPC-MS analyses. DB and ER carried out protein analysis and identification. JF and CV performed the genome sequencing and assembly and provided nucleotide sequence data. FLPG elaborated GC-MS data, conceived fragmentation patterns and commented on the manuscript. HPEK participated in the design of the study and commented on the manuscript. AS and PFXC participated in the design of the study, commented on the manuscript and supervised the Ph.D. thesis of BAK from which large parts of this study originated.
Electronic supplementary material
13568_2011_8_MOESM1_ESM.TIFF
Additional file 1: Proposed fragmentation pattern for trimethylsilylated 4-hydroxymuconic semialdehyde and its methylated analogon. 1, proposed GC-MS fragmentation pattern of 4-hydroxymuconic semialdehyde; 2, proposed GC-MS fragmentation pattern of the ring-cleavage product of 2-methyl-hydroquinone with cleavage sites between the ring bonds C(1)-C(6), C(3)-C(4) and C(5)-C(6), respectively; 3, proposed GC-MS fragmentation pattern of the ring-cleavage product of 2-methyl-hydroquinone with cleavage sites between the ring bonds C(1)-C(2); 4, GC-MS fragmentation pattern of 2-hydroxy-6-(2-hydroxyphenyl)-6oxo-2,4-hexadienoic acid (Kohler et al. 1993). (TIFF 211 KB)
13568_2011_8_MOESM2_ESM.PDF
Additional file 2: Multiple sequence alignment performed by ClustalW 2 of the sequence of HqdA with sequences retrieved by BLAST search. Shown is the original multiple sequence alignment from which Figure 4A has been rendered. (PDF 41 KB)
13568_2011_8_MOESM3_ESM.PDF
Additional file 3: Multiple sequence alignment performed by ClustalW 2 of the sequence of HqdB with sequences retrieved by BLAST search. Shown is the original multiple sequence alignment from which Figure 4B has been rendered. (PDF 75 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Kolvenbach, B.A., Lenz, M., Benndorf, D. et al. Purification and characterization of hydroquinone dioxygenase from Sphingomonas sp. strain TTNP3. AMB Expr 1, 8 (2011). https://doi.org/10.1186/2191-0855-1-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2191-0855-1-8