
Abstract
Presenilins (PSs) are the catalytic core of γ-secretase complex. However, the mechanism of FAD-associated PS mutations in AD pathogenesis still remains elusive. Here we review the general biology and mechanism of γ-secretase and focus on the catalytic components – presenilins and their biological functions and contributions to the AD pathogenesis. The functions of presenilins are divided into γ-secretase dependent and γ-secretase independent ones. The γ-secretase dependent functions of presenilins are exemplified by the sequential cleavages in the processing of APP and Notch; the γ-secretase independent functions of presenilins include stabilizing β-catenin in Wnt signaling pathway, regulating calcium homeostasis and their interaction with synaptic transmission.
Similar content being viewed by others


Introduction
Alzheimer’s disease is the most common neurodegenerative disorder leading to dementia, accounting for two thirds of dementia in elderly populations. The majority of AD cases are late-onset and sporadic without defined causes, whereas less than 5% of cases are familial with early-onset and caused by gene mutations. Genetic studies have shown that four genes confer susceptibility to AD: amyloid-β precursor protein (APP) on chromosome 21 [1–6], Presenilin 1 (PS1) on chromosome 14 [7–10], Presenilin 2 (PS2) on chromosome 1 [11–13] and apolipoprotein E (ApoE) on chromosome 19 [14, 15]. Neuritic plaques, neurofibrillary tangles (NTFs) and neuronal loss are pathological hallmarks of AD. However, the mechanism underlying AD pathogenesis remains elusive and there is no effective prevention or treatment to this devastating disorder so far.
Neuritic plaques are formed by extracellular deposits of amyloid β protein (Aβ) [16]. Aβ is derived from proteolytic processing of APP and consists primarily of 40- and 42-amino acid residues, with the more hydrophobic Aβ42 as the major component in neuritic plaques [16, 17]. NFTs are intraneuronal inclusions composed of hyperphosphorylated forms of the microtubule-associated protein Tau [18–21]. Aβ-containing neuritic plaques are the unique pathological feature in AD brains whereas NTFs could also be detected in other dementia subtypes like frontotemporal dementia with Parkinsonism caused by mutations on MAPT gene [22]. Current prevailing “amyloid hypothesis” in AD suggests that the accumulation of Aβ, particularly the more hydrophobic and aggregation-prone Aβ42, being soluble oligomers [23–29] or aggregate fibril form, initiates neuronal dysfunction, resulting in neurodegeneration in AD [30].
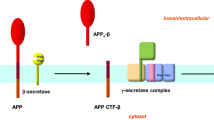
The central event of “amyloid hypothesis” is APP processing. APP undergoes posttranslational proteolytic processing by α, β and γ-secretases (Figure 1). The majority of APP is constitutively processed by α-secretase within the Aβ domain in a non-amyloidogenic pathway [31]. In the amyloidogenic pathway, APP undergoes sequential cleavages by β- and γ-secretase to generate Aβ. A transmembrane aspartic protease BACE1 was identified as β-secretase [32–35]. BACE1 processes APP at the Asp1 site of Aβ domain to generate APP C99 fragment [34, 36]. The C99 fragment is further processed by γ-secretase at the intramembrane Val40 and Ala42 sites to generate Aβ40 and Aβ42, respectively. The second cleavage, which takes place within the hydrophobic transmembrane domain (TMD) and is termed as regulated intramembrane proteolysis (RIP) [37], has been attributed to γ-secretase with presenilins as the catalytic component [38–45].

APP processing pathways. Under the physiological conditions, the majority of APP undergoes the non-amyloidogenic pathway. The α-secretase processes APP within the Aβ domain to generate C83 and this cleavage abolishes Aβ generation. In the amyloidogenic pathway, β-secretase processes APP at Asp1 site to generate C99 fragment, which is the substrate for γ-secretase for Aβ generation.
As the catalytical component of γ-secretase, the first part of this review will focus on the contribution of presenilins to γ-secretase and its role in AD pathogenesis in the scenario of “Amyloid hypothesis”. The rest of this review will discuss diverse biological functions of presenilins independent of γ-secretase activity. Its well-established role in β-catenin/Wnt-signaling and calcium homeostasis as well as the contribution to the AD pathogenesis will be addressed.
Presenilins and γ-secretase
Presenilins
Presenilins have two homologs, PS1 and PS2, with 67% identical sequence [11]. mRNAs of both Presenilins are ubiquitously detected in many human and mouse tissues, including brain, heart, kidney and muscle [46]. PS1 and PS2 are highly conserved and functionally redundant with SEL-12 as their homolog in Caenorhabditis elegans[47].
PS1 is a multi-transmembrane protein with nine-transmembrane topology (Figure 2) [48, 49], and abundantly present in the ER and trans-Golgi network [50–53]. Under physiological condition, the majority of PS1 undergoes endoproteolysis within the large hydrophobic loop in the cytoplasmic side to generate N-terminal fragment (NTF) and C-terminal fragment (CTF) [54]. The endoproteolytic cleavage takes place at heterogeneous sites from amino acid 292 to 299 [55–57]. While some studies reported an independent protease as the “presenilinase” [58, 59], growing evidence supported the hypothesis that PS undergoes autoendoproteolysis [43, 60–64]. The endoproteolysis event might be important to render the γ-secretase activity to PS NTF/CTF heterodimer by removing the auto-inhibitory effect of the large hydrophobic loop [64, 65]. However, it is not clear whether endoproteolysis is an absolute requirement for the maturation of presenilins since some presenilin mutants are enzymatically active in the absence of endoproteolysis, as are the cases in FAD-associated PS1ΔE9 and PS2 M292D [57, 66].

Presenilin 1 structure. This diagram shows the amino acid sequence of PS1 and the distribution of the FAD-associated mutations. Blue circles represent the FAD-associated mutations and red circles indicate the two catalytic active aspartates.
γ-Secretase complex assembling
γ-secretase is essential for cleavage of APP C99 to generate Aβ [67]. γ-secretase is a multi-unit enzymatic complex, including presenilin NTF/CTF heterodimer, nicastrin, Aph-1 and Pen-2 [39, 41–45]. Presenilins are the first molecules identified to be associated with γ-secretase in vivo and in vitro. PS1 knockout mice showed markedly reduced γ-secretase cleavage of APP [38] and knockout of both PS1 and PS2 completely abolished γ-secretase activity [40, 68]. Using anti-PS antibody, Yu et al. identified Nicastrin, an integral transmembrane protein with a large N-terminal domain, as the second γ-secretase component [39]. However, expression of both presenilins and nicastrin don’t suffice to restore γ-secretase activity, indicating the existence of other components. Further gene screening studies on the glp-1 (Notch homolog) deficient phenotype of C.elegans discovered Aph-1 and Pen-2 as another two components of γ-secretase [41, 42]. Aph-1 is a 30 kDa multi-transmembrane protein like presenilin, whereas Pen-2 is a 12 kDa hairpin-like transmembrane protein. Co-expression of presenilin, Aph-1, Pen-2 and nicastrin increases γ-secretase activity in transfected cells and the four proteins together are sufficient to reconstitute γ-secretase activity in yeast [44, 69].
Previous studies demonstrated that the minimal molecular weight of γ-secretase complex was around 200–250 kDa, implying a 1:1:1:1 stoichiometry of PS/Nicastrin/Aph-1/Pen-2 in γ-secretase complex [44]. Though it was accepted that the four molecules were the minimal γ-secretase complex assembling, recent report suggested that the PS/Pen-2/Aph-1 complex was sufficient for the catalytic activity in the absence of Nicastrin [70]. Another study demonstrated that PS1ΔE9 alone had partial γ-secretase activity and PS1ΔE9/Pen-2 was sufficient to restore full γ-secretase activity [71]. These studies suggest the complexity of γ-secretase complex assembling. Given the stoichiometry of γ-secretase complex and the existence of PS and Aph-1 homologs, there are at least six different forms of γ-secretase complex that could be assembled [72, 73]. PS1-involved complex or PS2-involved complex processed APP C99 differentially and showed distinct susceptibility to certain γ-secretase inhibitors [74, 75], indicating different γ-secretase complexes with possible distinct functions.
Structure of γ-Secretase complex
The catalytic core of γ-secretase complex is presenilins. Presenilins, together with signal peptides peptidases (SPPs), belong to aspartyl intramembrane cleaving proteases (I-CLiPs) [76]. The two catalytic aspartate residues (Asp257 in transmemberane 6 (TM6) and Asp385 in TM7) are located at NTF and CTF of presenilins, respectively. Mutations on either aspartate abolish the enzymatic activity of γ-secretase complex [60]. With a large highly glycosylated ectodomain, nicastrin has been implicated to function as the initial recognition of substrates [77]. Electronic microscopic analysis and single particle imaging revealed the existence of intramembrane water-accessible cylindrical chamber in gamma-secretase with a low-density cavity from extracellular side [78, 79]. Parallel substituted cysteine accessible method (SCAM) and cross-link experiment confirmed that TM6, TM7 and TM9 of PS formed the intramembrane chamber with two catalytic aspartates residing oppositely on TM6 and TM7, respectively [80–84]. The constitutive autoendoproteolysis of PS removes the inhibitory allosteric effect of the large hydrophobic loop from the catalytic chamber structure in PS [64, 65]. With direct interaction between γ-secretase components [85, 86], Nct/Aph-1 subunits and Pen-2 tighten the relative loose PS TM6/TM7/TM9 intramembrane cavity and rearrange the PALP motif of TM9 to the proximity of the catalytic center, thus activate the γ-secretase complex [87] (Figure 3). Recently, Li et al. reported the crystal structure of a presenilin/SPP homologue (PSH) from the archaeon Methanoculleus marisnigri JR1 and predicted the structure of presenilin based on the conserved sequence between the two homologues [88]. They confirmed the existence of the water permissible cavity but also revealed some differences in TM7 and TM9 compared with the NMR structure of PS1 CTF. The work shed new light on elucidation of the crystal structure of presenilin.

γ-secretase complex and the formation of the catalytic pore of PS1. PS1 transmembrane domains (TMDs) are shown as columns with numbers. Without the assistance of other subunits, PS1 forms a relatively open pore structure within the membrane. Upon the binding of subunits, the PALP motif moves to the proximity of the catalytic center, and the catalytic structure is activated by the structural changes in TMDs of PS1.
