
Abstract
Two major types of amyloidosis are primary amyloidosis or amyloid light chain amyloidosis and secondary amyloidosis. Although amyloidosis involves a variety of organ systems including skin, the occurrence of bullous skin lesions is rare. Little is known about the mechanism of blister formation. These blisters are often hemorrhagic and typically occur in the oral mucosa. Only a few case reports have described skin involvement in systemic amyloidosis. The manifestation of bullous lesions on the breast in association with primary amyloidosis has not been previously reported. Therefore, we report a case of cutaneous hemorrhagic bullous of the breast secondary to primary systemic amyloidosis, which may be important for medical oncologists to be aware of this uncommon presentation of plasma cell dysrasias. Furthermore, this case only partially responded to the commonly used multiple myeloma-type regimen, the skin lesions responded completely to a five-drug combination chemotherapy regimen, utilizing immunomodulators, liposomal doxorubicin, cyclophosphamide, bortezomib, and dexamethasone, suggesting that a more aggressive modality of chemotherapy may be necessary to treat such cases.
Similar content being viewed by others


Background
Primary and secondary amyloidosis are the two major types of amyloidosis. The primary systemic amyloidosis, also known as light chain (AL) amyloidosis, is mostly related to a plasma cell dyscrasia. The secondary (AA) amyloidosis is derived from serum amyloid A subunit protein, an acute-phase protein that is produced in response to inflammatory conditions [1]. There is no identifiable, underlying cause of AL amyloidosis [2]. The fibrils of AL amyloidosis are composed of polymerized immunoglobulin light chain or light chain fragments [3]. Amyloid protein is resistant to proteolysis and has a three dimensional configuration as a beta pleated sheet [4]. Although cutaneous involvement in primary systemic amyloidosis is relatively common [5], the occurrence of bullous skin lesions is rare [2, 3].
Only a few cases of cutaneous involvement with systemic light chain amyloidosis have been reported in the literature. Skin involvement in the form of hemorrhagic bullous is much rarer. To the best of our knowledge, hemorrhagic bullous presentation of amyloidosis on the breast skin has not been reported in the literature. This uncommon presentation of amyloidosis was only partially responsive to the commonly used combination chemotherapy regimens, but it responded completely to a five-drug combination regimen. This suggests that a more aggressive approach, with combination of multiple immunomodulators and chemotherapy agents may be required to achieve a meaningful response in similar cases.
Case report
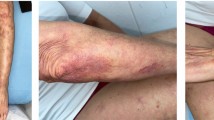
A 51-year-old African American female with no significant past medical history presented in April 2009 with a 1-month history of hemorrhagic skin lesions on both breasts. The patient had no other symptoms. She was not taking any medication, and her social and family histories were noncontributory. Physical examination revealed extensive bullous ulcerating and hemorrhagic skin lesions involving posterior aspects of both breasts and upper abdominal skin bilaterally (Figure 1). An initial diagnostic skin biopsy of the skin lesion revealed abundant amyloid deposits with positive congo red stain and positive apple-green birefringence under polarized light microscopy. These findings were consistent with pathologic diagnosis of bullous amyloidosis of skin (Figure 2). A direct immunofluorescence study of the specimen with a panel of four immunoglobulins (IgG, IgA, IgM, and C3) was negative. No circulating antibody against basement membrane zone antibody was detected by indirect immunofluorescence study. Initial hematologic workup included serum protein electrophoresis, which had a normal pattern without any M-spike in the gamma region; serum immunofixation was negative for any monoclonal gammopathy, and quantitative immunoglobulin assay was consistent with mild panhypogammaglobulinemia. However, a serum-free light chain assay revealed a very high level of kappa light chain of 6090 mg/dl and lambda light chain of 0.05 mg/dL; urine light chain assay was confirmatory with a very high level of kappa light chain of 6220 mg/dl. The patient also had a mild, normochromic, normocytic anemia with hemoglobin of 11.2 g/dl and normal red blood cell indices. Bone marrow aspiration and biopsy showed infiltration of the marrow with a monoclonal population of plasma cells, comprising 50% of total cells. Flow cytometry of an aspirated bone marrow specimen yielded a monoclonal population of CD138 positive, IgG κ plasma myeloma cells. Conventional cytogenetic examination showed a normal female karyotype of 46 XX; however, FISH was positive for monosomy of chromosome 13 (loss of both RB1 and LAMP1) in 10.3% of cells, and t [6, 7], indicating overexpression of BCL1, and cyclin D1 (CCND1/IGH) rearrangement in 5.8% of the cells. Bone survey revealed multiple lytic bone lesions, including the left greater femoral trochanter, right humeral head, and T12 vertebral bodies. Therefore, a diagnosis of bullous hemorrhagic skin lesions, associated with primary systemic light chain amyloidosis was made.

