
Abstract
JAK-STAT (Janus associated kinase-signal transducer and activator of transcription) pathway plays a critical role in transduction of extracellular signals from cytokines and growth factors involved in hematopoiesis, immune regulation, fertility, lactation, growth and embryogenesis. JAK family contains four cytoplasmic tyrosine kinases, JAK1-3 and Tyk2. Seven STAT proteins have been identified in human cells, STAT1-6, including STAT5a and STAT5b. Negative regulators of JAK–STAT pathways include tyrosine phosphatases (SHP1 and 2, CD45), protein inhibitors of activated STATs (PIAS), suppressors of cytokine signaling (SOCS) proteins, and cytokine-inducible SH2-containing protein (CIS). Dysregulation of JAK-STAT pathway have been found to be key events in a variety of hematological malignancies. JAK inhibitors are among the first successful agents reaching clinical application. Ruxolitinib (Jakafi), a non-selective inhibitor of JAK1 & 2, has been approved by FDA for patients with intermediate to high risk primary or secondary myelofibrosis. This review will also summarize early data on selective JAK inhibitors, including SAR302503 (TG101348), lestaurtinib (CEP701), CYT387, SB1518 (pacritinib), LY2784544, XL019, BMS-911543, NS-018, and AZD1480.
Similar content being viewed by others


Introduction
JAK-STAT (Janus associated kinase-signal transducer and activator of transcription) pathway is one of the critical intracellular signaling cascades in transduction of extracellular signals to the nucleus to control gene expression. A variety of cytokines and growth factors complete their physiological tasks through JAK-STAT pathway, including hematopoiesis, immune-regulation, fertility, lactation, growth and embryogenesis [1–6].
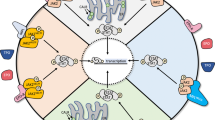
JAK family contains four cytoplasmic tyrosine kinases, JAK1-3 and Tyk2 [7]. These kinases bind to the juxta-membrane region of cytokine receptors [8]. Each molecule contains seven JAK homology domains (JH1-7). The carboxyl JH1 domain contains the catalytic activity, whereas N-terminal JH7 domain is responsible for receptor binding. JH2 domain has significant homology to JH1 but lacks enzymatic activity and therefore is a pseudo-kinase domain. Binding of a ligand to its receptor results in receptor dimerization, leading to activation of the JAK kinase activity. Subsequently activated JAKs phosphorylate cytoplasmic domain of the receptor [9]. Activated JAK-cytokine receptor complex recruits and phosphorylates specific cytoplasmic transcription factors called STAT proteins [10]. Seven STAT proteins have been identified in human cells, STAT1-6, including STAT5a and STAT5b [7]. Phosphorylation of specific STAT proteins results in their dimerization and subsequent translocation into the nucleus to interact with various regulatory elements for gene expression (Figure 1) [11, 12].

Signaling cascade of JAK-STAT pathway. Binding of the ligand to cytokine receptor induces receptor dimerization and activation of receptor associated JAK kinase, which in turn phosphorylates STAT proteins. After forming a homodimer, STAT proteins translocate to the nucleus to control gene expression. Negative regulation of the JAK-STAT pathway is provided by phospho-tyrosine phosphatases (PTP), member for SOCS family proteins and PIAS proteins (shown in red).
Regulation of JAK-STAT pathway
Three major mechanisms have been implicated to negatively regulate the JAK-STAT pathway [11]. Figure 1 depicts the positive and negative regulation of the JAK-STAT pathway.
Tyrosine phosphatases
Src homology-2 (SH2) containing tyrosine phosphatase and CD45 tyrosine phosphatase play a major role in modulating JAK-STAT pathway. SH2 containing tyrosine phosphatases include SHP1 and SHP2 (shatterproof 1 & 2). Their SH2 domains allow attachment to the phospho-tyrosine residues present on activated receptors, JAKs or STAT proteins, leading to dephosphorylation of the substrates. SHP1 is mainly expressed in hematopoietic cells, epithelial and smooth muscle cells whereas SHP2 is ubiquitous. The expression of SHP1 gene was found to be silenced by promoter hypermethylation in various hematological malignancies [10]. CD45 transmembrane tyrosine phosphatase is the second type of tyrosine phosphatase that negatively regulates JAK-STAT pathway. CD45, the leukocyte common antigen, is expressed in hematopoietic cells. It has two phosphatase domains in its intracellular region, though only one is active. Mice deficient for CD45 show hyper-activation of JAK1 and JAK3 [10].
Protein inhibitors of activated STATs (PIAS)
PIAS family consists of PIAS1, PIAS3, PIASx and PIASy. Biochemical assays have shown that PIAS3 and PIASx interact with STAT3 and STAT4 respectively, while PIAS1 and PIASy with STAT1 [13, 14]. PIAS1 and PIAS3 exert negative regulation by blocking the DNA binding of STAT1 and STAT3, respectively [15, 16]. On the other hand, PIASx and PIASy repress the transcriptional activity of STAT1 and STAT4 by recruiting co-repressor molecules such as histone deacetylases [17].
Suppressors of cytokine signaling (SOCS) proteins
The suppressor of cytokine signaling (SOCS) family is composed of SOCS-1 to SOCS-7 and cytokine-inducible SH2-containing protein (CIS). SOCS proteins are cytokine-induced negative feedback-loop regulators of JAK-STAT signaling. Each member protein in this family has a central SH2 domain, variable amino-terminal domain and a carboxy-terminal of 40 amino acids module known as SOCS box. The central SH2 domain directs the target binding of each CIS/SOCS protein [18]. Three mechanisms have been proposed by which a SOCS protein provides negative modulation. First, SOCS binds the phospho-tyrosine residues on the receptors and physically blocks the STATs from binding to its receptors. Second, SOCS proteins can bind directly to its specific JAKs or to the receptors and inhibit the corresponding JAK kinase activity. Third, SOCS associate with the elongin BC complex and cullin 2, accelerating the ubiquination of JAKs and presumably the receptors [19].
Endosomal degradation of JAK/receptor complex through receptor mediated endocytosis appears to be another mechanism involved in the regulation of the pathway. STAT5ß dimerizes with the full-length, wild-type protein, STAT5α, upon activation. This leads to negative regulation of the JAK-STAT signaling cascade in addition to the major mechanisms described above [20].
JAK-STAT pathway and hematological malignancies
Dysregulation in JAK-STAT pathway have been described in a variety of hematological malignancies, especially in myeloid disorders. These events may play key roles in initiation and progression of respective diseases.
Myeloproliferative Neoplasms (MPN)
Ph negative MPNs
Short interfering RNA and JAK2 inhibitor (AG490) have been used to study the activity of JAK2. JAK2 was shown to be central to the erythropoietin-independent erythroid proliferation in polycythemia vera (PV). A common mutation (V617F) in the JH2 pseudo-kinase domain of JAK2 was shown in 80% of the patients [21]. This results in constitutive activation of STAT5 and a malignant phenotype [22]. Other investigators confirmed the fact and showed that 30-50% of patients with essential thrombocythemia (ET) and primary myelofibrosis (PMF) also harbor the same mutation [23–25].
