
Abstract
Genetic modification has long provided an approach for “reverse genetics”, analyzing gene function and linking DNA sequence to phenotype. However, traditional genome editing technologies have not kept pace with the soaring progress of the genome sequencing era, as a result of their inefficiency, time-consuming and labor-intensive methods. Recently, invented genome modification technologies, such as ZFN (Zinc Finger Nuclease), TALEN (Transcription Activator-Like Effector Nuclease), and CRISPR/Cas9 nuclease (Clustered Regularly Interspaced Short Palindromic Repeats/Cas9 nuclease) can initiate genome editing easily, precisely and with no limitations by organism. These new tools have also offered intriguing possibilities for conducting functional large-scale experiments. In this review, we begin with a brief introduction of ZFN, TALEN, and CRISPR/Cas9 technologies, then generate an extensive prediction of effective TALEN and CRISPR/Cas9 target sites in the genomes of a broad range of taxonomic species. Based on the evidence, we highlight the potential and practicalities of TALEN and CRISPR/Cas9 editing in non-model organisms, and also compare the technologies and test interesting issues such as the functions of candidate domesticated, as well as candidate genes in life-environment interactions. When accompanied with a high-throughput sequencing platform, we forecast their potential revolutionary impacts on evolutionary and ecological research, which may offer an exciting prospect for connecting the gap between DNA sequence and phenotype in the near future.
Similar content being viewed by others


Introduction
Genetic modification has long provided the ability to use “reverse genetics” as an approach for analyzing gene function and linking DNA sequence to phenotype. Different functional experiments demand different modifications of gene function, which includes gene sequence modification, such as knockin and knockout, and gene expression modifications, such as RNA interference (RNAi). For the past few decades, functional genes have been successfully integrated into endogenous genomes and over-expressed through transposon-mediated modification, similar to T-DNA and p-elements. Furthermore, scientists are able to knockdown genes using RNA interference [1–3] and carry out gene targeting by site-specific recombinase technology, such as Cre/loxP [4], Flp/FRT [5], and φC31-mediated systems [6]. Among these different forms of genetic modification, gene targeting is thought to be the most straightforward, and thus be a “gold standard” for the exploration of gene function in vivo; because, compared with gene targeting, the expression level induced by a transposon is severely affected by the random insertion positions of genes. Similarly, RNAi has temporary knockdown effects, unpredictable off-target influence and too much background noise [1]. However, because RNAi is inefficient, time-consuming and labor-intensive, until now, traditional gene-targeting technology has only been able to be applied in rare model systems, such as Drosophila[7] and mouse [8], characterized by short generation times and easy inbreeding.
Recent years have witnessed a breakthrough in gene targeting technology. ZFN (Zinc Finger Nuclease) [9, 10], TALEN (Transcription Activator-Like Effector Nuclease) [11] and CRISPR/Cas9 nuclease (Clustered Regularly Interspaced Short Palindromic Repeats) [12, 13] systems now make it possible for scientists to easily, efficiently and cheaply modify the genome. ZFN, TALEN and CRISPR/Cas9 can be assembled in a few days by regular cloning methods, commercial kits or commercial services [14–19] (Additional file 1: Table S1). These technologies can introduce novel mutations in any gene efficiently, sometimes exceeding a frequency of 50% [20, 21] and in a variety of organisms (Additional file 1: Table S2). The emergence and rapid development of such techniques has raised great interest in their applications in either model or non-model organisms. In this review, we highlight the potential and practicalities of TALEN and CRISPR/Cas9 in non-model organisms, and also compare the technologies and test interesting issues, such as the functions of candidate domesticated genes, as well as candidate genes in life-environment interactions. Accompanied with a high-throughput sequencing platform, TALEN and CRISPR/Cas9 will undoubtedly have revolutionary impacts on evolutionary and ecological research, which may offer an exciting prospect for elucidating the gap between DNA sequence and phenotype in the near future.