γ-Secretase substrates and sequential cleavages
APP and Notch as classical substrates
γ-Secretase preferentially processes type I integral membrane protein after the ectodomain is shedded [89]. It is intriguing that many of its classical substrates function in the signaling pathways, cell adhesion and migration, neuritis outgrowth and synapse formation, and many of these events are often disrupted during AD pathogenesis [90]. The number of substrates is growing to over 80, including APP, Notch, neuregulin, ErbB4, E-cadherins and N-cadherins, CD44 and growth hormone receptor [40, 91–98].
APP and Notch are two most well known γ-secretase substrates. γ-Secretase is named after it function as the enzyme to process APP at the γ-cleavage site to generate Aβ, which is currently believed to play an essential role in the “Amyloid cascades” in AD pathogenesis. Notch is a type I transmembrane cell surface receptor that mediates cell fate decisions in both vertebrates and invertebrates [99, 100]. After it is cleaved by furin, cell surface Notch receptor heterodimers bind to the DSL (Delta/Serrate/LAG-2) family ligands on the surface of neighboring cells, then the transmembrane-intracellular fragment of Notch undergoes further proteolysis to release Notch intracellular domain (NICD) from the membrane to the nucleus to activate target genes [101, 102]. Presenilins have been shown to play an essential role in Notch signaling. PS-deficient mice exhibit Notch-knockout phenotype [103, 104]. Knockout of PS abolishes intramembrane γ-secretase cleavage of Notch as well as the following release of NICD [40, 91, 105], and FAD-associated PS mutations impair the generation of NICD [91]. Although it is reported that the impaired Notch-signaling is involved in synaptic plasticity and late-onset cognitive decline [106–110], the contribution of Notch- signaling to AD pathogenesis remain to be elucidated.
γ-secretase cleavages at ϵ-site and γ-site
γ-Secretase can process substrates at multiple cleavage sites. γ-secretase cleaves the transmembrane domain of APP at two positions: the γ-site to generate Aβ and the downstream ϵ-site to produce the APP intracellular domain (AICD) [111]. Cleavage at the γ-site is heterogeneous, producing Aβ of 39–43 residues, whereas the cutting at the ϵ-site produces AICD of 50 residues almost exclusively. The same phenomenon occurs in Notch processing: heterogeneous cleavages at the S4 site (γ-site) to generate Nβ and homogeneous cleavage at the S3 site (ϵ-site) to generate NICD [112]. Recent independent studies supported the notion that the ϵ-cleavage occurs prior to γ-cleavage [113–115]. Qi-Takahara and colleagues first detected Aβ49, the proteolytic counterpart to AICD50-99[113]. Later Ihara and colleagues demonstrated that ϵ-cleavage occurs first and produces Aβ48 and Aβ49 for later γ-cleavage, based on the presence of the induction period for the generation for tripeptides/tetrapeptides detected by liquid chromatography tandem mass spectrometry (LC-MS/MS) in cell-free γ-scretase system [116]. The various Aβ species (ranging from 49- to 40-amino acids) and corresponding tripeptides released from the trimming of Aβ48/49 were identified using LC-MS/MS, further confirming the sequential γ-cleavage from the ϵ-site to γ-site [113, 116, 117].
The effect of FAD-associated presenilins mutations on γ-cleavages
Presenilin mutations are the main cause reasons of early-onset FAD. Presenilin mutations result in the production of the more hydrophobic Aβ42 either in conditioned medium in vitro assay [67, 118] or in APP/PS1 transgenic mice [119]. It still remains elusive how PS1 mutations affect the enzymatic activity on ϵ- and/or γ-site to initiate the AD pathogenesis. Considering the significant role of Notch in neurogenesis and impaired Notch-signaling in the scenario of presenilins mutations, the contribution of Notch signaling has always been a debating topic in AD field.
γ-Secretase processes its substrates at γ- and ϵ-site, generating distinct products exemplified as Aβ and NICD. Aβ plays central role in AD pathogenesis; whereas NICD is the nuclear transcription factor activator involving in evolutionarily conserved pathway, mediating short-range intercellular communication and cell-fate determination in development as well as in adulthood. Presenilins mutations affect the production of Aβ as well as the generation of NICD, indicating that presenilins mutations influence both γ- and ϵ-cleavages. However, recent studies indicate that γ and ϵ cleavages are distinct enzymatic events with their own enzymatic kinetics and pharmaceutical characterization, and they can be differentially affected by the FAD-associated PS mutations. Fukumori and colleagues reported that the inhibition of endocytosis of PS1 altered AICD formation without changing Aβ42/Aβ40, implying that the efficiency of γ and ϵ cleavage of γ-secretase are different event in plasma membrane and endosome, respectively [120]. Parallel study on TMP21 directly pointed out that TMP21 acted as γ-secretase modulator but affected γ cleavage only [121]. The artificial PSΔE10 impaired the normal Aβ generation but spared the intracellular domain production in both APP and Notch, supporting the possibility that γ- and ϵ-cleavages are dissociated [122]. Using in vitro enzyme kinetics assays, De Strooper and his team demonstrated that PS mutations consistently exhibited impaired γ-cleavage activity with altered Aβ42/40 ratio, but the effect on the ICD-producing ϵ-cleavage was varied and substrate-specific. For instance, PSM139V displayed enhanced ϵ-cleavage in N-cadherin but unchanged in APP, Notch and Erb4 [123]. All those studies indicated that the inefficient processing on ϵ-cleavage and the impaired Notch-signaling are not essential for AD pathogenesis.
Presenilins beyond γ-secretase
Recently, mounting evidence has supported that presenilins carry out multiple functions beyond the catalytic functions of γ-secretase. The conditional knock-out of presenilins in excitatory neurons demonstrated age-dependent neurodegeneration, indicating an essential role of presenilins in neurodegeneration independent of amyloid cascade [124, 125]. However, given the fundamental function of Notch, it is hard to exclude that the phenotypes in conditional PS knock-out mice is due to the impaired Notch signaling. In the moss Physcomitrella patens (P. patens), which lacks Notch signaling, presenilin-deficit phenotype could be rescued by wild type presenilins as well as PS mutants without γ-secretase activity, indicating other functions of PS beyond γ-secretase activity [126]. Moreover, presenilins-knock-out mice exhibited more severe somite phenotype than mice lacking canonical Notch-signaling and mice deficient of Nicastrin, Aph-1 or Pen-2, which could still develop anterior somite [127]. In summary, all these clues point to the existence of independent functions of presenilins beyond γ-secretase.
FAD-associated presenilins mutations exhibit not only significant heterogeneity on clinical features like age of onset, neurological and psychiatric symptoms, but also on neuropathology including greater NTF formation, altered neuritic plaque composition, presence of Pick body, and neuropathological lesion in basal ganglia and brainstem [128–130]. It is also reported that presenilins are involved in Wnt signaling, cell adhesion, calcium homeostasis, protein degradation and apoptosis, raising the possibility that γ-secretase-independent function of presenilins might contribute to the presenilins mutations-associated heterogeneity. The subsequent sections of this review will focus on these issues (Figure 4).

Presenilin functions. This diagram divides the functions of presenilins into γ-secretase-dependent and γ-secretase-independent ones by the vertical dash line. The γ-secretase activity of presenilins are exemplified by APP processing and Notch processing. Other γ-secretase independent functions of presenilins include stabilizing β-catenin in Wnt signaling pathway, regulating calcium homeostasis and its interaction with synaptic transmission. APP, amyloid β precursor protein; AICD, APP intracellular domain; NICD, Notch intracellular domain; Nct, Nicastrin; IP3, Inositol trisphosphate receptor; SERCA, Sarco/endoplasmic reticulum Ca2+−ATPase; Ryan R, Ryanodine receptor; GSK, Glycogen synthase kinase.
PS1 and β-catenin
β-Catenin in Wnt-signaling and cell-cell adhesion
β-Catenin is a signal transducer protein in Wnt-signaling pathway as well as a cell adhesion molecule [131]. β-Catenin carries out two distinct functions according to its cellular location: the membrane β-catenin forms complex with E-cadherin as cell-cell adhesion molecule; whereas the cytoplasmic β-catenin is involved in Wnt-signaling pathway to regulate gene expression. In the absence of Wnt ligand, β-Catenin undergoes phosphorylation by Glycogen Synthase Kinase-3β (GSK3β) with the assistance of Axin/APC complex, and then the phosphorylated β-catenin is constitutively degraded in ubiquitin proteasome pathway. Binding of Wnt to its receptor Frizzled and co-receptor LRP5/6 blocks phosphorylation of β-catenin by GSK3β, precluding the degradation of β-catenin. β-catenin is translocated into nuclear to activate transcriptional factor like T-cell-specific transcriptional factor 1 (TCF), to regulate target genes like cyclin D1, c-myc, and metalloproteases. On the other hand, membrane β-catenin acts as a bridge to link cadherins to α-catenin, and the latter binds to actin network, to stabilize adherence junctions as well as the cytoskeleton [131].
PS1/β-catenin interaction and AD
PS1 negatively regulates β-catenin level via physically interacting with β-catenin through the cytosolic-loop structure of PS1 [132–136]. This function of PS1 is γ-secretase-independent since the D257A mutant could rescue the turnover of β-catenin as wild type PS1 does [137]. Though evidence supports the idea that PS1 works as a scaffold to facilitate β-catenin phosphorylation, the underlying mechanism remains to be elucidated [136–138].
It has been reported that the stabilization of β-catenin contributes to the development of skin cancer in PS1-deficient mice [139]. However, the precise role of β-catenin in AD pathogenesis is not clear. Most studies on sporadic AD indicated that reduced Wnt/β-catenin signaling might make a contribution to AD pathogenesis [140]. However, the studies on FAD were conflicting, some PS mutations were found to stabilize β-catenin while others destabilized it [132, 136, 137, 141–143].