Allelic burden of JAK2V617F was found to play an important role in determining a distinct clinico-pathologic phenotype from the same mutation [22, 26]. It is suggested by the fact that homozygous JAK2V617F mutation is 30% in PV compared to 2-4% in ET [22, 26]. Transgenic mouse models and clinical cases support the gene-dosage model for MPN pathogenesis. It was observed that high allele burden leads to PV phenotype, whereas reduction in allelic burden leads to ET [22, 27–29].
In patients with PV who are negative for JAK2V617F, Scott et al., used a candidate gene approach to search for alleles that can activate JAK signaling. Their work demonstrated at least 8 distinct mutations involving residues 538 to 543 in exon 12 region of JAK2 [29].
Irrespective of JAK2V617F status, patients with PV exhibits high STAT3 and 5 activities, patients with ET have high STAT3 activity, whereas patients with PMF showed low activities of both STAT3 and 5. It is evident that preferential activation of JAK2–STAT5 or JAK2–STAT3 signaling determines PV, ET and PMF phenotype. However patients with JAK2V617F-negative ET or PMF may harbor mutations in other genes in JAK–STAT pathway [30].
Atypical MPNs
Rare JAK2 gene rearrangement can result in atypical form of chronic myeloid leukemia (aCML). These gene rearrangements include PCM1-JAK2, TEL-JAK2 and BCR-JAK2 [31–33]. Rarely, JAK2V617F mutation can present phenotypically as chronic myelomonocytic leukemia /aCML.
PCM1-JAK2 gene rearrangement was discovered in the translocation, t(8;9)(p22;p24). It is reported in few cases with atypical CML, chronic eosinophilic leukemia, AML (M2, M6), pre B-ALL and T-cell lymphoma.
Lymphoma and lymphoid leukemia
T-cell lymphoma and leukemia
JAK1 gene plays a unique role in lymphohematopoiesis. Flex et al., showed several acquired JAK1 missense mutations in adult lymphoblastic leukemia, especially in T-cell precursor type where it accounted for 18% of the cases. It was associated with advanced age and poor prognosis [34–36].
Translocation between short-arm of chromosome 9 (harboring JAK2 gene) and 12, t(9;12)(p24;p13) results in the formation of a fusion protein, TEL-JAK2. This results in constitutive JAK2 activation. It was found in a case of childhood T-cell acute lymphoblastic leukemia [37] and in a case with pre-B-cell ALL [33].
Adult T-cell Leukemia (HTLV-1 positive)
Human T-cell lymphotropic virus-1 encoded Tax protein suppresses apoptosis through constitutive activation of the NFκB pathway. This is sufficient for immortalization of CD4+ lymphocytes. HTLV-1 infected T-cells over-express IL-2, IL-15 as well as IL-2 and IL-15 receptors by Tax-induced NFκB activation. These in turn activate JAK3-STAT5 pathway leading to lymphocyte proliferation and adult T-cell lymphoma /leukemia [38].
T-cell Large Granular Lymphocytic Leukemia, Anaplastic Large cell Lymphoma, Angioimmunoblastic Lymphoma and Sezary Syndrome
Dysregulation of STAT proteins also contributes to the pathogenesis of various types of lymphoid malignancies. Schade et al., reported increase activity of STAT3 in T-cell large granular lymphocytic leukemia [39]. Constitutive activation of STAT3 & 5 was also found to be an important event in the pathogenesis of anaplastic large cell lymphoma, T-cell angioimmunoblastic lymphoma and Sezary syndrome [40, 41].
B-Cell lymphoma and leukemia
Acute lymphoblastic leukemia
Kearney et al., performed sequencing of exon 14 of JAK2 gene in patients with Down syndrome associated ALL. An acquired somatic point mutation invovling residue R683 was found in 28% of cases [42]. A different mutation in JAK2 gene in a patient with Down syndrome associated precursor B-cell lymphoblastic leukemia was reported by Malinge et al., This mutation was in-frame deletion of five-amino acid residues from position 682 to 686, named as IREED mutation, in the pseudokinase domain of JAK2. This IREED mutant remains constitutively active and induces cytokine independence in Ba/F3 cells [43].
Another JAK2 mutation involves fusion with SSBP2 in t(5;9)(q14.1;p24.1) translocation. It was detected in a patient with pre B-cell ALL [44].
Chronic lymphocytic leukemia
Constitutive activation of STAT1 & 3 was noticed in transformed B-cells in patients with chronic lymphocytic leukemia (CLL). Activated STAT1 & 3 proteins in malignant B-cells were phosporylated at serine instead of tyrosine residue. It remained uncertain whether activated STAT involved directly in the pathophysiology of this disease or represented a pure epi-phenomenon [45].
Primary mediastinal B-cell lymphoma
A bi-allelic mutation in SOCS-1 has been reported in primary mediastinal B-cell lymphoma. This results in delayed turnover of phosphorylated JAK2 and increased activity of STAT5, which may explain the uncontrolled growth of malignant cells [46].
Hodgkin lymphoma
Gene rearrangement by translocation involving JAK2, t(4;9)(q21;p24), has been reported in 2 cases of classic Hodgkin lymphoma by a group from Belgium [47]. This gene re-arrangement creates a novel fusion product, SEC31A-JAK2. This oncogenic SEC31A-JAK2 has constitutively active tyrosine kinase activity. Mutations of SOCS-1 have also been identified in Hodgkin lymphoma [48].
Myeloid leukemia
Acute myeloid leukemia
Increased activity of STAT3 has been reported in 20-50% of AML patients while STAT5 and STAT1 activation is uncommon [49, 50]. The pathologic value of activated STAT3 in AML is unclear. It was also found in acute promyelocytic leukemia (APL). An interstitial deletion within chromosome 17 has been described to result in STAT5b-RARα fusion protein. This was implied to serve as a key pathogenic event in a patient with APL [51]. Transformation from severe congenital neutropenia due to truncated G-CSF receptor to AML also involves dysregulation of STAT proteins. Truncated G-CSF receptor lacks a crucial binding site for SOCS-3, thus relieving inhibition of STAT5, but not of STAT3. This favors myeloid proliferation over differentiation. These truncated receptors are also resistant to down modulation through ubiquination [52].
Rare point mutations, one in JAK2 (T875N) and three in JAK3 (A572V, V722I, P132T), have been described in patients with acute megakaryocytic luekemia [52]. PCM1-JAK2 gene rearrangement mentioned above was found in M2 and M6 AML [36].
Myelodysplastic syndrome (MDS)
Perturbation in JAK-STAT pathway is rare in MDS. Only 4.2% patients with MDS were found to harbor JAK2V617F mutation in a large screening series [53]. Of interest is a disease entity called refractory anemia with ringed sideroblast associated with marked thrombocytosis (RARS-t). These patients have features of both MDS and MPN. It was recently shown that 6 out of 9 (67%) patients with RARS-t have JAK2V617F mutation [54].
Multiple myeloma
Constitutive expression of STAT3 has been seen frequently in patients with multiple myeloma and lymphoma [55]. However, overexpression of anti-apoptotic proteins of Bcl-2, Bcl-XL and Mcl-1 may not rely on STAT3 activation. The role of this pathway in pathogenesis of myeloma remains to be determined [56].