Extensive hemorrhagic bullae with desquamation and surrounding purpura in skin of both breasts, prior to treatment.

A hematoxylin and eosin stained section demonstrates collections of dull eosinophilic fissured material within the dermis consistent with amyloid, which, on Congo red stain, showed apple-green birefringence in polarized light and confirmed amyloid deposits (not shown).
The patient was initially treated with an RVD regimen, which included a combination of bortezomib, with a dose of 1.3 mg/m2 on days 1, 4, 8, 11, plus lenalidomide, with dose of 25 mg daily on days 1–14, and dexamethasone, with a dose of 40 mg twice a week, repeated on every 21 days cycles for a total of four cycles. This unfortunately resulted in only a partial response, which included some improvement of the skin lesions and reduction of kappa light chain only to 2250 mg/dl (PR, as per NCCN guideline; more than 50% reduction in the serum free light chain levels). Because of significant persistent skin lesions, a search for enrolling the patient in a clinical trial was attempted; unfortunately, there was none available at the time. Therefore, a more aggressive treatment plan was initiated. This was included a combination of liposomal doxorubicin, with a dose of 40 mg/m2 on day one, cyclophosphamide, with a dose of 750 mg/m2 on day one, bortezomib, with a dose of 1.3 mg/m2, on days 1, 8, and 15, lenalidomide , 15 mg daily on days 1–21 , and dexamethasone, with a dose of 40 mg twice a week, repeated on every 28 days cycles for a total of six cycles. This treatment resulted in a very good partial response rate, with significant reduction of the kappa light chain to 208 mg/dl after two cycles of treatment (VGPR, as per NCCN guidelines; more than 90% reduction in the serum free light chain levels). Continuation of the above regimen for a total of six cycles resulted in further reduction of kappa light chain to 114 mg/dl and complete disappearance of hemorrhagic bullous skin lesions (Figure 3). It is important to mention that although skin rashes are common in the patients treated with lenalidomide, this patient did not report any such adverse side effects with the treatment. The patient was then successfully consolidated with high dose chemotherapy, followed by autologous hematopoietic stem cell transplantation. She has been followed closely, and she has continued to be in complete remission (CR) up to the date of this report.

Complete resolution of skin lesions, Post-therapy.
Discussion
Bullous pemphigoid (and linear IgA disease) is a heterogeneous group of autoimmune blistering disorders of the subepiderm and is associated with autoantibodies, predominantly IgG, against the transmembrane hemidesmosomal protein BP180/type XVII collagen [8]. Mucous membrane pemphigoid is characterized by subepithelial separation and deposition of immunoglobulins and complement along the basement membrane zone. This disease is diagnosed with direct immunofluorescence testing showing a linear deposition of immunoglobulins and/or complement along the basement membrane zone and indirect immunofluorescence testing showing circulating IgG (and sometimes IgA) autoantibodies along the basement membrane zone [9].
Light chain (AL) amyloidosis is a plasma cell dyscrasia characterized by the pathologic production and deposition of monoclonal light chain fibrillar proteins in tissues, resulting in organ dysfunction. The goal of treatment of AL amyloidosis is eradication of the monoclonal plasma cell population and suppression of the pathologic levels of light chains. The use of novel agents, such as thalidomide, lenalidomide and bortezomib alone or in combination with steroids and alkylating agents has shown efficacy and continues to be explored [10]. HevyLite™ is a new, recently developed method that facilitates separate quantification of the kappa and lambda-bounded amounts of a given immunoglobulin. This new assay provides precise quantification of heavy and light chain and their ratio of the involved immunoglobulin more accurately than the other currently available methods; moreover, they carry prognostic information regarding survival in multiple myeloma patients [11].
Primary systemic amyloidosis, a disease characterized by infiltration of different organs [4], and associated amyloidosis cause clinically detectable cutaneous changes in approximately 10-40% of patients [5]; The occurrence of bullous skin lesions, however, is rare [2, 3]. The bullae may be intradermal or subepidermal [2]. The bullae form of cutaneous amyloidosis results from cleavage of the amyloid deposits. These blisters are often hemorrhagic and most commonly occur in the oral mucosa [5].
In this reported case, skin lesions did not reveal immunoglobulin deposits, and indirect immunofluorescence staining for basement membrane protein was negative. Skin biopsy showed positive congo red staining, and amyloid deposits were identified with a positive apple-green birefringence under polarized light microscopy. These findings, along with bone marrow infiltration by abundant plasma cells, multiple lytic bone lesions, and the presence of light chain protein, confirmed a diagnosis of bullous skin lesion secondary to primary systemic amyloidosis.