Presenilins and calcium regulation
Interaction of presenilins and calcium channels
Presenilins mutations have been reported to connect with abnormal intracellular calcium signaling, and presenilins mutations promote the release of Ca2+ from overloaded ER stores through IP3 receptor [144]. Presenilins could interact with IP3 receptors to regulate IP3 channel activity. Using patch-clamp techniques, PS1M146L and PS2N141I prolonged IP3 channel opening and increased Ca2+ leak permeability [145]. Studies on primary neurons from PS1M146V-expressing mice revealed that mutant PS1 increased the expression and recruitment of ryanodine receptor (RyanR) to regulate the IP3R Ca2+ signaling in primary neurons [146–148]. Apart from IP3 receptor and RyanR, prensenilins were also reported to interact with sarco-/endoplasmic reticulum Ca2+ ATPase (SERCA) to regulate intracellular calcium signaling [149]. These studies suggested that presenilins and their mutants regulate intracellular Ca2+ signaling via interacting with various calcium channel-related proteins.
Presenilins themselves as Ca2+ leak channels
Presenilins themselves were reported to function as low-conductance, passive ER Ca2+ leak channels, which is independent of γ-secretase activity [150]. Many FAD mutants (e.g. PS1M146V and PS2N141I) disrupt or abolish the Ca2+ leak channel activity, leading to overload of Ca2+ in ER [151]. It has been reported that presenilin transmembrane domain 7 and 9 contribute to the forming of the ion conductance pore, and transmembrane water-filled catalytic cavity of presenilin constitutes the Ca2+ leak channel [152]. However, Ca2+ channel function of presenilins has been challenged by another group, showing that FAD PS mutants regulate calcium level by regulating IP3R channel gating [145, 153]. Further studies on the crystal structure of presenilin would be helpful to elucidate whether presenilins themselves act as Ca2+ channels.
Dysregulation of autophagy in AD and PS-associated calcium abnormality
The accumulation of autophagic vacuoles (AVs) has been observed in dystrophic neurites around the amyloid plaques for decades [154–156]. Autophagy serves as cellular processing for dysfunctional cellular organelles and toxic protein degradation, important for cell survival under stress like nutrient deprivation. Autophagy mainly involves two steps: generation of autophagosome containing dysfunctional cellular organelles and degradation of the contents via fusing with lysosome or late endosome [157, 158].
It has been well established that PS deficiency impairs the turnover of long-lived proteins like telencephalin(TLN) and α-synuclein [159, 160], which results from the lysosome fusion failure and autophagy deficit. In PS1−/− hippocampal neurons, TLN is accumulated in intracellular membrane organelles containing Apg12p and LC3, the autophagic vacuole markers, in both ultrastructure and immunostaining experiments [159, 161]. The accumulation of TLN could be rescued by PS1WT, FAD-associated PS1 mutants or dominant-negative PS1 mutant (PS1D257A), indicating that the PS-associated autophagic vacuoles accumulation was independent of γ-secretase activity. The accumulation of autophagic vacuoles may be caused either by increased production of autophagic vacuoles, resulting from accelerated autophagy activity; or by reduced consumption, resulting from dysfunctional fusion with the lysosome/endosome. Given that the formation of TLN-positive autophagosome triggered by microbeads was normal, the authors proposed that observed accumulation of TLN-positive autophagic vacuoles was correlated with failed lysosome fusion, which was in accordance with mounting evidence supporting lysosome deficit as the underlying cause of the autophagy deficit in AD [160, 162].
Other hypotheses explaining PS1-related autophagic vacuole accumulation involves the lysosome acidification deficiency. Lee et al. reported that PS1 acted as chaperone protein to facilitate the glycosylation of V-ATPase subunit V0a1, which helped V-ATPase traffic to lysosome and completed lysosome acidification [163]. The failed acidification of lysosome in PS1−/− blastocyst-derived cell line (BD15) repressed the fusion of lysosome with intermediate AVs, resulting in accumulation of AVs. However, later studies argued that lysosome acidification appeared to be unimpaired in PS1−/−/PS2−/− stem cells and the glycosylation of V0a1 subunit was unaffected. Nevertheless, Coen et al. demonstrated that the calcium loading of lysosome in PS1−/− or PS1−/−/PS2−/− cells was significantly less than wild type cells, which could be rescued by PS1 mutant without γ-secretase activity, indicating the γ-secretase-independent property. Given that PS1 itself could act as ER Ca2+ leak channels, they proposed that the accumulation of autophagic vacuoles often observed in AD could be interpreted by impaired PS1-related calcium abnormality [164–166].
It is well known that PS deficiency is related with AV abnormality [160, 165], however, the relationship between autophagy and FAD-associated PS1 mutations is not well defined. For example, Esselens et al. reported that FAD-associated PS1 mutations rescued PS1 deficiency-related TLN-positive autophagy deficit; whereas Lee et al. reported autophagy deficit in PS FAD mutations human fibroblast. Thus, it needs further investigation to clarify the contribution of FAD-associated PS1 mutations to autophagy deficit of AD.
Correlation of Cotton wool plaques (CWP) and abnormal Calcium signaling
CWP are large, non-cored and diffuse amyloid plaques, which are composed primarily of Aβ42 without surrounding neuritic dystrophy and glial activation in Alzheimer cases [167]. CWP is often associated with spastic paraparesis (SP) [168], both of which were reported in a subset of PS1 mutants like PS1M233T, PS1R278T and PS1ΔE9[169, 170]. The mechanism underlying these unique clinical and pathological phenotypes is unknown. It is well established that Ca2+ release from intracellular stores is increased in both sporadic and familial AD [171–173], and thus it is proposed that the disturbed Ca2+ regulation in FAD is correlated with CWP [174, 175]. Over 20 PS1 mutations have been analyzed and though all PS1 mutations show increased Aβ42/40 ratio, their effects on calcium signaling are various. It’s very illuminating to correlate calcium dysfunction with FAD variant phenotypes, but the underlying mechanism needs further investigation.
Presenilins and synaptic transmission
Another pathological aspect of Alzheimer’s Disease is the failure of synaptic transmission and further disturbance in the neural circuit. Many believe that independent of plaque formation, impairment of synaptic function is what accounts for AD pathogenesis [176]. It has been reported that Aβ plays an important role in maintaining efficient synaptic transmission and stabilizing the neural circuit [30, 177]. Recently presenilins stand out to be a candidate participating in the release of neurotransmitter and synaptic scaling independent of their γ-secretase function. It was reported that presenilins are essential for regulating neurotransmitter release like glutamate [178]. Presynaptic knockout of presenilins leads to inhibition of theta burst-induced long-term potentiation. Moreover, the inhibition effect is probably mediated by depletion of endoplasmic reticulum Ca2+ storage and blockade of intracellular Ca2+ release [178]. PS1 was also proposed to regulate homeostatic synaptic scaling [179]. PS1 knockout and PSM146V neurons fail to scale up synaptic strengths in response to tetrodotoxin treatment, which can be rescued by viral expression of wild type PS1. Furthermore, γ-secretase inhibitor does not influence the effect of presenilins on synaptic scaling, suggesting that this function is independent of γ-secretase in AD pathogenesis. On the other hand, synaptic activity can in turn modulate the activity of PS1 such as regulating Aβ40/42 ratio via altering PS1 conformation, thereby forming bidirectional interaction [180]. Using a Cer-PS1-Cit FRET sensor, the group discovered that spike bursts trigger PS1 conformational change through vesicle exocytosis. More importantly, the conformational change of wild type PS1 upregulates Aβ40/42 ratio, which is uniformly decreased in almost all cases of FAD mutations. Overall, mounting evidence points to a role of presenilins in synaptic transmission. However, the underlying mechanism is still not clear. The comprehensive interaction between presenilins and synaptic activity could result from presenilin’s functions independent and/or dependent of γ-secretase activity.
Conclusion
γ-secretase sequentially processes its substrates at ϵ- and γ-sites and the enzymatic activities on two cleavages are distinct. As the catalytic component of the γ-secretase complex, FAD-associated presenilins affect γ-secretase activity on the γ-site but the effects on ϵ-cleavage vary. These studies suggest the possibility of development of γ-secretase modulators sparing the Notch signaling in the future. It has long been observed that presenilins are involved in functions independent of the γ-secretase activity, like interaction with β-catenin/Wnt signaling, calcium regulation and autophagy degradation. However, its contribution to AD pathogenesis is not clear. Further studies are needed to clearly define the function of presenilins and its role in AD pathogenesis.
Abbreviations
- AD:
-
Alzheimer’s disease
- PS:
-
Pensenilin
- APP:
-
amyloid β precursor proteins
- BACE1:
-
Beta-site APP cleaving enzyme 1
- Aβ:
-
Amyloid β protein.