Novel inhibitors of JAK-STAT pathway
The specific and profound involvement of JAK-STAT pathway in various hematological malignancies, especially Bcr-Abl negative MPNs, makes them ideal targets for pharmacological intervention [57]. Several strategies have been proposed to inhibit or modulate this pathway [58, 59]. JAK inhibitors are among the first successful agents reaching clinical application [60, 61].
JAK inhibitors
There are at least 10 different JAK inhibitors undergoing various phases of clinical trials (Table 1). Ruxolitinib (Jakafi manufactured by Incyte, Inc) has been approved by FDA for patients with intermediate- to high- risk primary or secondary myelofibrosis.
Ruxolitinib
Ruxolitinib (INCB018424) is a non-selective JAK1 & 2 inhibitor. Multiple phase I/II studies in various hematological malignancies are underway. Among these, results are most mature for PMF and post –ET /PV myelofibrosis.
Phase I/II studies
A total of 153 patients with myelofibrosis were recruited in a phase I/II study. Dose limiting toxicity was thrombocytopenia with maximum tolerated dose of 25 mg twice daily. Effect on JAK2V617F allele burden or bone marrow fibrosis was insignificant [62]. After a longer follow up, it was described that abrupt discontinuation can lead to serious adverse events like acute relapse of constitutional symptoms, rapid and painful enlargement of spleen and shock. By the time of last report on the follow up of these patients 35% of patient died and 10% transformed to leukemia [63, 64].
Eghtedar et al., reported a phase I/II study of ruxolitinib in patients with relapsed /refractory AML. This study has shown 7% complete remission rate. It was noticed that patients who achieved complete remission were those with post-MPN AML [65].
Phase III studies
Ruxolitinib has undergone 2 separate randomized controlled trials, referred to as the COntrolled MyeloFibrosis study with ORal JAK inhibitor Treatment (COMFORT) I & II (Table 2) [66, 67]. Patients were randomized between ruxolitinib vs placebo (COMFORT I) or best available therapy (COMFORT II). Patients with PMF or post –ET /PV myelofibrosis must have high or intermediate international prognostic scores, palpable spleen ≥5 cm below the left costal margin, and platelet count equal or greater than 100 × 109 /L. Patients were recruited irrespective of JAK2 gene mutation status. Differences in the design of these trials and major outcomes are summarized in Table 2.
These trials support the palliative role of ruxolitinib which significantly reduced spleen volume and constitutional symptoms. The responses were not specific for JAK2 mutation.
SAR302503 (TG101348)
SAR302503 is a selective JAK2 inhibitor with activity on both wild type and mutant JAK2 [68, 69]. Fifty nine patients were enrolled in a multi-center phase I/II trial with high-or- intermediate-risk primary or post-PV/ET myelofibrosis. Patients with platelet count equal or greater than 50 x 109 /L were eligible. Reversible increase in serum amylase was the dose limiting toxicity. Serious adverse events included mainly myelosuppression and gastrointestinal toxicity. A spleen response of 47% was seen after one year of treatment. JAK2V617F allele burden was significantly decreased at 6 months in mutation-positive patients (n = 51; P = .04). The response was higher in patients with allele burden greater than 20% (n = 23; P < .01) [70]. A multinational, randomized, placebo controlled phase III trial has been launched recently for patients with high risk myelofibrosis.
Lestaurtinib (CEP701)
Lestaurtinib is an orally available indolocarbazole derivative. It was originally identified as an inhibitor of the neurotropin receptor TrkA. It was found to also inhibit FLT3, JAK2 and JAK3. Due to its strong activity against FLT3, it is under investigation in patients with acute leukemia. A phase I/II study of lestaurtinib was done in 22 patients with JAK2-mutated myelofibrosis. The patients received 80 mg twice-daily. The response rate was 27%. The median duration of response was more than 14 months. However, bone marrow fibrosis or JAK2V617F allele burden was not significantly altered [71].
In another phase 2 study, lestaurtinib was given to 39 high-risk patients with JAK2V617F-positive PV (n=27) or ET (n=12). After 18 weeks of treatment, a significant decrease in spleen size was reported in 83% of the patients. Six patient developed thrombosis. The increase in the thrombotic events remains a concern for this agent [72].
CYT387
CYT387 is an ATP-competitive small molecule JAK1/JAK2 inhibitor [73–76]. It has an IC50 of 11 and 18 nM for JAK1 and JAK2, respectively. Unlike lestaurtinib, CYT387 has significantly less activity against other kinases, including JAK3 (IC50=0.16 μM).
One hundred sixty three patients with high- or intermediate- risk myelofibrosis were enrolled in a phase I/II trial of CYT387. The median duration of treatment was 6.6 months. Dose limiting toxicities included hyper-lipasemia and headaches. Maximum tolerated dose was 300 mg/day. The response rates for reduction of spleen size, anemia, and constitutional symptom were 45%, 50%, and more than 50%, respectively. It is interesting to note that responses were also seen in patients who had previously failed ruxolitinib or SAR302503. Surprisingly, 58% of transfusion-dependent patients became transfusion-independent. Transient light headedness and hypotension were the common adverse events. Treatment-related severe anemia was infrequent (< 1%). This property separates it from ruxolitinib and SAR302503 [77, 78].
SB1518 (pacritinib)
Pacritinib is another orally bioavailable ATP-competitive small molecule inhibitor of JAK2 and the mutant JAK2V617F [79–82]. It inhibitis JAK-STAT signaling pathway through caspase-dependent apoptosis. In a phase I/II trial, 34 patients with primary or post PV/ET myelofibrosis were enrolled. The median time on study was 8.2 months. Patients with low blood counts were not excluded. Most common adverse events were diarrhea, nausea and vomiting. The primary end point was spleen response, which was 32%. The median duration of spleen response has not been reached at the recent update [83].
In another phase I trial, 30 patients with relapsed/refractory lymphoma were enrolled. The dose was escalated from 200–600 mg/day. Severe adverse events included cytopenia, internal hemorrhage, various infections and abdominal distention. No patient achieved complete remission. Three patients had partial remission and 13 patients had stable disease [84]. Phase II trial is ongoing.
LY2784544
LY2784544 is a selective mutant JAK2 kinase inhibitor with an IC50 of 68 nM. This molecule selectively inhibited the JAK2 V617F-STAT5 signaling at a concentration that was 41-fold lower than that needed for the wild type (WT) JAK2 signaling (IC50 = 55 nM for V617F vs. 2260 nM for WT ) [60]. A phase I/II trial recruited 19 patients with Ph negative MPN. Maximum tolerated dose was 120 mg/day. Spleen response was 22% for patients with MF. JAK2V617F allele burden was not significantly decreased. Main adverse events included diarrhea, nausea, anemia and electrolyte imbalance [85].
XL019
XL019 represents another oral selective JAK2 inhibitor (IC50= 2nM for JAK2, 130 nM for JAK1, 250 nM for JAK3, 340 nM for TYK2) [86, 87]. A phase I study recruited 21 patients with myelofibrosis. 16 patients had JAK2V617F, one had MPL W515F. Dose ranged from 25 to 300 mg. Surprisingly, there was no hematological adverse events. Improvement in clinical outcome observed in all patients with JAK2V617F or MPL activating mutations [86]. Despite these results, further development of clinical trials using XL019 was aborted out of the concerns for neurologic toxicities [88].