Review
Principles of genome editing technologies
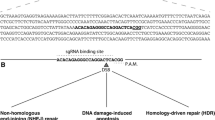
In terms of the principles of different genome editing technologies, such as ZFN, TALEN and CRISPR/Cas9, there are several good reviews published elsewhere [22–25] and we will not present a redundant review here, but we aim to provide a summary of the actual mechanisms for further discussion (Figure 1). Briefly, all three new genome-editing techniques, (i.e. ZFN, TALEN and CRISPR/Cas9) achieve precise and efficient genome modification through similar mechanisms — by inducing targeted DNA to generate double strand breaks (DSB), followed-by DSBs being corrected by error-prone non-homologous end joining (NHEJ) [26] and homologous recombination (HR) [27] (Figure 1A), where NHEJ and HR are the two key DNA repair mechanisms in eukaryotic cells. However, ZFN, TALEN and CRISPR/Cas9 systems each behave differently due to the way each system recognizes and breaks the target DNA in vivo[9, 11, 28–30]. It is notable that both ZFN and TALEN systems stimulate DSBs by a non-specific FokI nuclease domain fused to their binding domains; whereas the CRISPR/Cas9 system acts via a ribonucleo-protein complex, in which the target recognition lobe of Cas9 interacts with sgRNA which could be modified to have the homologous sequence with target DNA and direct specific binding [12, 30]. Besides the differences in mechanism of action, ZFN, TALEN, and CRISPR/Cas9 have other unique and advantageous features (Table 1). ZFN has limited target sites because of its 3-nucleotides recognizing model, and the system is also more expensive and difficult to assemble. The TALEN and CRISPR/Cas9 techniques are considered to be the ideal gene-targeting technologies, because they are easier to assemble, are more efficient, and have more abundant target-specific recognition sites and activations compared with a similar range of cell types and organisms. Thus, we will mainly focus on TALEN and CRISPR/Cas9 here.

Mechanism of ZFN, TALEN and CRISPR/ Cas9. A. ZFN, TALEN and CRISPR/Cas9 achieve precise and efficient genome modification by inducing targeted DNA DSBs, which would be corrected by NHEJ and HR repair mechanisms. NHEJ-mediated repair leads to the introduction of variable length insertion or deletion. HR-mediated repair could lead to point mutation and gene replacement, in the present of donor DNA. B. TALEs and Cas9 protein fused with effector proteins such as VP64, Mxi1 could regulate expressions of endogenous genes. Additionally, TALEs fused with histone-deacetylating epigenetic effectors could regulate epigenetics status of endogenous genes. CRISPR, Clustered Regularly Interspaced Short Palindromic Repeats; dCas9, inactive Cas9 protein; DSB, Double Strand Breaks; NHEJ, Error-prone Nonhomologous End Joining; HR, Homologous Recombination; InDel, Insertion and Deletion; PAM, Protospacer Adjacent Motif; RNA Pol II, RNA Polymerase II; sgRNA, single guide RNA; TALE, Transcription Activator-Like Effector; TALEN, Transcription Activator-Like Effector Nuclease; ZFN, Zinc Finger Nuclease.
The functional effects of utilizing TALEN and CRISPR/Cas9 in model organisms can be classified into disruption, substitution and regulation (Figure 1B). Disruption by both small indels and large segmentation deletions have been successfully used in gene knockout in a broad series of model species, such as human cells [31], Drosophila[32], mice [33], cynomolgus [34, 35], zebrafish [21], Xenopus tropicalis[36], and Arabidopsis [37]. Substitution by which single or multiple transgenes can be efficiently integrated into target sites, resulting in single nucleotide substitution and gene knock-in, have been successfully applied in mice [38] and zebrafish [39]. In contrast, regulation by CRISPR/Cas9 system or TALEN will not involve incision of the genome, but regulate the expression level of target endogenous genes. For example, inactive Cas9 protein (dCas9) fused with Mxi1, a mammalian transcriptional repressor domain, could result in the knock-down expression of an endogenous gene [40]. Alternatively, inactive Cas9 fused to an activator protein, such as VP64, may increase the expression of ZFP42 and POU5F1[41]. Similarly, TALEs (Transcription Activator-Like Effector) fused with effector proteins, such as VP64[42], may regulate endogenous genes. One interesting feature is that TALEs fused with histone-deacetylating epigenetic effectors could regulate the epigenetic status in vivo[43, 44]. Although, regulation effects are not so widely applied in genome modification as disruption and substitution.