References
Goldgaber D, Lerman MI, McBride OW, Saffiotti U, Gajdusek DC: Characterization and chromosomal localization of a cDNA encoding brain amyloid of Alzheimer’s disease. Science 1987, 235: 877-880. 10.1126/science.3810169
Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, Multhaup G, Beyreuther K, Muller-Hill B: The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature 1987, 325: 733-736. 10.1038/325733a0
Robakis NK, Ramakrishna N, Wolfe G, Wisniewski HM: Molecular cloning and characterization of a cDNA encoding the cerebrovascular and the neuritic plaque amyloid peptides. Proc Natl Acad Sci U S A 1987, 84: 4190-4194. 10.1073/pnas.84.12.4190
St George-Hyslop PH, Tanzi RE, Polinsky RJ, Haines JL, Nee L, Watkins PC, Myers RH, Feldman RG, Pollen D, Drachman D, et al.: The genetic defect causing familial Alzheimer’s disease maps on chromosome 21. Science 1987, 235: 885-890. 10.1126/science.2880399
Tanzi RE, Gusella JF, Watkins PC, Bruns GA, St George-Hyslop P, Van Keuren ML, Patterson D, Pagan S, Kurnit DM, Neve RL: Amyloid beta protein gene: cDNA, mRNA distribution, and genetic linkage near the Alzheimer locus. Science 1987, 235: 880-884. 10.1126/science.2949367
Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L, et al.: Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 1991, 349: 704-706. 10.1038/349704a0
Schellenberg GD, Bird TD, Wijsman EM, Orr HT, Anderson L, Nemens E, White JA, Bonnycastle L, Weber JL, Alonso ME, et al.: Genetic linkage evidence for a familial Alzheimer’s disease locus on chromosome 14. Science 1992, 258: 668-671. 10.1126/science.1411576
St George-Hyslop P, Haines J, Rogaev E, Mortilla M, Vaula G, Pericak-Vance M, Foncin JF, Montesi M, Bruni A, Sorbi S, et al.: Genetic evidence for a novel familial Alzheimer’s disease locus on chromosome 14. Nat Genet 1992, 2: 330-334. 10.1038/ng1292-330
Li J, Ma J, Potter H: Identification and expression analysis of a potential familial Alzheimer disease gene on chromosome 1 related to AD3. Proc Natl Acad Sci U S A 1995, 92: 12180-12184. 10.1073/pnas.92.26.12180
Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K, Tsuda T, Mar L, Foncin JF, Bruni AC, Montesi MP, Sorbi S, Rainero I, Pinessi L, Nee L, Chumakov I, Pollen D, Brookes A, Sanseau P, Polinsky RJ, Wasco W, Da Silva HA, Haines JL, Perkicak-Vance MA, Tanzi RE, Roses AD, Fraser PE, Rommens JM, St George-Hyslop PH: Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 1995, 375: 754-760. 10.1038/375754a0
Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, Yu CE, Jondro PD, Schmidt SD, Wang K, et al.: Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science 1995, 269: 973-977. 10.1126/science.7638622
Levy-Lahad E, Wijsman EM, Nemens E, Anderson L, Goddard KA, Weber JL, Bird TD, Schellenberg GD: A familial Alzheimer’s disease locus on chromosome 1. Science 1995, 269: 970-973. 10.1126/science.7638621
Rogaev EI, Sherrington R, Rogaeva EA, Levesque G, Ikeda M, Liang Y, Chi H, Lin C, Holman K, Tsuda T, et al.: Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature 1995, 376: 775-778. 10.1038/376775a0
Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA: Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261: 921-923. 10.1126/science.8346443
Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, Roses AD: Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci U S A 1993, 90: 1977-1981. 10.1073/pnas.90.5.1977
Glenner GG, Wong CW: Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun 1984, 120: 885-890. 10.1016/S0006-291X(84)80190-4
Iwatsubo T, Odaka A, Suzuki N, Mizusawa H, Nukina N, Ihara Y: Visualization of A beta 42(43) and A beta 40 in senile plaques with end-specific A beta monoclonals: evidence that an initially deposited species is A beta 42(43). Neuron 1994, 13: 45-53. 10.1016/0896-6273(94)90458-8
Grundke-Iqbal I, Iqbal K, Quinlan M, Tung YC, Zaidi MS, Wisniewski HM: Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J Biol Chem 1986, 261: 6084-6089.
Kosik KS, Joachim CL, Selkoe DJ: Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc Natl Acad Sci U S A 1986, 83: 4044-4048. 10.1073/pnas.83.11.4044
Goedert M, Wischik CM, Crowther RA, Walker JE, Klug A: Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: identification as the microtubule-associated protein tau. Proc Natl Acad Sci U S A 1988, 85: 4051-4055. 10.1073/pnas.85.11.4051
Iqbal K, Grundke-Iqbal I, Smith AJ, George L, Tung YC, Zaidi T: Identification and localization of a tau peptide to paired helical filaments of Alzheimer disease. Proc Natl Acad Sci U S A 1989, 86: 5646-5650. 10.1073/pnas.86.14.5646
Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, de Graaff E, Wauters E, van Baren J, Hillebrand M, Joosse M, Kwon JM, Nowotny P, Che LK, Norton J, Morris JC, Reed LA, Trojanowski J, Basun H, et al.: Association of missense and 5'-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 1998, 393: 702-705. 10.1038/31508
Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH: Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci 2005, 8: 79-84. 10.1038/nn1372
Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH: A specific amyloid-beta protein assembly in the brain impairs memory. Nature 2006, 440: 352-357. 10.1038/nature04533
Cheng IH, Scearce-Levie K, Legleiter J, Palop JJ, Gerstein H, Bien-Ly N, Puolivali J, Lesne S, Ashe KH, Muchowski PJ, Mucke L: Accelerating amyloid-beta fibrillization reduces oligomer levels and functional deficits in Alzheimer disease mouse models. J Biol Chem 2007, 282: 23818-23828. 10.1074/jbc.M701078200
Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL: Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci 2007, 27: 2866-2875. 10.1523/JNEUROSCI.4970-06.2007
Selkoe DJ: Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behav Brain Res 2008, 192: 106-113. 10.1016/j.bbr.2008.02.016
Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ: Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med 2008, 14: 837-842. 10.1038/nm1782
Tomiyama T, Matsuyama S, Iso H, Umeda T, Takuma H, Ohnishi K, Ishibashi K, Teraoka R, Sakama N, Yamashita T, Nishitsuji K, Ito K, Shimada H, Lambert MP, Klein WL, Mori H: A mouse model of amyloid beta oligomers: their contribution to synaptic alteration, abnormal tau phosphorylation, glial activation, and neuronal loss in vivo. J Neurosci 2010, 30: 4845-4856. 10.1523/JNEUROSCI.5825-09.2010
Hardy J, Selkoe DJ: The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 2002, 297: 353-356. 10.1126/science.1072994
Esch FS, Keim PS, Beattie EC, Blacher RW, Culwell AR, Oltersdorf T, McClure D, Ward PJ: Cleavage of amyloid beta peptide during constitutive processing of its precursor. Science 1990, 248: 1122-1124. 10.1126/science.2111583
Hussain I, Powell D, Howlett DR, Tew DG, Meek TD, Chapman C, Gloger IS, Murphy KE, Southan CD, Ryan DM, Smith TS, Simmons DL, Walsh FS, Dingwall C, Christie G: Identification of a novel aspartic protease (Asp 2) as beta-secretase. Mol Cell Neurosci 1999, 14: 419-427. 10.1006/mcne.1999.0811
Sinha S, Anderson JP, Barbour R, Basi GS, Caccavello R, Davis D, Doan M, Dovey HF, Frigon N, Hong J, Jacobson-Croak K, Jewett N, Keim P, Knops J, Lieberburg I, Power M, Tan H, Tatsuno G, Tung J, Schenk D, Seubert P, Suomensaari SM, Wang S, Walker D, Zhao J, McConlogue L, John V: Purification and cloning of amyloid precursor protein beta-secretase from human brain. Nature 1999, 402: 537-540. 10.1038/990114
Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, Collins F, Treanor J, Rogers G, Citron M: Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 1999, 286: 735-741. 10.1126/science.286.5440.735
Yan R, Bienkowski MJ, Shuck ME, Miao H, Tory MC, Pauley AM, Brashier JR, Stratman NC, Mathews WR, Buhl AE, Carter DB, Tomasselli AG, Parodi LA, Heinrikson RL, Gurney ME: Membrane-anchored aspartyl protease with Alzheimer’s disease beta-secretase activity. Nature 1999, 402: 533-537. 10.1038/990107
Li Y, Zhou W, Tong Y, He G, Song W: Control of APP processing and Abeta generation level by BACE1 enzymatic activity and transcription. FASEB J 2006, 20: 285-292. 10.1096/fj.05-4986com
Brown MS, Ye J, Rawson RB, Goldstein JL: Regulated intramembrane proteolysis: a control mechanism conserved from bacteria to humans. Cell 2000, 100: 391-398. 10.1016/S0092-8674(00)80675-3
De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, Von Figura K, Van Leuven F: Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 1998, 391: 387-390. 10.1038/34910
Yu G, Nishimura M, Arawaka S, Levitan D, Zhang L, Tandon A, Song YQ, Rogaeva E, Chen F, Kawarai T, Supala A, Levesque L, Yu H, Yang DS, Holmes E, Milman P, Liang Y, Zhang DM, Xu DH, Sato C, Rogaev E, Smith M, Janus C, Zhang Y, Aebersold R, Farrer LS, Sorbi S, Bruni A, Fraser P, St George-Hyslop P: Nicastrin modulates presenilin-mediated notch/glp-1 signal transduction and betaAPP processing. Nature 2000, 407: 48-54. 10.1038/35024009
Zhang Z, Nadeau P, Song W, Donoviel D, Yuan M, Bernstein A, Yankner BA: Presenilins are required for gamma-secretase cleavage of beta-APP and transmembrane cleavage of Notch-1. Nat Cell Biol 2000, 2: 463-465. 10.1038/35017108
Francis R, McGrath G, Zhang J, Ruddy DA, Sym M, Apfeld J, Nicoll M, Maxwell M, Hai B, Ellis MC, Parks AL, Xu W, Li J, Gurney M, Myers RL, Himes CS, Hiebsch R, Ruble C, Nye JS, Curtis D: aph-1 and pen-2 are required for Notch pathway signaling, gamma-secretase cleavage of betaAPP, and presenilin protein accumulation. Dev Cell 2002, 3: 85-97. 10.1016/S1534-5807(02)00189-2
Goutte C, Tsunozaki M, Hale VA, Priess JR: APH-1 is a multipass membrane protein essential for the Notch signaling pathway in Caenorhabditis elegans embryos. Proc Natl Acad Sci U S A 2002, 99: 775-779. 10.1073/pnas.022523499
Edbauer D, Winkler E, Regula JT, Pesold B, Steiner H, Haass C: Reconstitution of gamma-secretase activity. Nat Cell Biol 2003, 5: 486-488. 10.1038/ncb960
Kimberly WT, LaVoie MJ, Ostaszewski BL, Ye W, Wolfe MS, Selkoe DJ: Gamma-secretase is a membrane protein complex comprised of presenilin, nicastrin, Aph-1, and Pen-2. Proc Natl Acad Sci U S A 2003, 100: 6382-6387. 10.1073/pnas.1037392100
Takasugi N, Tomita T, Hayashi I, Tsuruoka M, Niimura M, Takahashi Y, Thinakaran G, Iwatsubo T: The role of presenilin cofactors in the gamma-secretase complex. Nature 2003, 422: 438-441. 10.1038/nature01506
Lee MK, Slunt HH, Martin LJ, Thinakaran G, Kim G, Gandy SE, Seeger M, Koo E, Price DL, Sisodia SS: Expression of presenilin 1 and 2 (PS1 and PS2) in human and murine tissues. J Neurosci 1996, 16: 7513-7525.