NS-018, BMS-911543 and AZD1480
These JAK inhibitors are also selective JAK inhibitors at very early stage of drug development [48, 89–91]. NS-018 was shown to be highly selective against JAK2 with IC50 <1 nM [89]. It had 30–50 fold greater selectivity for JAK2 over other JAK-family kinases, such as JAK1, JAK3 and Tyk2. BMS-911543 is a functionally selective JAK2 inhibitor [90]. AZD1480 has been shown both in vitro and in vivo to inhibit the growth of multiple malignancies by inhibiting phosphorylation of JAK2, STAT3 and MAPK signaling proteins [91–99]. Clinical investigations on these agents are underway.
Conclusion and future directions
Currently the benefits of therapy with JAK inhibitors in MPNs are palliative in nature. This is suggested by inability of ruxolitinib to influence the natural history of the diseases and by the fact that the responses are unrelated to JAK mutation status. JAK2V617F mutation may be a key event but not necessarily a primary factor driving the tumorigenesis of MPNs. Other molecular events may precede the acquisition of JAK2V617F mutation in these disorders. Novel selective JAK2 inhibitors are the active focus for further clinical investigations [60, 61, 88].
In addition to JAK inhibitors, targeting STAT signaling is emerging as an attractive approach to inhibit tumorigenesis. Various strategies have been employed to influence STAT signaling at multiple levels, though these approaches are still at early stage and only supported by pre-clinical data [13, 100–107]. Further advancement in understanding the molecular events and refining the targets can potentially lead to further clinical benefit. Simultaneous intervention at multiple levels of the signaling cascade by combining different targeting agents may offer additional advantages and is worthwhile studying.
References
Ferrajoli A, Faderl S, Ravandi F, Estrov Z: The JAK-STAT pathway: a therapeutic target in hematological malignancies. Curr Cancer Drug Targets 2006,6(8):671–679. 10.2174/156800906779010227
Moriggl R, Sexl V, Piekorz R, Topham D, Ihle JN: Stat5 activation is uniquely associated with cytokine signaling in peripheral T cells. Immunity 1999,11(2):225–230. 10.1016/S1074-7613(00)80097-7
Friedrich K, Kammer W, Erhardt I, Brandlein S, Sebald W, Moriggl R: Activation of STAT5 by IL-4 relies on Janus kinase function but not on receptor tyrosine phosphorylation, and can contribute to both cell proliferation and gene regulation. Int Immunol 1999,11(8):1283–1294. 10.1093/intimm/11.8.1283
Moriggl R, Topham DJ, Teglund S, Sexl V, McKay C, Wang D, Hoffmeyer A, van Deursen J, Sangster MY, Bunting KD, et al.: Stat5 is required for IL-2-induced cell cycle progression of peripheral T cells. Immunity 1999,10(2):249–259. 10.1016/S1074-7613(00)80025-4
Bradley HL, Hawley TS, Bunting KD: Cell intrinsic defects in cytokine responsiveness of STAT5-deficient hematopoietic stem cells. Blood 2002,100(12):3983–3989. 10.1182/blood-2002-05-1602
Bunting KD, Bradley HL, Hawley TS, Moriggl R, Sorrentino BP, Ihle JN: Reduced lymphomyeloid repopulating activity from adult bone marrow and fetal liver of mice lacking expression of STAT5. Blood 2002,99(2):479–487. 10.1182/blood.V99.2.479
Ward AC, Touw I, Yoshimura A: The Jak-Stat pathway in normal and perturbed hematopoiesis. Blood 2000,95(1):19–29.
Wells JA, de Vos AM: Hematopoietic receptor complexes. Annu Rev Biochem 1996, 65: 609–634. 10.1146/annurev.bi.65.070196.003141
Carter-Su C, Smit LS: Signaling via JAK tyrosine kinases: growth hormone receptor as a model system. Recent Prog Horm Res 1998, 53: 61–82. discussion 82–63
Valentino L, Pierre J: JAK/STAT signal transduction: regulators and implication in hematological malignancies. Biochem Pharmacol 2006,71(6):713–721. 10.1016/j.bcp.2005.12.017
Vera J, Rateitschak K, Lange F, Kossow C, Wolkenhauer O, Jaster R: Systems biology of JAK-STAT signalling in human malignancies. Prog Biophys Mol Biol 2011,106(2):426–434. 10.1016/j.pbiomolbio.2011.06.013
Levy DE, Darnell JE Jr: Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol 2002,3(9):651–662.
Shuai K, Liu B: Regulation of JAK-STAT signalling in the immune system. Nat Rev Immunol 2003,3(11):900–911. 10.1038/nri1226
Shuai K, Liu B: Regulation of gene-activation pathways by PIAS proteins in the immune system. Nat Rev Immunol 2005,5(8):593–605. 10.1038/nri1667
Chung CD, Liao J, Liu B, Rao X, Jay P, Berta P, Shuai K: Specific inhibition of Stat3 signal transduction by PIAS3. Science 1997,278(5344):1803–1805. 10.1126/science.278.5344.1803
Liu B, Liao J, Rao X, Kushner SA, Chung CD, Chang DD, Shuai K: Inhibition of Stat1-mediated gene activation by PIAS1. Proc Natl Acad Sci U S A 1998,95(18):10626–10631. 10.1073/pnas.95.18.10626
Arora T, Liu B, He H, Kim J, Murphy TL, Murphy KM, Modlin RL, Shuai K: PIASx is a transcriptional co-repressor of signal transducer and activator of transcription 4. J Biol Chem 2003,278(24):21327–21330. 10.1074/jbc.C300119200
Tamiya T, Kashiwagi I, Takahashi R, Yasukawa H, Yoshimura A: Suppressors of cytokine signaling (SOCS) proteins and JAK/STAT pathways: regulation of T-cell inflammation by SOCS1 and SOCS3. Arterioscler Thromb Vasc Biol 2011,31(5):980–985. 10.1161/ATVBAHA.110.207464
Rawlings JS, Rosler KM, Harrison DA: The JAK/STAT signaling pathway. J Cell Sci 2004,117(Pt 8):1281–1283.
Benekli M, Baumann H, Wetzler M: Targeting signal transducer and activator of transcription signaling pathway in leukemias. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 2009,27(26):4422–4432. 10.1200/JCO.2008.21.3264
James C, Ugo V, Le Couedic JP, Staerk J, Delhommeau F, Lacout C, Garcon L, Raslova H, Berger R, Bennaceur-Griscelli A, et al.: A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005,434(7037):1144–1148. 10.1038/nature03546
Levine RL, Gilliland DG: Myeloproliferative disorders. Blood 2008,112(6):2190–2198. 10.1182/blood-2008-03-077966
Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, Vassiliou GS, Bench AJ, Boyd EM, Curtin N, et al.: Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005,365(9464):1054–1061.
Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, Tichelli A, Cazzola M, Skoda RC: A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Eng J Med 2005,352(17):1779–1790. 10.1056/NEJMoa051113
Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ, Boggon TJ, Wlodarska I, Clark JJ, Moore S, et al.: Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005,7(4):387–397. 10.1016/j.ccr.2005.03.023
Tefferi A, Lasho TL, Patnaik MM, Finke CM, Hussein K, Hogan WJ, Elliott MA, Litzow MR, Hanson CA, Pardanani A: JAK2 germline genetic variation affects disease susceptibility in primary myelofibrosis regardless of V617F mutational status: nullizygosity for the JAK2 46/1 haplotype is associated with inferior survival. Leukemia: official journal of the Leukemia Society of America, Leukemia Research Fund, UK 2010,24(1):105–109. 10.1038/leu.2009.225
Passamonti F, Rumi E: Clinical relevance of JAK2 (V617F) mutant allele burden. Haematologica 2009,94(1):7–10. 10.3324/haematol.2008.001271
Spivak JL, Silver RT: The revised World Health Organization diagnostic criteria for polycythemia vera, essential thrombocytosis, and primary myelofibrosis: an alternative proposal. Blood 2008,112(2):231–239. 10.1182/blood-2007-12-128454
Scott LM, Tong W, Levine RL, Scott MA, Beer PA, Stratton MR, Futreal PA, Erber WN, McMullin MF, Harrison CN, et al.: JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Eng J Med 2007,356(5):459–468. 10.1056/NEJMoa065202
Teofili L, Martini M, Cenci T, Petrucci G, Torti L, Storti S, Guidi F, Leone G, Larocca LM: Different STAT-3 and STAT-5 phosphorylation discriminates among Ph-negative chronic myeloproliferative diseases and is independent of the V617F JAK-2 mutation. Blood 2007,110(1):354–359. 10.1182/blood-2007-01-069237
Bousquet M, Quelen C, De Mas V, Duchayne E, Roquefeuil B, Delsol G, Laurent G, Dastugue N, Brousset P: The t(8;9)(p22;p24) translocation in atypical chronic myeloid leukaemia yields a new PCM1-JAK2 fusion gene. Oncogene 2005,24(48):7248–7252. 10.1038/sj.onc.1208850
Griesinger F, Hennig H, Hillmer F, Podleschny M, Steffens R, Pies A, Wormann B, Haase D, Bohlander SK: A BCR-JAK2 fusion gene as the result of a t(9;22)(p24;q11.2) translocation in a patient with a clinically typical chronic myeloid leukemia. Genes Chromosomes Cancer 2005,44(3):329–333. 10.1002/gcc.20235
Peeters P, Raynaud SD, Cools J, Wlodarska I, Grosgeorge J, Philip P, Monpoux F, Van Rompaey L, Baens M, Van den Berghe H, et al.: Fusion of TEL, the ETS-variant gene 6 (ETV6), to the receptor-associated kinase JAK2 as a result of t(9;12) in a lymphoid and t(9;15;12) in a myeloid leukemia. Blood 1997,90(7):2535–2540.
Flex E, Petrangeli V, Stella L, Chiaretti S, Hornakova T, Knoops L, Ariola C, Fodale V, Clappier E, Paoloni F, et al.: Somatically acquired JAK1 mutations in adult acute lymphoblastic leukemia. J Exp Med 2008,205(4):751–758. 10.1084/jem.20072182
Murati A, Gelsi-Boyer V, Adelaide J, Perot C, Talmant P, Giraudier S, Lode L, Letessier A, Delaval B, Brunel V, et al.: PCM1-JAK2 fusion in myeloproliferative disorders and acute erythroid leukemia with t(8;9) translocation. Leukemia: official journal of the Leukemia Society of America, Leukemia Research Fund, UK 2005,19(9):1692–1696. 10.1038/sj.leu.2403879
Reiter A, Walz C, Watmore A, Schoch C, Blau I, Schlegelberger B, Berger U, Telford N, Aruliah S, Yin JA, et al.: The t(8;9)(p22;p24) is a recurrent abnormality in chronic and acute leukemia that fuses PCM1 to JAK2. Cancer Res 2005,65(7):2662–2667. 10.1158/0008-5472.CAN-04-4263
Lacronique V, Boureux A, Valle VD, Poirel H, Quang CT, Mauchauffe M, Berthou C, Lessard M, Berger R, Ghysdael J, et al.: A TEL-JAK2 fusion protein with constitutive kinase activity in human leukemia. Science 1997,278(5341):1309–1312. 10.1126/science.278.5341.1309
Ratner L: JAK blockade and HTLV. Blood 2011,117(6):1771–1772. 10.1182/blood-2010-12-322008
Schade AE, Wlodarski MW, Maciejewski JP: Pathophysiology defined by altered signal transduction pathways: the role of JAK-STAT and PI3K signaling in leukemic large granular lymphocytes. Cell Cycle 2006,5(22):2571–2574. 10.4161/cc.5.22.3449
Meier C, Hoeller S, Bourgau C, Hirschmann P, Schwaller J, Went P, Pileri SA, Reiter A, Dirnhofer S, Tzankov A: Recurrent numerical aberrations of JAK2 and deregulation of the JAK2-STAT cascade in lymphomas. Modern pathology: an official journal of the United States and Canadian Academy of Pathology, Inc 2009,22(3):476–487. 10.1038/modpathol.2008.207
Zhang Q, Nowak I, Vonderheid EC, Rook AH, Kadin ME, Nowell PC, Shaw LM, Wasik MA: Activation of Jak/STAT proteins involved in signal transduction pathway mediated by receptor for interleukin 2 in malignant T lymphocytes derived from cutaneous anaplastic large T-cell lymphoma and Sezary syndrome. Proc Natl Acad Sci U S A 1996,93(17):9148–9153. 10.1073/pnas.93.17.9148
Kearney L, Gonzalez De Castro D, Yeung J, Procter J, Horsley SW, Eguchi-Ishimae M, Bateman CM, Anderson K, Chaplin T, Young BD, et al.: Specific JAK2 mutation (JAK2R683) and multiple gene deletions in Down syndrome acute lymphoblastic leukemia. Blood 2009,113(3):646–648. 10.1182/blood-2008-08-170928
Malinge S, Ben-Abdelali R, Settegrana C, Radford-Weiss I, Debre M, Beldjord K, Macintyre EA, Villeval JL, Vainchenker W, Berger R, et al.: Novel activating JAK2 mutation in a patient with Down syndrome and B-cell precursor acute lymphoblastic leukemia. Blood 2007,109(5):2202–2204. 10.1182/blood-2006-09-045963
Poitras JL, Dal Cin P, Aster JC, Deangelo DJ, Morton CC: Novel SSBP2-JAK2 fusion gene resulting from a t(5;9)(q14.1;p24.1) in pre-B acute lymphocytic leukemia. Genes Chromosomes Cancer 2008,47(10):884–889. 10.1002/gcc.20585
Frank DA, Mahajan S, Ritz J: B lymphocytes from patients with chronic lymphocytic leukemia contain signal transducer and activator of transcription (STAT) 1 and STAT3 constitutively phosphorylated on serine residues. J Clin Invest 1997,100(12):3140–3148. 10.1172/JCI119869
Melzner I, Bucur AJ, Bruderlein S, Dorsch K, Hasel C, Barth TF, Leithauser F, Moller P: Biallelic mutation of SOCS-1 impairs JAK2 degradation and sustains phospho-JAK2 action in the MedB-1 mediastinal lymphoma line. Blood 2005,105(6):2535–2542. 10.1182/blood-2004-09-3701
Van Roosbroeck K, Cox L, Tousseyn T, Lahortiga I, Gielen O, Cauwelier B, De Paepe P, Verhoef G, Marynen P, Vandenberghe P, et al.: JAK2 rearrangements, including the novel SEC31A-JAK2 fusion, are recurrent in classical Hodgkin lymphoma. Blood 2011,117(15):4056–4064. 10.1182/blood-2010-06-291310