Applications of genome editing technologies in non-model organisms
TALEN and CRISPR/Cas9 have provided an easy approach to manipulate the genomes of both model and non-model organisms. Model organisms only represent a small percentage of natural life, considering that numerous adaptation, morphological and behavioral traits are absent in model species. For example, naked mole rats have an extraordinary long lifespan, high fecundity, the ability to live in areas with low oxygen levels [45], and eusocial ants have highly organized society castes and specialized behavior for workers and queens [46]. To better understand the underlying genetic mechanisms of evolutionary adaptations and biodiversity, a highly efficient applicable system for non-model organisms would be highly in demand for evolutionary and ecological studies. The now prevalent applications of high-throughput sequencing technology and invention of TALEN and CRISPR/Cas9 have finally made it practical to unveil the mysteries of non-model organisms. Successful genetic modification has been obtained in non-model organisms, such as silkworm [47], cattle [48], Brassica oleracea[49], Anopheles gambiae[50], Aedes aegypti[51], medaka [52], liverwort [53] and wheat [54, 55].
To explore the potential application of these technologies on a broader range of taxonomic species, we collected 26 genome sequences from 26 new model organisms, comprising mammals, birds, insects and reptiles, as well as predicted TALEN and CRISPR/Cas9 target sites in gene and promoter regions. Promoter regions are defined as the 2 kb regions found upstream and downstream of genes. The N20-NGG sequence pattern is used to identify the CRISPR/Cas9 target sits. Then, these target sites are BLAST (using the Basic Local Alignment Search Tool) against both gene and promoter regions to eliminate off-target sites using strict criteria, where the candidate editable site is defined only when the seed region (12 bps adjacent to Protospacer Adjacent Motif) is unique. TALEN target sites are predicted by the TAL Effector-Nucleotide Targeter [56]. For over 80% (22 out of 26) of these species, approximately 50-84% of gene coding sequences and 50-97% of promoters could be successfully targeted by both TALEN and CRISPR/Cas9, as predicted by bioinformatics calculations through rigorous criteria (Table 2, Table 3) and an additional file shows the prediction information in more details [see Additional file 2]. This indicates that, genetic modification could be carried out in these organisms through TALEN and CRISPR/Cas9, demonstrating its revolutionary potential in evolutionary and ecological studies.
Testing the functional roles of genetic innovations
Genetic innovations play essential roles in the evolution of lineage-specific phenotype and adaptation innovation. The origin of new genes and their novel functions have been considered as an important source of genetic innovation, and have attracted the attentions of evolutionary biologists for quite some time. Numerous novel genes have been predicted by bioinformatic analyses, such as the identification of 308 new genes in different Drosophila species [57] and 75 de novo genes in mice and rats [58]. Until now, only a few new genes have been extensively studied with solid experimental evidence, mainly in Drosophila[59–66], yeast [67] and nematodes [68]. Among these new genes, the Drosophila nsr gene [59], CG11700 gene [60] and Zeus gene [62] are reported to be primarily expressed in male reproductive tissues and have effects on male fecundity. In addition, sphinx has been proven to be responsible for male courtship behavior [63, 64]. Furthermore, the Umbrea gene [65], p24-2 gene [66] and eud-1 gene [68] are essential for organismal development. These functional experiments indicate that new genes may play significant roles in important biological processes or phenotypes. Although the important functions on new genes have been revealed in some cases, a systemic experimental testing of new gene function is still lacking. Therefore, the relevant functional information of a large number of genes, including new genes, are still to be explored. Large-scale functional studies on new genes by RNAi are reported, providing candidates with important functions for further study using complementary techniques such as the mutagenesis approaches discussed in this review [69]. Furthermore, new genes are phylogenetic [70] and species-specific findings and follow-up conclusions in one system need to be tested in additional organisms. Another important genetic innovation is non-coding elements. Less than 2% of a genome sequence encodes protein [71], while the remaining genome sequence was initially thought to be non-functional. However, a growing number of non-coding transcripts have functional roles in gene regulation, such as siRNA [72] and long non-coding RNAs [73]. Efforts have been made to annotate the non-coding regions of the genome [74], and in a recent study of 29 mammalian genomes, 3.5% of the non-coding regions were shown to be under purifying selection, indicating possible regulatory roles in the genomes [75]. Although many lineage-specific highly conserved elements on non-coding regions have been proven to have essential regulatory functions in development, a greater number of non-coding regions are in need of functional verification.