Levitan D, Greenwald I: Facilitation of lin-12-mediated signalling by sel-12, a Caenorhabditis elegans S182 Alzheimer’s disease gene. Nature 1995, 377: 351-354. 10.1038/377351a0
Laudon H, Hansson EM, Melen K, Bergman A, Farmery MR, Winblad B, Lendahl U, von Heijne G, Naslund J: A nine-transmembrane domain topology for presenilin 1. J Biol Chem 2005, 280: 35352-35360. 10.1074/jbc.M507217200
Spasic D, Tolia A, Dillen K, Baert V, De Strooper B, Vrijens S, Annaert W: Presenilin-1 maintains a nine-transmembrane topology throughout the secretory pathway. J Biol Chem 2006, 281: 26569-26577. 10.1074/jbc.M600592200
Walter J, Capell A, Grunberg J, Pesold B, Schindzielorz A, Prior R, Podlisny MB, Fraser P, Hyslop PS, Selkoe DJ, Haass C: The Alzheimer’s disease-associated presenilins are differentially phosphorylated proteins located predominantly within the endoplasmic reticulum. Mol Med 1996, 2: 673-691.
Culvenor JG, Maher F, Evin G, Malchiodi-Albedi F, Cappai R, Underwood JR, Davis JB, Karran EH, Roberts GW, Beyreuther K, Masters CL: Alzheimer’s disease-associated presenilin 1 in neuronal cells: evidence for localization to the endoplasmic reticulum-Golgi intermediate compartment. J Neurosci Res 1997, 49: 719-731. 10.1002/(SICI)1097-4547(19970915)49:6<719::AID-JNR6>3.0.CO;2-A
Annaert WG, Levesque L, Craessaerts K, Dierinck I, Snellings G, Westaway D, George-Hyslop PS, Cordell B, Fraser P, De Strooper B: Presenilin 1 controls gamma-secretase processing of amyloid precursor protein in pre-golgi compartments of hippocampal neurons. J Cell Biol 1999, 147: 277-294. 10.1083/jcb.147.2.277
Kim SH, Lah JJ, Thinakaran G, Levey A, Sisodia SS: Subcellular localization of presenilins: association with a unique membrane pool in cultured cells. Neurobiol Dis 2000, 7: 99-117. 10.1006/nbdi.1999.0280
Thinakaran G, Borchelt DR, Lee MK, Slunt HH, Spitzer L, Kim G, Ratovitsky T, Davenport F, Nordstedt C, Seeger M, Hardy J, Levey AI, Gandy SE, Jenkins NA, Copeland NG, Price DL, Sisodia SS: Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo. Neuron 1996, 17: 181-190. 10.1016/S0896-6273(00)80291-3
Podlisny MB, Citron M, Amarante P, Sherrington R, Xia W, Zhang J, Diehl T, Levesque G, Fraser P, Haass C, Koo EH, Seubert P, St George-Hyslop P, Teplow DB, Selkoe DJ: Presenilin proteins undergo heterogeneous endoproteolysis between Thr291 and Ala299 and occur as stable N- and C-terminal fragments in normal and Alzheimer brain tissue. Neurobiol Dis 1997, 3: 325-337. 10.1006/nbdi.1997.0129
Shirotani K, Takahashi K, Ozawa K, Kunishita T, Tabira T: Determination of a cleavage site of presenilin 2 protein in stably transfected SH-SY5Y human neuroblastoma cell lines. Biochem Biophys Res Commun 1997, 240: 728-731. 10.1006/bbrc.1997.7730
Jacobsen H, Reinhardt D, Brockhaus M, Bur D, Kocyba C, Kurt H, Grim MG, Baumeister R, Loetscher H: The influence of endoproteolytic processing of familial Alzheimer’s disease presenilin 2 on abeta42 amyloid peptide formation. J Biol Chem 1999, 274: 35233-35239. 10.1074/jbc.274.49.35233
Campbell WA, Reed ML, Strahle J, Wolfe MS, Xia W: Presenilin endoproteolysis mediated by an aspartyl protease activity pharmacologically distinct from gamma-secretase. J Neurochem 2003, 85: 1563-1574. 10.1046/j.1471-4159.2003.01799.x
Nyabi O, Bentahir M, Horre K, Herreman A, Gottardi-Littell N, Van Broeckhoven C, Merchiers P, Spittaels K, Annaert W, De Strooper B: Presenilins mutated at Asp-257 or Asp-385 restore Pen-2 expression and Nicastrin glycosylation but remain catalytically inactive in the absence of wild type Presenilin. J Biol Chem 2003, 278: 43430-43436. 10.1074/jbc.M306957200
Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, Selkoe DJ: Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature 1999, 398: 513-517. 10.1038/19077
Xia W: Relationship between presenilinase and gamma-secretase. Drug News Perspect 2003, 16: 69-74. 10.1358/dnp.2003.16.2.740248
Xia W: From presenilinase to gamma-secretase, cleave to capacitate. Curr Alzheimer Res 2008, 5: 172-178. 10.2174/156720508783954712
Ahn K, Shelton CC, Tian Y, Zhang X, Gilchrist ML, Sisodia SS, Li YM: Activation and intrinsic gamma-secretase activity of presenilin 1. Proc Natl Acad Sci U S A 2010, 107: 21435-21440. 10.1073/pnas.1013246107
Fukumori A, Fluhrer R, Steiner H, Haass C: Three-amino acid spacing of presenilin endoproteolysis suggests a general stepwise cleavage of gamma-secretase-mediated intramembrane proteolysis. J Neurosci 2010, 30: 7853-7862. 10.1523/JNEUROSCI.1443-10.2010
Knappenberger KS, Tian G, Ye X, Sobotka-Briner C, Ghanekar SV, Greenberg BD, Scott CW: Mechanism of gamma-secretase cleavage activation: is gamma-secretase regulated through autoinhibition involving the presenilin-1 exon 9 loop? Biochemistry 2004, 43: 6208-6218. 10.1021/bi036072v
Steiner H, Romig H, Grim MG, Philipp U, Pesold B, Citron M, Baumeister R, Haass C: The biological and pathological function of the presenilin-1 Deltaexon 9 mutation is independent of its defect to undergo proteolytic processing. J Biol Chem 1999, 274: 7615-7618. 10.1074/jbc.274.12.7615
Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird TD, Hardy J, Hutton M, Kukull W, Larson E, Levy-Lahad E, Viitanen M, Peskind E, Poorkaj P, Schellenberg G, Tanzi R, Wasco W, Lannfelt L, Selkoe D, Younkin S: Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat Med 1996, 2: 864-870. 10.1038/nm0896-864
Herreman A, Serneels L, Annaert W, Collen D, Schoonjans L, De Strooper B: Total inactivation of gamma-secretase activity in presenilin-deficient embryonic stem cells. Nat Cell Biol 2000, 2: 461-462. 10.1038/35017105
Luo WJ, Wang H, Li H, Kim BS, Shah S, Lee HJ, Thinakaran G, Kim TW, Yu G, Xu H: PEN-2 and APH-1 coordinately regulate proteolytic processing of presenilin 1. J Biol Chem 2003, 278: 7850-7854. 10.1074/jbc.C200648200
Zhao G, Liu Z, Ilagan MX, Kopan R: Gamma-secretase composed of PS1/Pen2/Aph1a can cleave notch and amyloid precursor protein in the absence of nicastrin. J Neurosci 2010, 30: 1648-1656. 10.1523/JNEUROSCI.3826-09.2010
Ahn CK, Woo SH, Park JM: Surface solubilization of phenanthrene by surfactant sorbed on soils with different organic matter contents. J Hazard Mater 2010, 177: 799-806. 10.1016/j.jhazmat.2009.12.104
Shirotani K, Edbauer D, Prokop S, Haass C, Steiner H: Identification of distinct gamma-secretase complexes with different APH-1 variants. J Biol Chem 2004, 279: 41340-41345. 10.1074/jbc.M405768200
Shirotani K, Tomioka M, Kremmer E, Haass C, Steiner H: Pathological activity of familial Alzheimer’s disease-associated mutant presenilin can be executed by six different gamma-secretase complexes. Neurobiol Dis 2007, 27: 102-107. 10.1016/j.nbd.2007.04.011
Mastrangelo P, Mathews PM, Chishti MA, Schmidt SD, Gu Y, Yang J, Mazzella MJ, Coomaraswamy J, Horne P, Strome B, Pelly H, Levesque G, Ebeling C, Jiang Y, Nixon RA, Rozmahel R, Fraser PE, St George-Hyslop P, Carlson GA, Westaway D: Dissociated phenotypes in presenilin transgenic mice define functionally distinct gamma-secretases. Proc Natl Acad Sci U S A 2005, 102: 8972-8977. 10.1073/pnas.0500940102
Bentahir M, Nyabi O, Verhamme J, Tolia A, Horre K, Wiltfang J, Esselmann H, De Strooper B: Presenilin clinical mutations can affect gamma-secretase activity by different mechanisms. J Neurochem 2006, 96: 732-742. 10.1111/j.1471-4159.2005.03578.x
Wolfe MS, Kopan R: Intramembrane proteolysis: theme and variations. Science 2004, 305: 1119-1123. 10.1126/science.1096187
Shah S, Lee SF, Tabuchi K, Hao YH, Yu C, LaPlant Q, Ball H, Dann CE III, Sudhof T, Yu G: Nicastrin functions as a gamma-secretase-substrate receptor. Cell 2005, 122: 435-447. 10.1016/j.cell.2005.05.022
Lazarov VK, Fraering PC, Ye W, Wolfe MS, Selkoe DJ, Li H: Electron microscopic structure of purified, active gamma-secretase reveals an aqueous intramembrane chamber and two pores. Proc Natl Acad Sci U S A 2006, 103: 6889-6894. 10.1073/pnas.0602321103
Osenkowski P, Li H, Ye W, Li D, Aeschbach L, Fraering PC, Wolfe MS, Selkoe DJ: Cryoelectron microscopy structure of purified gamma-secretase at 12 A resolution. J Mol Biol 2009, 385: 642-652. 10.1016/j.jmb.2008.10.078
Sato C, Morohashi Y, Tomita T, Iwatsubo T: Structure of the catalytic pore of gamma-secretase probed by the accessibility of substituted cysteines. J Neurosci 2006, 26: 12081-12088. 10.1523/JNEUROSCI.3614-06.2006
Tolia A, Chavez-Gutierrez L, De Strooper B: Contribution of presenilin transmembrane domains 6 and 7 to a water-containing cavity in the gamma-secretase complex. J Biol Chem 2006, 281: 27633-27642. 10.1074/jbc.M604997200
Sato C, Takagi S, Tomita T, Iwatsubo T: The C-terminal PAL motif and transmembrane domain 9 of presenilin 1 are involved in the formation of the catalytic pore of the gamma-secretase. J Neurosci 2008, 28: 6264-6271. 10.1523/JNEUROSCI.1163-08.2008
Takagi S, Tominaga A, Sato C, Tomita T, Iwatsubo T: Participation of transmembrane domain 1 of presenilin 1 in the catalytic pore structure of the gamma-secretase. J Neurosci 2010, 30: 15943-15950. 10.1523/JNEUROSCI.3318-10.2010
Watanabe N, Image I II, Takagi S, Tominaga A, Image Image I, Tomita T, Iwatsubo T: Functional analysis of the transmembrane domains of presenilin 1: participation of transmembrane domains 2 and 6 in the formation of initial substrate-binding site of gamma-secretase. J Biol Chem 2010, 285: 19738-19746. 10.1074/jbc.M110.101287
Kaether C, Capell A, Edbauer D, Winkler E, Novak B, Steiner H, Haass C: The presenilin C-terminus is required for ER-retention, nicastrin-binding and gamma-secretase activity. EMBO J 2004, 23: 4738-4748. 10.1038/sj.emboj.7600478
Steiner H, Winkler E, Haass C: Chemical cross-linking provides a model of the gamma-secretase complex subunit architecture and evidence for close proximity of the C-terminal fragment of presenilin with APH-1. J Biol Chem 2008, 283: 34677-34686. 10.1074/jbc.M709067200
Takeo K, Watanabe N, Tomita T, Iwatsubo T: Contribution of the gamma-Secretase Subunits to the Formation of Catalytic Pore of Presenilin 1 Protein. J Biol Chem 2012, 287: 25834-25843. 10.1074/jbc.M111.336347
Li X, Dang S, Yan C, Gong X, Wang J, Shi Y: Structure of a presenilin family intramembrane aspartate protease. Nature 2013, 493: 56-61.