Weniger MA, Melzner I, Menz CK, Wegener S, Bucur AJ, Dorsch K, Mattfeldt T, Barth TF, Moller P: Mutations of the tumor suppressor gene SOCS-1 in classical Hodgkin lymphoma are frequent and associated with nuclear phospho-STAT5 accumulation. Oncogene 2006,25(18):2679–2684. 10.1038/sj.onc.1209151
Ghoshal Gupta S, Baumann H, Wetzler M: Epigenetic regulation of signal transducer and activator of transcription 3 in acute myeloid leukemia. Leuk Res 2008,32(7):1005–1014. 10.1016/j.leukres.2007.11.035
Steensma DP, McClure RF, Karp JE, Tefferi A, Lasho TL, Powell HL, DeWald GW, Kaufmann SH: JAK2 V617F is a rare finding in de novo acute myeloid leukemia, but STAT3 activation is common and remains unexplained. Leukemia: official journal of the Leukemia Society of America, Leukemia Research Fund, UK 2006,20(6):971–978. 10.1038/sj.leu.2404206
Maurer AB, Wichmann C, Gross A, Kunkel H, Heinzel T, Ruthardt M, Groner B, Grez M: The Stat5-RARalpha fusion protein represses transcription and differentiation through interaction with a corepressor complex. Blood 2002,99(8):2647–2652. 10.1182/blood.V99.8.2647
Constantinescu SN, Girardot M, Pecquet C: Mining for JAK-STAT mutations in cancer. Trends Biochem Sci 2008,33(3):122–131. 10.1016/j.tibs.2007.12.002
Levine RL, Loriaux M, Huntly BJ, Loh ML, Beran M, Stoffregen E, Berger R, Clark JJ, Willis SG, Nguyen KT, et al.: The JAK2V617F activating mutation occurs in chronic myelomonocytic leukemia and acute myeloid leukemia, but not in acute lymphoblastic leukemia or chronic lymphocytic leukemia. Blood 2005,106(10):3377–3379. 10.1182/blood-2005-05-1898
Szpurka H, Tiu R, Murugesan G, Aboudola S, Hsi ED, Theil KS, Sekeres MA, Maciejewski JP: Refractory anemia with ringed sideroblasts associated with marked thrombocytosis (RARS-T), another myeloproliferative condition characterized by JAK2 V617F mutation. Blood 2006,108(7):2173–2181. 10.1182/blood-2006-02-005751
Zl W, Yq S, Yf S, Zhu J: High nuclear expression of STAT3 is associated with unfavorable prognosis in diffuse large B-cell lymphoma. J Hematol Oncol 2011,4(1):31. 10.1186/1756-8722-4-31
Quintanilla-Martinez L, Kremer M, Specht K, Calzada-Wack J, Nathrath M, Schaich R, Hofler H, Fend F: Analysis of signal transducer and activator of transcription 3 (Stat 3) pathway in multiple myeloma: Stat 3 activation and cyclin D1 dysregulation are mutually exclusive events. Am J Pathol 2003,162(5):1449–1461. 10.1016/S0002-9440(10)64278-2
Zhao A, Gao R, Zhao Z: Development of a highly sensitive method for detection of JAK2V617F. J Hematol Oncol 2011,4(1):40. 10.1186/1756-8722-4-40
Tognon R, Gasparotto E, Neves R, Nunes N, Ferreira A, Palma P, Kashima S, Covas D, Santana M, Souto E, et al.: Deregulation of apoptosis-related genes is associated with PRV1 overexpression and JAK2 V617F allele burden in Essential Thrombocythemia and Myelofibrosis. J Hematol Oncol 2012,5(1):2. 10.1186/1756-8722-5-2
Zhao W, Du Y, Ho W, Fu X, Zhao Z: JAK2V617F and p53 mutations coexist in erythroleukemia and megakaryoblastic leukemic cell lines. Experimental Hematology & Oncology 2012,1(1):15. 10.1186/2162-3619-1-15
Passamonti F, Maffioli M, Caramazza D: New generation small-molecule inhibitors in myeloproliferative neoplasms. Curr Opin Hematol 2012,19(2):117–123. 10.1097/MOH.0b013e32834ff575
Randhawa J, Ostojic A, Vrhovac R, Atallah E, Verstovsek S: Splenomegaly in myelofibrosis–new options for therapy and the therapeutic potential of Janus kinase 2 inhibitors. J Hematol Oncol 2012,5(1):43. 10.1186/1756-8722-5-43
Verstovsek S, Kantarjian H, Mesa RA, Pardanani AD, Cortes-Franco J, Thomas DA, Estrov Z, Fridman JS, Bradley EC, Erickson-Viitanen S, et al.: Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Eng J Med 2010,363(12):1117–1127. 10.1056/NEJMoa1002028
Tefferi A, Litzow MR, Pardanani A: Long-term outcome of treatment with ruxolitinib in myelofibrosis. N Eng J Med 2011,365(15):1455–1457. 10.1056/NEJMc1109555
Verstovsek S, Kantarjian HM, Estrov Z, Cortes JE, Thomas DA, Kadia T, Pierce S, Jabbour E, Borthakur G, Rumi E, et al.: Long-term outcomes of 107 patients with myelofibrosis receiving JAK1/JAK2 inhibitor ruxolitinib: survival advantage in comparison to matched historical controls. Blood 2012,120(6):1202–1209. 10.1182/blood-2012-02-414631
Eghtedar A, Verstovsek S, Estrov Z, Burger J, Cortes J, Bivins C, Faderl S, Ferrajoli A, Borthakur G, George S, et al.: Phase 2 study of the JAK kinase inhibitor ruxolitinib in patients with refractory leukemias, including postmyeloproliferative neoplasm acute myeloid leukemia. Blood 2012,119(20):4614–4618. 10.1182/blood-2011-12-400051
Harrison C, Kiladjian JJ, Al-Ali HK, Gisslinger H, Waltzman R, Stalbovskaya V, McQuitty M, Hunter DS, Levy R, Knoops L, et al.: JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Eng J Med 2012,366(9):787–798. 10.1056/NEJMoa1110556
Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, Catalano JV, Deininger M, Miller C, Silver RT, et al.: A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Eng J Med 2012,366(9):799–807. 10.1056/NEJMoa1110557
Wernig G, Kharas MG, Okabe R, Moore SA, Leeman DS, Cullen DE, Gozo M, McDowell EP, Levine RL, Doukas J, et al.: Efficacy of TG101348, a selective JAK2 inhibitor, in treatment of a murine model of JAK2V617F-induced polycythemia vera. Cancer Cell 2008,13(4):311–320. 10.1016/j.ccr.2008.02.009
Lasho TL, Tefferi A, Hood JD, Verstovsek S, Gilliland DG, Pardanani A: TG101348, a JAK2-selective antagonist, inhibits primary hematopoietic cells derived from myeloproliferative disorder patients with JAK2V617F, MPLW515K or JAK2 exon 12 mutations as well as mutation negative patients. Leukemia: official journal of the Leukemia Society of America, Leukemia Research Fund, UK 2008,22(9):1790–1792. 10.1038/leu.2008.56
Pardanani A, Gotlib JR, Jamieson C, Cortes JE, Talpaz M, Stone RM, Silverman MH, Gilliland DG, Shorr J, Tefferi A: Safety and efficacy of TG101348, a selective JAK2 inhibitor, in myelofibrosis. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 2011,29(7):789–796. 10.1200/JCO.2010.32.8021
Santos FP, Kantarjian HM, Jain N, Manshouri T, Thomas DA, Garcia-Manero G, Kennedy D, Estrov Z, Cortes J, Verstovsek S: Phase 2 study of CEP-701, an orally available JAK2 inhibitor, in patients with primary or post-polycythemia vera/essential thrombocythemia myelofibrosis. Blood 2010,115(6):1131–1136. 10.1182/blood-2009-10-246363
Moliterno AR, Hexner E, Roboz GJ, Carroll M, Luger S, Mascarenhas J, Hoffman R, Bensen-Kennedy DM: An Open-Label Study of CEP-701 in Patients with JAK2 V617F-Positive PV and ET: Update of 39 Enrolled Patients. ASH Annual Meeting Abstracts 2009,114(22):753.