Insufficient data are available in the literature for the optimal treatment of amyloidosis with underlying plasma cell disorder; therefore, most treatment strategies used in systemic light chain amyloidosis are derived from multiple myeloma regimens. Intermediate or high-dose melphalan followed by stem cell transplantation is one of the therapeutic options listed by the National Comprehensive Cancer Network panel. In some cases, AL amyloidosis is considered to be a treatable disease, and a substantial proportion of patients now attain a long survival; a 22% survival rate of more than 10 years has been reported by Gertz et al. [4]. Treating systemic AL amyloidosis has recently evolved with the use of autologous stem cell transplantation and the introduction of the newer thalidomide analogue. The thalidomide analog, lenalidomide, and the proteasome inhibitor, bortezomib, are both active in advanced and refractory multiple myeloma. The ability of these drugs in combination with dexamethasone to rapidly reduce the level of the monoclonal protein is being examined in several clinical trials. However, this option may not be applicable to all cases, especially those involving hemorrhagic bullous amyloidosis (Table 1). Multiple different single-agent or combination regimens have been used with variable responses, mostly unsatisfactory. Steroids and alkylating agents and even the CHOP regimen (cyclophosphamide, hydroxydaunomycin, Oncovin, and prednisone) have been used as part of the treatment. Lenalidomide is an appealing therapeutic option for subjects with advanced AL amyloidosis and refractory to both melphalan and bortezomib [12].
It is important to know that 43% of the patients with amyloidosis, who are treated with lenalidomide, may develop skin rashes. The rashes are characterized as morbilliform, urticarial, dermatitic, acneiform, and undefined. Severe rashes require permanent discontinuation of lenalidomide [19].
This unique reported case of AL amyloidosis presented with hemorrhagic bullous skin lesions, high levels of light chain protein only, presence of t(11,14) by cytogenic study, overexpression of cyclin D1, and only partial response to the standard multiple myeloma regimen, which includes lenalidomide and bortezomib, and dexamethasone (RVD). All these features represent a poor prognosis [20–22]. A more aggressive treatment approach with combination of five agents resulted in achieving meaningful success. This included combination of lenalidomide, bortezomib, liposomal doxorubicin, cyclophosphamide, and dexamethasone. The overall response included a very good partial response (VGPR), associated with a great than 90% reduction in the kappa light chain level, and complete resolution of the breast skin lesions. This case illustrates that bullous hemorrhagic skin lesions, associated with primary systemic amyloidosis may need more aggressive treatment such as the five-drug combination that we have used.
Consent
The patient of the interest in this manuscript has been aware of our intention to publish her case. She has consented to publishing the manuscript without including any pertinent information, which may disclose her identity. We have done our best to protect her identity in this paper.
References
Stoopler ET, Alawi F, Laudenbach JM, Sollecito TP: Bullous amyloidosis of the oral cavity: A rare clinical presentation and review. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2006,101(6):734. 10.1016/j.tripleo.2006.01.003
Johnson TM, Rapini RP, Hebert AA, Lowe L, Verani R, Evanoff G: Bullous amyloidosis. Cutis 1989,43(4):346.
Robert C, Aractingi S, Prost C, Verola O, Blanchet-Bradon C, Blanc F, Bagot M, Dubbertret L, Fermand J-P: Bullous amyloidosis, report of 3 cases and review of the literature. Medicine 1993,72(1):38.
Gertz Morie A, Giampaolo M, Treon Steven P: Amyloidosis and Waldenström’s Macroglobulinemia. Hematology 2004, 257: 82.
Beacham Bruce E, Greer Kenneth E, Andrews Brian S, Cooper Philip H: Bullous amyloidosis. Am Acad Dermatol 1980,3(5):506. 10.1016/S0190-9622(80)80117-4
Ruzicka T, Schmoeckel C, Ring J, Linke RP, Braun-Falco O: Bullous Amyloidosis. British J of Dermatol 1985, 113: 85. 10.1111/j.1365-2133.1985.tb02047.x
Grundmann JU, Bonnekoh B, Gollnick H: Extensive haemorrhagic-bullous skin manifestations of systemic AA-amyloidosis associated with IgImyeloma. Eur J Dermatol 2000,10(2):139.