Struhl G, Adachi A: Requirements for presenilin-dependent cleavage of notch and other transmembrane proteins. Mol Cell 2000, 6: 625-636. 10.1016/S1097-2765(00)00061-7
Bossy-Wetzel E, Schwarzenbacher R, Lipton SA: Molecular pathways to neurodegeneration. Nat Med 2004, 10(Suppl):S2-S9.
Song W, Nadeau P, Yuan M, Yang X, Shen J, Yankner BA: Proteolytic release and nuclear translocation of Notch-1 are induced by presenilin-1 and impaired by pathogenic presenilin-1 mutations. PNAS 1999, 96: 6959-6963. 10.1073/pnas.96.12.6959
Ni CY, Murphy MP, Golde TE, Carpenter G: gamma -Secretase cleavage and nuclear localization of ErbB-4 receptor tyrosine kinase. Science 2001, 294: 2179-2181. 10.1126/science.1065412
Kim DY, Ingano LA, Kovacs DM: Nectin-1alpha, an immunoglobulin-like receptor involved in the formation of synapses, is a substrate for presenilin/gamma-secretase-like cleavage. J Biol Chem 2002, 277: 49976-49981. 10.1074/jbc.M210179200
Lammich S, Okochi M, Takeda M, Kaether C, Capell A, Zimmer AK, Edbauer D, Walter J, Steiner H, Haass C: Presenilin-dependent intramembrane proteolysis of CD44 leads to the liberation of its intracellular domain and the secretion of an Abeta-like peptide. J Biol Chem 2002, 277: 44754-44759. 10.1074/jbc.M206872200
Marambaud P, Shioi J, Serban G, Georgakopoulos A, Sarner S, Nagy V, Baki L, Wen P, Efthimiopoulos S, Shao Z, Wisniewski T, Robakis NK: A presenilin-1/gamma-secretase cleavage releases the E-cadherin intracellular domain and regulates disassembly of adherens junctions. EMBO J 2002, 21: 1948-1956. 10.1093/emboj/21.8.1948
May P, Reddy YK, Herz J: Proteolytic processing of low density lipoprotein receptor-related protein mediates regulated release of its intracellular domain. J Biol Chem 2002, 277: 18736-18743. 10.1074/jbc.M201979200
Marambaud P, Wen PH, Dutt A, Shioi J, Takashima A, Siman R, Robakis NK: A CBP binding transcriptional repressor produced by the PS1/epsilon-cleavage of N-cadherin is inhibited by PS1 FAD mutations. Cell 2003, 114: 635-645. 10.1016/j.cell.2003.08.008
Haapasalo A, Kovacs DM: The many substrates of presenilin/gamma-secretase. J Alzheimers Dis 2011, 25: 3-28.
Artavanis-Tsakonas S, Matsuno K, Fortini ME: Notch signaling. Science 1995, 268: 225-232. 10.1126/science.7716513
Kopan R, Schroeter EH, Weintraub H, Nye JS: Signal transduction by activated mNotch: importance of proteolytic processing and its regulation by the extracellular domain. Proc Natl Acad Sci U S A 1996, 93: 1683-1688. 10.1073/pnas.93.4.1683
Struhl G, Fitzgerald K, Greenwald I: Intrinsic activity of the Lin-12 and Notch intracellular domains in vivo. Cell 1993, 74: 331-345. 10.1016/0092-8674(93)90424-O
Kidd S, Lieber T, Young MW: Ligand-induced cleavage and regulation of nuclear entry of Notch in Drosophila melanogaster embryos. Genes Dev 1998, 12: 3728-3740. 10.1101/gad.12.23.3728
Shen J, Bronson RT, Chen DF, Xia W, Selkoe DJ, Tonegawa S: Skeletal and CNS defects in Presenilin-1-deficient mice. Cell 1997, 89: 629-639. 10.1016/S0092-8674(00)80244-5
Wong PC, Zheng H, Chen H, Becher MW, Sirinathsinghji DJ, Trumbauer ME, Chen HY, Price DL, Van der Ploeg LH, Sisodia SS: Presenilin 1 is required for Notch1 and DII1 expression in the paraxial mesoderm. Nature 1997, 387: 288-292. 10.1038/387288a0
De Strooper B, Annaert W, Cupers P, Saftig P, Craessaerts K, Mumm JS, Schroeter EH, Schrijvers V, Wolfe MS, Ray WJ, Goate A, Kopan R: A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature 1999, 398: 518-522. 10.1038/19083
Sestan N, Artavanis-Tsakonas S, Rakic P: Contact-dependent inhibition of cortical neurite growth mediated by notch signaling. Science 1999, 286: 741-746. 10.1126/science.286.5440.741
Presente A, Andres A, Nye JS: Requirement of Notch in adulthood for neurological function and longevity. Neuroreport 2001, 12: 3321-3325. 10.1097/00001756-200110290-00035
Presente A, Shaw S, Nye JS, Andres AJ: Transgene-mediated RNA interference defines a novel role for notch in chemosensory startle behavior. Genesis 2002, 34: 165-169. 10.1002/gene.10149
Wang Y, Chan SL, Miele L, Yao PJ, Mackes J, Ingram DK, Mattson MP, Furukawa K: Involvement of Notch signaling in hippocampal synaptic plasticity. Proc Natl Acad Sci U S A 2004, 101: 9458-9462. 10.1073/pnas.0308126101
Salama-Cohen P, Arevalo MA, Grantyn R, Rodriguez-Tebar A: Notch and NGF/p75NTR control dendrite morphology and the balance of excitatory/inhibitory synaptic input to hippocampal neurones through Neurogenin 3. J Neurochem 2006, 97: 1269-1278. 10.1111/j.1471-4159.2006.03783.x
Weidemann A, Eggert S, Reinhard FB, Vogel M, Paliga K, Baier G, Masters CL, Beyreuther K, Evin G: A novel epsilon-cleavage within the transmembrane domain of the Alzheimer amyloid precursor protein demonstrates homology with Notch processing. Biochemistry 2002, 41: 2825-2835. 10.1021/bi015794o
Okochi M, Steiner H, Fukumori A, Tanii H, Tomita T, Tanaka T, Iwatsubo T, Kudo T, Takeda M, Haass C: Presenilins mediate a dual intramembranous gamma-secretase cleavage of Notch-1. EMBO J 2002, 21: 5408-5416. 10.1093/emboj/cdf541
Qi-Takahara Y, Morishima-Kawashima M, Tanimura Y, Dolios G, Hirotani N, Horikoshi Y, Kametani F, Maeda M, Saido TC, Wang R, Ihara Y: Longer forms of amyloid beta protein: implications for the mechanism of intramembrane cleavage by gamma-secretase. J Neurosci 2005, 25: 436-445. 10.1523/JNEUROSCI.1575-04.2005
Sato T, Tanimura Y, Hirotani N, Saido TC, Morishima-Kawashima M, Ihara Y: Blocking the cleavage at midportion between gamma- and epsilon-sites remarkably suppresses the generation of amyloid beta-protein. FEBS Lett 2005, 579: 2907-2912. 10.1016/j.febslet.2005.04.037
Kakuda N, Funamoto S, Yagishita S, Takami M, Osawa S, Dohmae N, Ihara Y: Equimolar production of amyloid beta-protein and amyloid precursor protein intracellular domain from beta-carboxyl-terminal fragment by gamma-secretase. J Biol Chem 2006, 281: 14776-14786. 10.1074/jbc.M513453200
Takami M, Nagashima Y, Sano Y, Ishihara S, Morishima-Kawashima M, Funamoto S, Ihara Y: gamma-Secretase: successive tripeptide and tetrapeptide release from the transmembrane domain of beta-carboxyl terminal fragment. J Neurosci 2009, 29: 13042-13052. 10.1523/JNEUROSCI.2362-09.2009
Funamoto S, Morishima-Kawashima M, Tanimura Y, Hirotani N, Saido TC, Ihara Y: Truncated carboxyl-terminal fragments of beta-amyloid precursor protein are processed to amyloid beta-proteins 40 and 42. Biochemistry 2004, 43: 13532-13540. 10.1021/bi049399k
Borchelt DR, Thinakaran G, Eckman CB, Lee MK, Davenport F, Ratovitsky T, Prada CM, Kim G, Seekins S, Yager D, Slunt HH, Wang R, Seeger M, Levey AI, Gandy SE, Copeland NG, Jenkins NA, Price DL, Younkin SG, Sisodia SS: Familial Alzheimer’s disease-linked presenilin 1 variants elevate Abeta1-42/1-40 ratio in vitro and in vivo. Neuron 1996, 17: 1005-1013. 10.1016/S0896-6273(00)80230-5
Jankowsky JL, Fadale DJ, Anderson J, Xu GM, Gonzales V, Jenkins NA, Copeland NG, Lee MK, Younkin LH, Wagner SL, Younkin SG, Borchelt DR: Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum Mol Genet 2004, 13: 159-170.