Sparidans RW, Durmus S, Xu N, Schinkel AH, Schellens JH, Beijnen JH: Liquid chromatography-tandem mass spectrometric assay for the JAK2 inhibitor CYT387 in plasma. J Chromatogr B Analyt Technol Biomed Life Sci 2012, 895–896: 174–177.
Monaghan KA, Khong T, Burns CJ, Spencer A: The novel JAK inhibitor CYT387 suppresses multiple signalling pathways, prevents proliferation and induces apoptosis in phenotypically diverse myeloma cells. Leukemia: official journal of the Leukemia Society of America, Leukemia Research Fund, UK 2011,25(12):1891–1899. 10.1038/leu.2011.175
Tyner JW, Bumm TG, Deininger J, Wood L, Aichberger KJ, Loriaux MM, Druker BJ, Burns CJ, Fantino E, Deininger MW: CYT387, a novel JAK2 inhibitor, induces hematologic responses and normalizes inflammatory cytokines in murine myeloproliferative neoplasms. Blood 2010,115(25):5232–5240. 10.1182/blood-2009-05-223727
Pardanani A, Lasho T, Smith G, Burns CJ, Fantino E, Tefferi A: CYT387, a selective JAK1/JAK2 inhibitor: in vitro assessment of kinase selectivity and preclinical studies using cell lines and primary cells from polycythemia vera patients. Leukemia: official journal of the Leukemia Society of America, Leukemia Research Fund, UK 2009,23(8):1441–1445. 10.1038/leu.2009.50
Pardanani A, Gotlib J, Gupta V, Roberts AW, Wadleigh M, Sirhan S, Litzow MR, Hogan WJ, Begna K, Smith G, et al.: An Expanded Multicenter Phase I/II Study of CYT387, a JAK- 1/2 Inhibitor for the Treatment of Myelofibrosis. ASH Annual Meeting Abstracts 2011,118(21):3849.
Pardanani AD, Caramazza D, George G, Lasho TL, Hogan WJ, Litzow MR, Begna K, Hanson CA, McClure RF, Bavisotto LM, et al.: Safety and efficacy of CYT387, a JAK-1/2 inhibitor, for the treatment of myelofibrosis. ASCO Meeting Abstracts 2011,29(15_suppl):6514.
Hart S, Goh KC, Novotny-Diermayr V, Tan YC, Madan B, Amalini C, Ong LC, Kheng B, Cheong A, Zhou J, et al.: Pacritinib (SB1518), a JAK2/FLT3 inhibitor for the treatment of acute myeloid leukemia. Blood cancer journal 2011,1(11):e44. 10.1038/bcj.2011.43
Poulsen A, William A, Blanchard S, Lee A, Nagaraj H, Wang H, Teo E, Tan E, Goh KC, Dymock B: Structure-based design of oxygen-linked macrocyclic kinase inhibitors: discovery of SB1518 and SB1578, potent inhibitors of Janus kinase 2 (JAK2) and Fms-like tyrosine kinase-3 (FLT3). J Comput Aided Mol Des 2012,26(4):437–450. 10.1007/s10822-012-9572-z
Hart S, Goh KC, Novotny-Diermayr V, Hu CY, Hentze H, Tan YC, Madan B, Amalini C, Loh YK, Ong LC, et al.: SB1518, a novel macrocyclic pyrimidine-based JAK2 inhibitor for the treatment of myeloid and lymphoid malignancies. Leukemia: official journal of the Leukemia Society of America, Leukemia Research Fund, UK 2011,25(11):1751–1759. 10.1038/leu.2011.148
William AD, Lee AC, Blanchard S, Poulsen A, Teo EL, Nagaraj H, Tan E, Chen D, Williams M, Sun ET, et al.: Discovery of the macrocycle 11-(2-pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6). 1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene (SB1518), a potent Janus kinase 2/fms-like tyrosine kinase-3 (JAK2/FLT3) inhibitor for the treatment of myelofibrosis and lymphoma. J Med Chem 2011,54(13):4638–4658. 10.1021/jm200326p
Komrokji RS, Wadleigh M, Seymour JF, Roberts AW, To LB, Zhu HJ, Mesa RA: Results of a phase 2 study of pacritinib (SB1518), a novel oral JAK2 inhibitor, in patients with primary, post-polycythemia vera, and post-essential thrombocythemia myelofibrosis. ASH Annual Meeting Abstracts 2011,118(21):282.
Younes A, Fanale MA, McLaughlin P, Copeland A, Zhu J, de Castro Faria S: Phase I Study of a Novel Oral JAK-2 Inhibitor SB1518 In Patients with Relapsed Lymphoma: Evidence of Clinical and Biologic Activity In Multiple Lymphoma Subtypes. ASH Annual Meeting Abstracts 2010,116(21):2830.
Verstovsek S, Mesa RA, Rhoades SK, Giles JLK, Pitou C, Jones E, Walgren RA, Prchal JT: Phase I Study of the JAK2 V617F Inhibitor, LY2784544, in Patients with Myelofibrosis (MF), Polycythemia Vera (PV), and Essential Thrombocythemia (ET). ASH Annual Meeting Abstracts 2011,118(21):2814.
Shah NP, Olszynski P, Sokol L, Verstovsek S, Hoffman R, List AF, Cortes J, Kantarjian HM, Gilliland DG, Clary DO, et al.: A Phase I Study of XL019, a Selective JAK2 Inhibitor, in Patients with Primary Myelofibrosis, Post-Polycythemia Vera, or Post-Essential Thrombocythemia Myelofibrosis. ASH Annual Meeting Abstracts 2008,112(11):98.
Paquette R, Sokol L, Shah NP, Silver RT, List AF, Clary DO, Bui LA, Talpaz M: A Phase I Study of XL019, a Selective JAK2 Inhibitor, in Patients with Polycythemia Vera. ASH Annual Meeting Abstracts 2008,112(11):2810.