More importantly, with the advantages of genome sequencing technology, large batches of genome information of non-model organisms with evolutionary and/or ecological importance will be available which may provide new gene resources with potential impact during diverse evolutionary processes. Scientists are already initiating ambitious large-scale genome sequencing projects, such as the Genome 10 K Project (G10K), 5,000 Insect Genome Project (i5K ) and Bird 10 K project (B10K ), to name a few. Large scale data obtained from those sequencing projects make it possible to discover genetic innovations [76, 77]. Efficient and high-throughput functional testing on candidate genes will be urgently demanded, and new knock-out technologies, such as TALEN and CRISPR/Cas9, will be able to shed light in these situations. Genome-editing technologies have also been successfully used in various model and non-model organisms with no genome limitations and high efficiency. By using these technologies, large-scale high-throughput gene knock-out and screen experiments have been achieved in human cells [78, 79]. The predicted new genes or constrained non-coding elements could be precisely modified one-by-one to verify their functional roles, meanwhile, more related genes involved in the same pathways with a new gene could be modified at the same time. Consequently, this strategy would help to understand the functional roles and, accelerate the study of new genes, as well as extend research to different species.
Testing the functions of candidate domesticated genes
Humans have domesticated hundreds of plants and animals species as sources of food and materials over the past 12,000 years [80–84]. Domestication is an evolutionary process driven by artificial selection, and the underlying mechanisms are still unclear. Research on domestication has not only been curiosity driven, but also driven by cultural and economic importance. Given the advantages and uptake of new sequencing technologies, genome and genomic polymorphic data of increasing numbers of domesticated species have been made publicly available. By comparing the genomes of domesticated and wild species, numerous candidate genes are predicted to be involved in the domestication process. Xu reported 73 candidate domestication genes in both japonica and indica rice [85]. There are also 516 reported candidate domestication genes for pigs [86] and 354 for silkworm [87]. However, a great obstacle lies between this invaluable gene information and functional certainty. Traditional genetic modification technology cannot handle large numbers of genes in such a short period of time, and TALEN and CRISPR/Cas9 methods may provide a bridge to overcome this. Taking rice as an example, TALEN and CRISPR/Cas9 could precisely knock out genes [88, 89], and also mediate the epigenetic status of genes [44]. Additionally, two or more genes could be modified at the same time [33]. Besides rice, more and more domesticated species genomes have been modified or are undergoing experiments through TALEN and CRISPR/Cas9, such as maize [90] silkworm [47], pig [91, 92] goat and cattle [93]. This technology foresees a large-scale genetic modification platform, which would tremendously promote domestication research, accompanied with high-throughput sequencing and analyses platforms. By combining both progressive platforms, important genes or economic-trait-related genes would be discovered and identified much more easily and quickly. Furthermore, it gives us a powerful tool to unveil the mechanism of artificial selection, and shortens the time period when precious economic-trait related genes can be transformed for agriculture and industrial productions.
Testing the role of candidate genes in life-environment interaction
Previous Genome-Wide Association Studies (GWAS), Quantitative Trait Locus (QTL) and related studies have revealed a mass of candidate genes corresponding to phenotypic changes and ecological adaptations [94]. However, many of the SNPs and candidate genes identified by GWAS are reported to be false positives. Meanwhile, the QTL mapping is often inaccurate, resulting in too many candidate genes. Therefore, it has been difficult and time-consuming to confirm the actual trait related genes from numerous candidates. But now, the TALEN and CRISPR/Cas9 systems makes this possible. The application of genome engineering technologies in non-model organisms can move our understanding of ecological adaptation much deeper by experimentally testing the functional effects of these genes. Previous adaptation studies have focused on model organisms which are often short-lived, weedy or commensal. Nowadays, next-generation sequencing (NGS) platforms now are providing sufficient data for non-model organisms, some of which are ideal ecological study models with their main traits involved in adaption, such as heliconius melpomene[95], oyster [96], Coregonus spp., and Salmonidae[97]. Numerous candidate genes underlying adaption have been predicted though bioinformatics-based approaches, such GWAS, QTLs analysis and population genetic studies [98–101]. TALEN and CRISPR/Cas9 systems could mediate candidate genes in non-model species efficiently, which provide the final proof of ecological importance for candidate genes of interest. One drawback is that adaptation-related traits are polygenic quantitative traits in most cases, meaning that a series of genes may be involved in the process. Modifying just one gene would not be efficient enough to verify the related phenotype. To get around this TALEN and CRISPR/Cas9 can mediate two or more genes at the same time [102], being more convenient and efficient than traditional technologies.