Kromminga A, Scheckenbach C, Georgi M, Hagel C, Arndt R, Christophers E, Brocker E-B, Zillikens D: Patients with bullous pemphigoid and linear iga disease show a dual iga and igg autoimmune response to BP180. J Autoimmun 2000,15(3):293. 10.1006/jaut.2000.0437
Sánchez AR, Rogers RS, Kupp LI, Sheridan PJ: Desquamative gingivitis associated with IgG/IgA pemphigoid presents a challenging diagnosis and treatment, a case report. J Periodontol 2004,75(12):1714. 10.1902/jop.2004.75.12.1714
Michael R, Heather L: Light chain (AL) amyloidosis: update on diagnosis and management. J Hematol Oncol 2011, 4: 47. 10.1186/1756-8722-4-47
Efstathios K, Panayiotis P, Harding Stephen J, Nikolitsa K, Dimitris M, Vassiliki B, Tatiana T, Maria D, George G, Ladan M, Bradwell Arthur R, Marie-Christine K: Ratio of involved/uninvolved immunoglobulin quantification by Hevylite™ assay: clinical and prognostic impact in multiple myeloma. Experimental Hematology Oncology 2012, 1: 9. 10.1186/2162-3619-1-9
Giovanni P, Paola R, Paolo M, Andrea F, Letizia Zenone B, Laura O, Francesca L, Silvia B, Vittorio P, Rosangela I, Giampaolo M: Salvage Therapy with Lenalidomide and Dexamethasone (LDex) In Patients with Advanced AL Amyloidosis Refractory to Both Melphalan and Bortezomib. Blood, ASH Annual Meeting Abstracts 2010, 116: 3062.
Bieber T, Ruzicka T, Linke RP, von Kries R, Goerz G, Braun-Falco O: Hemorrhagic Bullous Amyloidosis, A histologic, immunocytochemical, and ultrastructural study of two patients. Arch Dermatol 1988,124(11):1683. 10.1001/archderm.1988.01670110043009
Pramatarov K, Lazarova A, Mateev G, Popov A: Bullous hemorrhagic primary systemic amyloidosis. Intern J of Dermatol 1990,29(3):211. 10.1111/j.1365-4362.1990.tb03804.x
Ochiai T, Morishima T, Hao T, Takayama A: Bullous amyloidosis, the mechanism of blister formation revealed by electron microscopy. J Cutan Pathol 2001,28(8):407. 10.1034/j.1600-0560.2001.028008407.x
Comenzo RL, Gertz MA: Autologous stem cell transplantation for primary systemic amyloidosis. Blood 2002,99(12):4276. 10.1182/blood.V99.12.4276
Gertz MA, Lacy MQ, Dispenzieri A, Gastineau DA, Chen MG, Ansell SM, Inwards David J, Ivana NM, Micallef Ivana NM, Tefferi A, Litzow Mark R: Stem cell transplantation for the management of primary systemic amyloidosis. Am J Med 2002,113(7):549. 10.1016/S0002-9343(02)01208-1
Goodman Hugh JB, Gillmore Julian D, Lachmann Helen J, Wechalekar Ashutosh D, Bradwell Arthur R, Hawkins Philip N: Outcome of autologous stem cell transplantation for AL amyloidosis in the UK. Br J Haematol 2006,134(4):417. 10.1111/j.1365-2141.2006.06204.x
Sviggum Hans P, Davis Mark DP, Vincent RS, Dispenzieri A: Dermatologic adverse effects of lenalidomide therapy for amyloidosis and multiple myeloma. Arch Dermatol 2006,142(10):1298–1302. 10.1001/archderm.142.10.1298
Dispenzieri A, Lacy MQ, Katzmann JA, et al.: Absolute values of immunoglobulin free light chains are prognostic in patients with primary systemic amyloidosis undergoing peripheral blood stem cell transplantation. Blood 2006, 107: 3378–3383. 10.1182/blood-2005-07-2922
Comenzo RL, Hoffman JE, Hassoun H, Landau H, Iyer L, Zhou P: Pathobiologic associations of plasma cell (PC) overexpression of Cyclin D1 (CCND1) in systemic AL amyloidosis (AL) [abstract]. Amyloid 2010,17(s1):61.
Cohen Adam D, Comenzo Raymond L: Systemic Light-Chain Amyloidosis,Advances in Diagnosis, Prognosis, and Therapy. Hematology 2010, 287:.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
No authors report conflicts of interest with the pharmaceutical companies.
Authors’ contributions
AA, First Author, collected the patient information, reviewed the literature, and drafted the manuscript, and revised the final manuscript. JCW, Corresponding Author, designed the treatment protocol, and attended the patient; assisted and mentored in the writing the manuscript. MB, assisted in treating the patient, and assisted in collecting the relevant clinical data. GB, assisted in treating the patient, and assisted in collecting the relevant clinical data. GRC, assisted in treating the patient, and assisted in collecting the relevant clinical data. All authors have given approval for the final approval of the version to be published.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Agheli, A., Becker, M., Becker, G. et al. Response of hemorrhagic bullous skin lesions of the breast secondary to primary systemic amyloidosis to a five-drug combination chemotherapy: a case report and review of the literature. Exp Hematol Oncol 1, 19 (2012). https://doi.org/10.1186/2162-3619-1-19
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2162-3619-1-19