Fukumori A, Okochi M, Tagami S, Jiang J, Itoh N, Nakayama T, Yanagida K, Ishizuka-Katsura Y, Morihara T, Kamino K, Tanaka T, Kudo T, Tanii H, Ikuta A, Haass C, Takeda M: Presenilin-dependent gamma-secretase on plasma membrane and endosomes is functionally distinct. Biochemistry 2006, 45: 4907-4914. 10.1021/bi052412w
Chen F, Hasegawa H, Schmitt-Ulms G, Kawarai T, Bohm C, Katayama T, Gu Y, Sanjo N, Glista M, Rogaeva E, Wakutani Y, Pardossi-Piquard R, Ruan X, Tandon A, Checler F, Marambaud P, Hansen K, Westaway D, St George-Hyslop P, Fraser P: TMP21 is a presenilin complex component that modulates gamma-secretase but not epsilon-secretase activity. Nature 2006, 440: 1208-1212. 10.1038/nature04667
Wanngren J, Ottervald J, Parpal S, Portelius E, Stromberg K, Borgegard T, Klintenberg R, Jureus A, Blomqvist J, Blennow K, Zetterberg H, Lundkvist J, Rosqvist S, Karlstrom H: Second generation gamma-secretase modulators exhibit different modulation of Notch beta and Abeta production. J Biol Chem 2012, 287(39):32640-32650. 10.1074/jbc.M112.376541
Chavez-Gutierrez L, Bammens L, Benilova I, Vandersteen A, Benurwar M, Borgers M, Lismont S, Zhou L, Van Cleynenbreugel S, Esselmann H, Wiltfang J, Serneels L, Karran E, Gijsen H, Schymkowitz J, Rousseau F, Broersen K, De Strooper B: The mechanism of gamma-Secretase dysfunction in familial Alzheimer disease. EMBO J 2012, 31: 2261-2274. 10.1038/emboj.2012.79
Saura CA, Choi SY, Beglopoulos V, Malkani S, Zhang D, Shankaranarayana Rao BS, Chattarji S, Kelleher RJ III, Kandel ER, Duff K, Kirkwood A, Shen J: Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age-dependent neurodegeneration. Neuron 2004, 42: 23-36. 10.1016/S0896-6273(04)00182-5
Wines-Samuelson M, Schulte EC, Smith MJ, Aoki C, Liu X, Kelleher RJ III, Shen J: Characterization of age-dependent and progressive cortical neuronal degeneration in presenilin conditional mutant mice. PLoS One 2010, 5: e10195. 10.1371/journal.pone.0010195
Khandelwal A, Chandu D, Roe CM, Kopan R, Quatrano RS: Moonlighting activity of presenilin in plants is independent of gamma-secretase and evolutionarily conserved. Proc Natl Acad Sci U S A 2007, 104: 13337-13342. 10.1073/pnas.0702038104
Huppert SS, Ilagan MX, De Strooper B, Kopan R: Analysis of Notch function in presomitic mesoderm suggests a gamma-secretase-independent role for presenilins in somite differentiation. Dev Cell 2005, 8: 677-688. 10.1016/j.devcel.2005.02.019
Houlden H, Baker M, McGowan E, Lewis P, Hutton M, Crook R, Wood NW, Kumar-Singh S, Geddes J, Swash M, Scaravilli F, Holton JL, Lashley T, Tomita T, Hashimoto T, Verkkoniemi A, Kalimo H, Somer M, Paetau A, Martin JJ, Van Broeckhoven C, Golde T, Hardy J, Haltia M, Revesz T: Variant Alzheimer’s disease with spastic paraparesis and cotton wool plaques is caused by PS-1 mutations that lead to exceptionally high amyloid-beta concentrations. Ann Neurol 2000, 48: 806-808. 10.1002/1531-8249(200011)48:5<806::AID-ANA18>3.0.CO;2-F
Larner AJ, Doran M: Clinical phenotypic heterogeneity of Alzheimer’s disease associated with mutations of the presenilin-1 gene. J Neurol 2006, 253: 139-158. 10.1007/s00415-005-0019-5
Shepherd C, McCann H, Halliday GM: Variations in the neuropathology of familial Alzheimer’s disease. Acta Neuropathol 2009, 118: 37-52. 10.1007/s00401-009-0521-4
Huang H, He X: Wnt/beta-catenin signaling: new (and old) players and new insights. Curr Opin Cell Biol 2008, 20: 119-125. 10.1016/j.ceb.2008.01.009
Murayama M, Tanaka S, Palacino J, Murayama O, Honda T, Sun X, Yasutake K, Nihonmatsu N, Wolozin B, Takashima A: Direct association of presenilin-1 with beta-catenin. FEBS Lett 1998, 433: 73-77. 10.1016/S0014-5793(98)00886-2
Zhang Z, Hartmann H, Do VM, Abramowski D, Sturchler-Pierrat C, Staufenbiel M, Sommer B, van de Wetering M, Clevers H, Saftig P, De Strooper B, He X, Yankner BA: Destabilization of beta-catenin by mutations in presenilin-1 potentiates neuronal apoptosis. Nature 1998, 395: 698-702. 10.1038/27208
Cox RT, McEwen DG, Myster DL, Duronio RJ, Loureiro J, Peifer M: A screen for mutations that suppress the phenotype of Drosophila armadillo, the beta-catenin homolog. Genetics 2000, 155: 1725-1740.
Noll E, Medina M, Hartley D, Zhou J, Perrimon N, Kosik KS: Presenilin affects arm/beta-catenin localization and function in Drosophila. Dev Biol 2000, 227: 450-464. 10.1006/dbio.2000.9925
Soriano S, Kang DE, Fu M, Pestell R, Chevallier N, Zheng H, Koo EH: Presenilin 1 negatively regulates beta-catenin/T cell factor/lymphoid enhancer factor-1 signaling independently of beta-amyloid precursor protein and notch processing. J Cell Biol 2001, 152: 785-794. 10.1083/jcb.152.4.785
Kang DE, Soriano S, Frosch MP, Collins T, Naruse S, Sisodia SS, Leibowitz G, Levine F, Koo EH: Presenilin 1 facilitates the constitutive turnover of beta-catenin: differential activity of Alzheimer’s disease-linked PS1 mutants in the beta-catenin-signaling pathway. J Neurosci 1999, 19: 4229-4237.
Kang DE, Soriano S, Xia X, Eberhart CG, De Strooper B, Zheng H, Koo EH: Presenilin couples the paired phosphorylation of beta-catenin independent of axin: implications for beta-catenin activation in tumorigenesis. Cell 2002, 110: 751-762. 10.1016/S0092-8674(02)00970-4
Xia X, Qian S, Soriano S, Wu Y, Fletcher AM, Wang XJ, Koo EH, Wu X, Zheng H: Loss of presenilin 1 is associated with enhanced beta-catenin signaling and skin tumorigenesis. Proc Natl Acad Sci U S A 2001, 98: 10863-10868. 10.1073/pnas.191284198
De Ferrari GV, Moon RT: The ups and downs of Wnt signaling in prevalent neurological disorders. Oncogene 2006, 25: 7545-7553. 10.1038/sj.onc.1210064
Nishimura M, Yu G, Levesque G, Zhang DM, Ruel L, Chen F, Milman P, Holmes E, Liang Y, Kawarai T, Jo E, Supala A, Rogaeva E, Xu DM, Janus C, Levesque L, Bi Q, Duthie M, Rozmahel R, Mattila K, Lannfelt L, Westaway D, Mount HT, Woodgett J, St George-Hyslop P, et al.: Presenilin mutations associated with Alzheimer disease cause defective intracellular trafficking of beta-catenin, a component of the presenilin protein complex. Nat Med 1999, 5: 164-169. 10.1038/5526
Kawamura Y, Kikuchi A, Takada R, Takada S, Sudoh S, Shibamoto S, Yanagisawa K, Komano H: Inhibitory effect of a presenilin 1 mutation on the Wnt signalling pathway by enhancement of beta-catenin phosphorylation. Eur J Biochem 2001, 268: 3036-3041. 10.1046/j.1432-1327.2001.02197.x
Teo JL, Ma H, Nguyen C, Lam C, Kahn M: Specific inhibition of CBP/beta-catenin interaction rescues defects in neuronal differentiation caused by a presenilin-1 mutation. Proc Natl Acad Sci U S A 2005, 102: 12171-12176. 10.1073/pnas.0504600102
Ito E, Oka K, Etcheberrigaray R, Nelson TJ, McPhie DL, Tofel-Grehl B, Gibson GE, Alkon DL: Internal Ca2+ mobilization is altered in fibroblasts from patients with Alzheimer disease. Proc Natl Acad Sci U S A 1994, 91: 534-538. 10.1073/pnas.91.2.534
Cheung KH, Shineman D, Muller M, Cardenas C, Mei L, Yang J, Tomita T, Iwatsubo T, Lee VM, Foskett JK: Mechanism of Ca2+ disruption in Alzheimer’s disease by presenilin regulation of InsP3 receptor channel gating. Neuron 2008, 58: 871-883. 10.1016/j.neuron.2008.04.015
Stutzmann GE, Caccamo A, LaFerla FM, Parker I: Dysregulated IP3 signaling in cortical neurons of knock-in mice expressing an Alzheimer’s-linked mutation in presenilin1 results in exaggerated Ca2+ signals and altered membrane excitability. J Neurosci 2004, 24: 508-513. 10.1523/JNEUROSCI.4386-03.2004
Stutzmann GE, Smith I, Caccamo A, Oddo S, Laferla FM, Parker I: Enhanced ryanodine receptor recruitment contributes to Ca2+ disruptions in young, adult, and aged Alzheimer’s disease mice. J Neurosci 2006, 26: 5180-5189. 10.1523/JNEUROSCI.0739-06.2006
Chakroborty S, Goussakov I, Miller MB, Stutzmann GE: Deviant ryanodine receptor-mediated calcium release resets synaptic homeostasis in presymptomatic 3xTg-AD mice. J Neurosci 2009, 29: 9458-9470. 10.1523/JNEUROSCI.2047-09.2009
Green KN, Demuro A, Akbari Y, Hitt BD, Smith IF, Parker I, LaFerla FM: SERCA pump activity is physiologically regulated by presenilin and regulates amyloid beta production. J Cell Biol 2008, 181: 1107-1116. 10.1083/jcb.200706171
Tu H, Nelson O, Bezprozvanny A, Wang Z, Lee SF, Hao YH, Serneels L, De Strooper B, Yu G, Bezprozvanny I: Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer’s disease-linked mutations. Cell 2006, 126: 981-993. 10.1016/j.cell.2006.06.059
Nelson O, Tu H, Lei T, Bentahir M, de Strooper B, Bezprozvanny I: Familial Alzheimer disease-linked mutations specifically disrupt Ca2+ leak function of presenilin 1. J Clin Invest 2007, 117: 1230-1239. 10.1172/JCI30447
Nelson O, Supnet C, Tolia A, Horre K, De Strooper B, Bezprozvanny I: Mutagenesis mapping of the presenilin 1 calcium leak conductance pore. J Biol Chem 2011, 286: 22339-22347. 10.1074/jbc.M111.243063
Cheung KH, Mei L, Mak DO, Hayashi I, Iwatsubo T, Kang DE, Foskett JK: Gain-of-function enhancement of IP3 receptor modal gating by familial Alzheimer’s disease-linked presenilin mutants in human cells and mouse neurons. Sci Signal 2010, 3: ra22. 10.1126/scisignal.2000818
Suzuki K, Terry RD: Fine structural localization of acid phosphatase in senile plaques in Alzheimer’s presenile dementia. Acta Neuropathol 1967, 8: 276-284. 10.1007/BF00688828
Cataldo AM, Barnett JL, Berman SA, Li J, Quarless S, Bursztajn S, Lippa C, Nixon RA: Gene expression and cellular content of cathepsin D in Alzheimer’s disease brain: evidence for early up-regulation of the endosomal-lysosomal system. Neuron 1995, 14: 671-680. 10.1016/0896-6273(95)90324-0
Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, Cuervo AM: Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol 2005, 64: 113-122.