Resources for TALEN and CRISPR/Cas9 design and services
Efficient TALEN and CRISPR/Cas9 online design tools and services are vital for promoting their application. These resources have developed rapidly along with the emergence and development of the technologies themselves. To date, there are more than 38 online prediction software tools and 39 commercial service agencies (incomplete statistics) for TALEN and CRISPR/Cas9. For TALEN, classical software applications include TAL Effector-Nucleotide Targeter [56] and TALE Toolbox [103], both of which may help investigators design TALEN plasmids efficiently. For CRISPR/Cas9, 23 software tools have been released since last year, some of which may deal with multiple model organisms, such as E-CRISP [104] and CRISPR-PLANT [105]. As the demands for high-throughput TALEN and CRISPR/Cas9 design rapidly increase, some tools have been developed for local analysis, such as TAL Effector-Nucleotide Targeter and sgRNAcas9 [106]. We have summarized the accessible tools and service companies in Additional file 1: Table S1 and Table S2, which have also been added to a GitHub wiki [107] so that others are able to update and curate the list.
Future perspectives
TALEN and CRISPR/Cas9 systems are promising accurate genome editing tools, that have the potential to promote biological research. However, there are limitations to both techniques. With regards to TALEN, the plasmid is large, which would affect delivery efficiency to cells, and it is difficult to assemble repeat monomers. With regards to CRISPR/Cas9, the main weakness is the occasional high off-target effects, in special species or gene cases [108]. A few efforts could be made to minimize the impacts for both technologies. For TALENs, developing a new TALEN scaffold would diminish the plasmid size, and different kits have been invented enabling monomer assembly in a short time. For CRISPR/Cas9, firstly, a pair of Cas9 nicking variants that requires cooperatives to generate a DSB would reduce the likelihood of off-target effects [109, 110]. Recently, Cas9 protein and FokI protein have been combined to form a dimeric CRISPR/Cas9 RNA-guided FokI nucleases system, which could be useful in highly accurate genome editing applications [111]. Secondly, a strict screening strategy on the 8–12 seed region of sgRNA would help decrease undesired mutagenesis in an off-target region. Studies have shown that seed region accounts for most of CRISPR/Cas9 specificity [112], and a point mutation in a seed region would abrogate sgRNA:Cas9 recognition [113]. Hence, at least two mismatches of a seed region lying in the off-target sequence would improve targeting specificity [114]. Thirdly, a truncated sgRNA with less than 20 nucleotides complimentary to a target region would dramatically reduce the off-target effects by 5000 fold, without scarifying target efficiency [115].
Conclusion
Applications of genome editing are still in their early stages. Hopefully in the near future, the application and replacement of particular regulation methods can also be successful and in high-throughput manner, making the exploration of gene functions more precise and in-depth. Amazingly, epigenetic regulation of genes by this technology will possibly open a new means in the field of functional epigenetics. By taking advantages of these genome editing systems, we are now able to extend functional mechanistic studies to more research fields. Linked by technology, molecular biologists and ecologists will now be able to better cooperate to explore the interesting and important issues, such as animal social behavior, and mechanisms of biodiversity maintenance.
Abbreviations
- BLAST:
-
Basic Local alignment search tool
- CRISPR:
-
Clustered regularly interspaced short palindromic repeats
- DSB:
-
Double strand breaks
- GWAS:
-
Genome-wide association studies
- HR:
-
Homologous recombination
- NGS:
-
Next generation sequencing
- NHEJ:
-
Error-prone nonhomologous end joining
- PAM:
-
Protospacer adjacent motif
- QTL:
-
Quantitative trait locus
- TALEs:
-
Transcription activator-like effector
- TALEN:
-
Transcription activator-like effector nuclease
- ZFN:
-
Zinc finger nuclease.
References
Martin SE, Caplen NJ: Applications of RNA interference in mammalian systems. Annu Rev Genomics Hum Genet. 2007, 8: 81-108. 10.1146/annurev.genom.8.080706.092424.
Dietzl G, Chen D, Schnorrer F, Su KC, Barinova Y, Fellner M, Gasser B, Kinsey K, Oppel S, Scheiblauer S, Couto A, Marra V, Keleman K, Dickson BJ: A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature. 2007, 448 (7150): 151-156. 10.1038/nature05954.
Gonczy P, Echeverri C, Oegema K, Coulson A, Jones SJM, Copley RR, Duperon J, Oegema J, Brehm M, Cassin E, Hannak E, Kirkham M, Pichler S, Flohrs K, Goessen A, Leidel S, Alleaume AM, Martin C, Ozlu N, Bork P, Hyman AA: Functional genomic analysis of cell division in C. elegans using RNAi of genes on chromosome III. Nature. 2000, 408 (6810): 331-336. 10.1038/35042526.