Tefferi A: JAK inhibitors for myeloproliferative neoplasms: clarifying facts from myths. Blood 2012,119(12):2721–2730. 10.1182/blood-2011-11-395228
Nakaya Y, Shide K, Niwa T, Homan J, Sugahara S, Horio T, Kuramoto K, Kotera T, Shibayama H, Hori K, et al.: Efficacy of NS-018, a potent and selective JAK2/Src inhibitor, in primary cells and mouse models of myeloproliferative neoplasms. Blood cancer journal 2011,1(7):e29. 10.1038/bcj.2011.29
Purandare AV, McDevitt TM, Wan H, You D, Penhallow B, Han X, Vuppugalla R, Zhang Y, Ruepp SU, Trainor GL, et al.: Characterization of BMS-911543, a functionally selective small-molecule inhibitor of JAK2. Leukemia: official journal of the Leukemia Society of America, Leukemia Research Fund, UK 2012,26(2):280–288. 10.1038/leu.2011.292
Scuto A, Krejci P, Popplewell L, Wu J, Wang Y, Kujawski M, Kowolik C, Xin H, Chen L, Wang Y, et al.: The novel JAK inhibitor AZD1480 blocks STAT3 and FGFR3 signaling, resulting in suppression of human myeloma cell growth and survival. Leukemia: official journal of the Leukemia Society of America, Leukemia Research Fund, UK 2011,25(3):538–550. 10.1038/leu.2010.289
Derenzini E, Lemoine M, Buglio D, Katayama H, Ji Y, Davis RE, Sen S, Younes A: The JAK inhibitor AZD1480 regulates proliferation and immunity in Hodgkin lymphoma. Blood cancer journal 2011,1(12):e46. 10.1038/bcj.2011.46
Loveless ME, Lawson D, Collins M, Nadella MV, Reimer C, Huszar D, Halliday J, Waterton JC, Gore JC, Yankeelov TE: Comparisons of the efficacy of a Jak1/2 inhibitor (AZD1480) with a VEGF signaling inhibitor (cediranib) and sham treatments in mouse tumors using DCE-MRI, DW-MRI, and histology. Neoplasia 2012,14(1):54–64.
McFarland BC, Ma JY, Langford CP, Gillespie GY, Yu H, Zheng Y, Nozell SE, Huszar D, Benveniste EN: Therapeutic potential of AZD1480 for the treatment of human glioblastoma. Mol Cancer Ther 2011,10(12):2384–2393. 10.1158/1535-7163.MCT-11-0480
Deshpande A, Reddy MM, Schade GO, Ray A, Chowdary TK, Griffin JD, Sattler M: Kinase domain mutations confer resistance to novel inhibitors targeting JAK2V617F in myeloproliferative neoplasms. Leukemia: official journal of the Leukemia Society of America, Leukemia Research Fund, UK 2012,26(4):708–715. 10.1038/leu.2011.255
Xin H, Herrmann A, Reckamp K, Zhang W, Pal S, Hedvat M, Zhang C, Liang W, Scuto A, Weng S, et al.: Antiangiogenic and antimetastatic activity of JAK inhibitor AZD1480. Cancer Res 2011,71(21):6601–6610. 10.1158/0008-5472.CAN-11-1217
Mankan AK, Greten FR: Inhibiting signal transducer and activator of transcription 3: rationality and rationale design of inhibitors. Expert Opin Investig Drugs 2011,20(9):1263–1275. 10.1517/13543784.2011.601739
Ioannidis S, Lamb ML, Wang T, Almeida L, Block MH, Davies AM, Peng B, Su M, Zhang HJ, Hoffmann E, et al.: Discovery of 5-chloro-N2-[(1S)-1-(5-fluoropyrimidin-2-yl)ethyl]-N4-(5-methyl-1H-pyrazol-3-yl)p yrimidine-2,4-diamine (AZD1480) as a novel inhibitor of the Jak/Stat pathway. J Med Chem 2011,54(1):262–276. 10.1021/jm1011319
Hedvat M, Huszar D, Herrmann A, Gozgit JM, Schroeder A, Sheehy A, Buettner R, Proia D, Kowolik CM, Xin H, et al.: The JAK2 inhibitor AZD1480 potently blocks Stat3 signaling and oncogenesis in solid tumors. Cancer Cell 2009,16(6):487–497. 10.1016/j.ccr.2009.10.015
Coppelli FM, Grandis JR: Oligonucleotides as anticancer agents: from the benchside to the clinic and beyond. Curr Pharm Des 2005,11(22):2825–2840. 10.2174/1381612054546752
Jing N, Sha W, Li Y, Xiong W, Tweardy DJ: Rational drug design of G-quartet DNA as anti-cancer agents. Curr Pharm Des 2005,11(22):2841–2854. 10.2174/1381612054546761
Nelson EA, Sharma SV, Settleman J, Frank DA: A chemical biology approach to developing STAT inhibitors: molecular strategies for accelerating clinical translation. Oncotarget 2011,2(6):518–524.
Perrotti D, Neviani P: Protein phosphatase 2A (PP2A), a drugable tumor suppressor in Ph1(+) leukemias. Cancer Metastasis Rev 2008,27(2):159–168. 10.1007/s10555-008-9119-x
Redell MS, Ruiz MJ, Alonzo TA, Gerbing RB, Tweardy DJ: Stat3 signaling in acute myeloid leukemia: ligand-dependent and -independent activation and induction of apoptosis by a novel small-molecule Stat3 inhibitor. Blood 2011,117(21):5701–5709. 10.1182/blood-2010-04-280123
Santo L, Hideshima T, Cirstea D, Bandi M, Nelson EA, Gorgun G, Rodig S, Vallet S, Pozzi S, Patel K, et al.: Antimyeloma activity of a multitargeted kinase inhibitor, AT9283, via potent Aurora kinase and STAT3 inhibition either alone or in combination with lenalidomide. Clinical cancer research: an official journal of the American Association for Cancer Research 2011,17(10):3259–3271. 10.1158/1078-0432.CCR-10-3012
Turkson J, Kim JS, Zhang S, Yuan J, Huang M, Glenn M, Haura E, Sebti S, Hamilton AD, Jove R: Novel peptidomimetic inhibitors of signal transducer and activator of transcription 3 dimerization and biological activity. Mol Cancer Ther 2004,3(3):261–269.
Hayakawa F, Sugimoto K, Kurahashi S, Sumida T, Naoe T: A Novel STAT3 Inhibitor OPB-31121 Induces Tumor-Specific Growth Inhibition in a Wide Range of Hematopoietic Malignancies without Growth Suppression of Normal Hematopoietic Cells. ASH Annual Meeting Abstracts 2011,118(21):577.
Acknowledgment
This study was partly supported by New York Medical College Blood Diseases Fund.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interest
The authors have no relevant conflicts of interest.
Authors’ contributions
All authors have contributed to data preparation, drafting and revising the manuscripts. All authors have read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Furqan, M., Mukhi, N., Lee, B. et al. Dysregulation of JAK-STAT pathway in hematological malignancies and JAK inhibitors for clinical application. Biomark Res 1, 5 (2013). https://doi.org/10.1186/2050-7771-1-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2050-7771-1-5