Klionsky DJ, Emr SD: Autophagy as a regulated pathway of cellular degradation. Science 2000, 290: 1717-1721.
Levine B, Klionsky DJ: Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell 2004, 6: 463-477. 10.1016/S1534-5807(04)00099-1
Esselens C, Oorschot V, Baert V, Raemaekers T, Spittaels K, Serneels L, Zheng H, Saftig P, De Strooper B, Klumperman J, Annaert W: Presenilin 1 mediates the turnover of telencephalin in hippocampal neurons via an autophagic degradative pathway. J Cell Biol 2004, 166: 1041-1054. 10.1083/jcb.200406060
Wilson CA, Murphy DD, Giasson BI, Zhang B, Trojanowski JQ, Lee VM: Degradative organelles containing mislocalized alpha-and beta-synuclein proliferate in presenilin-1 null neurons. J Cell Biol 2004, 165: 335-346. 10.1083/jcb.200403061
Annaert WG, Esselens C, Baert V, Boeve C, Snellings G, Cupers P, Craessaerts K, De Strooper B: Interaction with telencephalin and the amyloid precursor protein predicts a ring structure for presenilins. Neuron 2001, 32: 579-589. 10.1016/S0896-6273(01)00512-8
Nixon RA, Yang DS: Autophagy failure in Alzheimer’s disease–locating the primary defect. Neurobiol Dis 2011, 43: 38-45. 10.1016/j.nbd.2011.01.021
Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, Wolfe DM, Martinez-Vicente M, Massey AC, Sovak G, Uchiyama Y, Westaway D, Cuervo AM, Nixon RA: Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 2010, 141: 1146-1158. 10.1016/j.cell.2010.05.008
Neely KM, Green KN, LaFerla FM: Presenilin is necessary for efficient proteolysis through the autophagy-lysosome system in a gamma-secretase-independent manner. J Neurosci 2011, 31: 2781-2791. 10.1523/JNEUROSCI.5156-10.2010
Coen K, Flannagan RS, Baron S, Carraro-Lacroix LR, Wang D, Vermeire W, Michiels C, Munck S, Baert V, Sugita S, Wuytack F, Hiesinger PR, Grinstein S, Annaert W: Lysosomal calcium homeostasis defects, not proton pump defects, cause endo-lysosomal dysfunction in PSEN-deficient cells. J Cell Biol 2012, 198: 23-35. 10.1083/jcb.201201076
Zhang X, Garbett K, Veeraraghavalu K, Wilburn B, Gilmore R, Mirnics K, Sisodia SS: A role for presenilins in autophagy revisited: normal acidification of lysosomes in cells lacking PSEN1 and PSEN2. J Neurosci 2012, 32: 8633-8648. 10.1523/JNEUROSCI.0556-12.2012
Tabira T, Chui DH, Nakayama H, Kuroda S, Shibuya M: Alzheimer’s disease with spastic paresis and cotton wool type plaques. J Neurosci Res 2002, 70: 367-372. 10.1002/jnr.10392
Karlstrom H, Brooks WS, Kwok JB, Broe GA, Kril JJ, McCann H, Halliday GM, Schofield PR: Variable phenotype of Alzheimer’s disease with spastic paraparesis. J Neurochem 2008, 104: 573-583.
Kwok JB, Taddei K, Hallupp M, Fisher C, Brooks WS, Broe GA, Hardy J, Fulham MJ, Nicholson GA, Stell R, St George Hyslop PH, Fraser PE, Kakulas B, Clarnette R, Relkin N, Gandy SE, Schofield PR, Martins RN: Two novel (M233T and R278T) presenilin-1 mutations in early-onset Alzheimer’s disease pedigrees and preliminary evidence for association of presenilin-1 mutations with a novel phenotype. Neuroreport 1997, 8: 1537-1542. 10.1097/00001756-199704140-00043
Crook R, Verkkoniemi A, Perez-Tur J, Mehta N, Baker M, Houlden H, Farrer M, Hutton M, Lincoln S, Hardy J, Gwinn K, Somer M, Paetau A, Kalimo H, Ylikoski R, Poyhonen M, Kucera S, Haltia M: A variant of Alzheimer’s disease with spastic paraparesis and unusual plaques due to deletion of exon 9 of presenilin 1. Nat Med 1998, 4: 452-455. 10.1038/nm0498-452
Gant JC, Sama MM, Landfield PW, Thibault O: Early and simultaneous emergence of multiple hippocampal biomarkers of aging is mediated by Ca2+ −induced Ca2+ release. J Neurosci 2006, 26: 3482-3490. 10.1523/JNEUROSCI.4171-05.2006
Foster TC: Calcium homeostasis and modulation of synaptic plasticity in the aged brain. Aging Cell 2007, 6: 319-325. 10.1111/j.1474-9726.2007.00283.x
Toescu EC, Verkhratsky A: The importance of being subtle: small changes in calcium homeostasis control cognitive decline in normal aging. Aging Cell 2007, 6: 267-273. 10.1111/j.1474-9726.2007.00296.x
Nelson O, Supnet C, Liu H, Bezprozvanny I: Familial Alzheimer’s disease mutations in presenilins: effects on endoplasmic reticulum calcium homeostasis and correlation with clinical phenotypes. J Alzheimers Dis 2010, 21: 781-793.
Supnet C, Bezprozvanny I: Presenilins as endoplasmic reticulum calcium leak channels and Alzheimer’s disease pathogenesis. Sci China Life Sci 2011, 54: 744-751. 10.1007/s11427-011-4201-y
Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L: Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proc Natl Acad Sci U S A 1999, 96: 3228-3233. 10.1073/pnas.96.6.3228
Palop JJ, Mucke L: Amyloid-beta-induced neuronal dysfunction in Alzheimer’s disease: from synapses toward neural networks. Nat Neurosci 2010, 13: 812-818. 10.1038/nn.2583
Zhang C, Wu B, Beglopoulos V, Wines-Samuelson M, Zhang D, Dragatsis I, Sudhof TC, Shen J: Presenilins are essential for regulating neurotransmitter release. Nature 2009, 460: 632-636. 10.1038/nature08177
Pratt KG, Zimmerman EC, Cook DG, Sullivan JM: Presenilin 1 regulates homeostatic synaptic scaling through Akt signaling. Nat Neurosci 2011, 14: 1112-1114. 10.1038/nn.2893
Dolev I, Fogel H, Milshtein H, Berdichevsky Y, Lipstein N, Brose N, Gazit N, Slutsky I: Spike bursts increase amyloid-beta 40/42 ratio by inducing a presenilin-1 conformational change. Nat Neurosci 2013, 16: 587-595. 10.1038/nn.3376
Acknowledgements
This work was supported by Canadian Institutes of Health Research (CIHR) Operating Grant CCI-117952. W.S. was the holder of the Tier 1 Canada Research Chair in Alzheimer's Disease. S. Z. was the recipient of the Chinese Scholarship Council award. M. Z. is supported by UBC 4YF Scholarship.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interest
The authors declared that they have no competing interest.
Authors’ contributions
SZ carried out literature search and drafted the manuscript. MZ wrote one section and critically revised the manuscript. FC drafted one of the figures and provided comments for the manuscript. WS was the supervisor of the research group, provided the guidance and instructions and critically revised the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Zhang, S., Zhang, M., Cai, F. et al. Biological function of Presenilin and its role in AD pathogenesis. Transl Neurodegener 2, 15 (2013). https://doi.org/10.1186/2047-9158-2-15
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2047-9158-2-15