Kilby NJ, Snaith MR, Murray JA: Site-specific recombinases: tools for genome engineering. Trends in Genet: TIG. 1993, 9 (12): 413-421. 10.1016/0168-9525(93)90104-P.
Dymecki SM: Flp recombinase promotes site-specific DNA recombination in embryonic stem cells and transgenic mice. Proc Natl Acad Sci U S A. 1996, 93 (12): 6191-6196. 10.1073/pnas.93.12.6191.
Groth AC, Fish M, Nusse R, Calos MP: Construction of transgenic Drosophila by using the site-specific integrase from phage φC31. Genetics. 2004, 166 (4): 1775-1782. 10.1534/genetics.166.4.1775.
Venken KJ, Bellen HJ: Transgenesis upgrades for Drosophila melanogaster. Development. 2007, 134 (20): 3571-3584. 10.1242/dev.005686.
Capecchi MR: Gene targeting in mice: functional analysis of the mammalian genome for the twenty-first century. Nat Rev Genet. 2005, 6 (6): 507-512. 10.1038/nrg1619.
Bibikova M, Beumer K, Trautman JK, Carroll D: Enhancing gene targeting with designed zinc finger nucleases. Science. 2003, 300 (5620): 764-10.1126/science.1079512.
Durai S, Mani M, Kandavelou K, Wu J, Porteus MH, Chandrasegaran S: Zinc finger nucleases: custom-designed molecular scissors for genome engineering of plant and mammalian cells. Nucleic Acids Res. 2005, 33 (18): 5978-5990. 10.1093/nar/gki912.
Bogdanove AJ, Voytas DF: TAL effectors: customizable proteins for DNA targeting. Science. 2011, 333 (6051): 1843-1846. 10.1126/science.1204094.
Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F: Multiplex genome engineering using CRISPR/Cas systems. Science. 2013, 339 (6121): 819-823. 10.1126/science.1231143.
Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM: RNA-guided human genome engineering via Cas9. Science. 2013, 339 (6121): 823-826. 10.1126/science.1232033.
Gonzalez B, Schwimmer LJ, Fuller RP, Ye Y, Asawapornmongkol L, Barbas CF: Modular system for the construction of zinc-finger libraries and proteins. Nat Protocols. 2010, 5 (4): 791-810. 10.1038/nprot.2010.34.
Kim S, Lee MJ, Kim H, Kang M, Kim JS: Preassembled zinc-finger arrays for rapid construction of ZFNs. Nat Methods. 2011, 8 (1): 7.
Li T, Huang S, Zhao X, Wright DA, Carpenter S, Spalding MH, Weeks DP, Yang B: Modularly assembled designer TAL effector nucleases for targeted gene knockout and gene replacement in eukaryotes. Nucleic Acids Res. 2011, 39 (14): 6315-6325. 10.1093/nar/gkr188.
Miller JC, Tan S, Qiao G, Barlow KA, Wang J, Xia DF, Meng X, Paschon DE, Leung E, Hinkley SJ, Dulay GP, Hua KL, Ankoudinova I, Cost GJ, Urnov FD, Zhang HS, Holmes MC, Zhang L, Gregory PD, Rebar EJ: A TALE nuclease architecture for efficient genome editing. Nat Biotechnol. 2011, 29 (2): 143-148. 10.1038/nbt.1755.
Reyon D, Tsai SQ, Khayter C, Foden JA, Sander JD, Joung JK: FLASH assembly of TALENs for high-throughput genome editing. Nat Biotechnol. 2012, 30 (5): 460-465. 10.1038/nbt.2170.
Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F: Genome engineering using the CRISPR-Cas9 system. Nat Protocols. 2013, 8 (11): 2281-2308. 10.1038/nprot.2013.143.
Dahlem TJ, Hoshijima K, Jurynec MJ, Gunther D, Starker CG, Locke AS, Weis AM, Voytas DF, Grunwald DJ: Simple methods for generating and detecting locus-specific mutations induced with TALENs in the zebrafish genome. PLoS Genet. 2012, 8 (8): e1002861-10.1371/journal.pgen.1002861.
Hwang WY, Fu YF, Reyon D, Maeder ML, Tsai SQ, Sander JD, Peterson RT, Yeh JRJ, Joung JK: Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat Biotechnol. 2013, 31 (3): 227-229. 10.1038/nbt.2501.
Sander JD, Joung JK: CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol. 2014, 32 (4): 347-355. 10.1038/nbt.2842.
Gaj T, Gersbach CA, Barbas CF: ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013, 31 (7): 397-405. 10.1016/j.tibtech.2013.04.004.
Belhaj K, Chaparro-Garcia A, Kamoun S, Nekrasov V: Plant genome editing made easy: targeted mutagenesis in model and crop plants using the CRISPR/Cas system. Plant Methods. 2013, 9 (1): 39-10.1186/1746-4811-9-39.
Sakuma T, Woltjen K: Nuclease-mediated genome editing: At the front-line of functional genomics technology. Dev Growth Differ. 2014, 56 (1): 2-13. 10.1111/dgd.12111.
Hefferin ML, Tomkinson AE: Mechanism of DNA double-strand break repair by non-homologous end joining. DNA Repair. 2005, 4 (6): 639-648. 10.1016/j.dnarep.2004.12.005.
van den Bosch M, Lohman PH, Pastink A: DNA double-strand break repair by homologous recombination. Biol Chem. 2002, 383 (6): 873-892.
Pabo CO, Peisach E, Grant RA: Design and selection of novel Cys2His2 zinc finger proteins. Annu Rev Biochem. 2001, 70: 313-340. 10.1146/annurev.biochem.70.1.313.
Mak AN, Bradley P, Cernadas RA, Bogdanove AJ, Stoddard BL: The crystal structure of TAL effector PthXo1 bound to its DNA target. Science. 2012, 335 (6069): 716-719. 10.1126/science.1216211.
Nishimasu H, Ran FA, Hsu PD, Konermann S, Shehata SI, Dohmae N, Ishitani R, Zhang F, Nureki O: Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell. 2014, 156 (5): 935-949. 10.1016/j.cell.2014.02.001.
Cho SW, Kim S, Kim JM, Kim JS: Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat Biotechnol. 2013, 31 (3): 230-232. 10.1038/nbt.2507.
Bassett AR, Tibbit C, Ponting CP, Liu JL: Highly efficient targeted mutagenesis of Drosophila with the CRISPR/Cas9 system. Cell reports. 2013, 4 (1): 220-228. 10.1016/j.celrep.2013.06.020.
Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, Jaenisch R: One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 2013, 153 (4): 910-918. 10.1016/j.cell.2013.04.025.
Liu H, Chen Y, Niu Y, Zhang K, Kang Y, Ge W, Liu X, Zhao E, Wang C, Lin S, Jing B, Si C, Lin Q, Chen X, Lin H, Pu X, Wang Y, Qin B, Wang F, Wang H, Si W, Zhou J, Tan T, Li T, Ji S, Xue Z, Luo Y, Cheng L, Zhou Q, Li S: TALEN-mediated gene mutagenesis in rhesus and cynomolgus monkeys. Cell Stem Cell. 2014, 14 (3): 323-328. 10.1016/j.stem.2014.01.018.
Niu Y, Shen B, Cui Y, Chen Y, Wang J, Wang L, Kang Y, Zhao X, Si W, Li W, Xiang AP, Zhou J, Guo X, Bi Y, Si C, Hu B, Dong G, Wang H, Zhou Z, Li T, Tan T, Pu X, Wang F, Ji S, Zhou Q, Huang X, Ji W, Sha J: Generation of gene-modified cynomolgus monkey via Cas9/RNA-mediated gene targeting in one-cell embryos. Cell. 2014, 156 (4): 836-843. 10.1016/j.cell.2014.01.027.
Lei Y, Guo X, Deng Y, Chen Y, Zhao H: Generation of gene disruptions by transcription activator-like effector nucleases (TALENs) in Xenopus tropicalis embryos. Cell & Biosci. 2013, 3 (1): 21-10.1186/2045-3701-3-21.
Jiang W, Zhou H, Bi H, Fromm M, Yang B, Weeks DP: Demonstration of CRISPR/Cas9/sgRNA-mediated targeted gene modification in Arabidopsis, tobacco, sorghum and rice. Nucleic Acids Res. 2013, 41 (20): e188-10.1093/nar/gkt780.
Yang H, Wang H, Shivalila CS, Cheng AW, Shi L, Jaenisch R: One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell. 2013, 154 (6): 1370-1379. 10.1016/j.cell.2013.08.022.
Auer TO, Duroure K, De Cian A, Concordet JP, Del Bene F: Highly efficient CRISPR/Cas9-mediated knock-in in zebrafish by homology-independent DNA repair. Genome Res. 2014, 24 (1): 142-153. 10.1101/gr.161638.113.
Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, Stern-Ginossar N, Brandman O, Whitehead EH, Doudna JA, Lim WA, Weissman JS, Qi LS: CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013, 154 (2): 442-451. 10.1016/j.cell.2013.06.044.
Mali P, Aach J, Stranges PB, Esvelt KM, Moosburner M, Kosuri S, Yang L, Church GM: CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat Biotechnol. 2013, 31 (9): 833-838. 10.1038/nbt.2675.
Crocker J, Stern DL: TALE-mediated modulation of transcriptional enhancers in vivo. Nat Methods. 2013, 10 (8): 762-767. 10.1038/nmeth.2543.
Konermann S, Brigham MD, Trevino AE, Hsu PD, Heidenreich M, Cong L, Platt RJ, Scott DA, Church GM, Zhang F: Optical control of mammalian endogenous transcription and epigenetic states. Nature. 2013, 500 (7463): 472-476.
Maeder ML, Angstman JF, Richardson ME, Linder SJ, Cascio VM, Tsai SQ, Ho QH, Sander JD, Reyon D, Bernstein BE, Costello JF, Wilkinson MF, Joung JK: Targeted DNA demethylation and activation of endogenous genes using programmable TALE-TET1 fusion proteins. Nat Biotechnol. 2013, 31 (12): 1137-1142. 10.1038/nbt.2726.
Kim EB, Fang X, Fushan AA, Huang Z, Lobanov AV, Han L, Marino SM, Sun X, Turanov AA, Yang P, Yim SH, Zhao X, Kasaikina MV, Stoletzki N, Peng C, Polak P, Xiong Z, Kiezun A, Zhu Y, Chen Y, Kryukov GV, Zhang Q, Peshkin L, Yang L, Bronson RT, Buffenstein R, Wang B, Han C, Li Q, Chen L: Genome sequencing reveals insights into physiology and longevity of the naked mole rat. Nature. 2011, 479 (7372): 223-227. 10.1038/nature10533.
Bonasio R, Zhang G, Ye C, Mutti NS, Fang X, Qin N, Donahue G, Yang P, Li Q, Li C, Zhang P, Huang Z, Berger SL, Reinberg D, Wang J, Liebig J: Genomic comparison of the ants Camponotus floridanus and Harpegnathos saltator. Science. 2010, 329 (5995): 1068-1071. 10.1126/science.1192428.
Wang Y, Li Z, Xu J, Zeng B, Ling L, You L, Chen Y, Huang Y, Tan A: The CRISPR/Cas System mediates efficient genome engineering in Bombyx mori. Cell Res. 2013, 23 (12): 1414-1416. 10.1038/cr.2013.146.
Xu L, Zhao P, Mariano A, Han R: Targeted myostatin gene editing in multiple mammalian species directed by a single pair of TALE nucleases. Molecular therapy Nucleic acids. 2013, 2: e112-10.1038/mtna.2013.39.
Sun Z, Li N, Huang G, Xu J, Pan Y, Wang Z, Tang Q, Song M, Wang X: Site-specific gene targeting using transcription activator-like effector (TALE)-based nuclease in Brassica oleracea. J Integr Plant Biol. 2013, 55 (11): 1092-1103. 10.1111/jipb.12091.
Smidler AL, Terenzi O, Soichot J, Levashina EA, Marois E: Targeted mutagenesis in the malaria mosquito using TALE nucleases. PLoS One. 2013, 8 (8): e74511-10.1371/journal.pone.0074511.
Aryan A, Anderson MA, Myles KM, Adelman ZN: TALEN-based gene disruption in the dengue vector Aedes aegypti. PLoS One. 2013, 8 (3): e60082-10.1371/journal.pone.0060082.
Ansai S, Sakuma T, Yamamoto T, Ariga H, Uemura N, Takahashi R, Kinoshita M: Efficient targeted mutagenesis in medaka using custom-designed transcription activator-like effector nucleases. Genetics. 2013, 193 (3): 739-749. 10.1534/genetics.112.147645.
Sugano SS, Shirakawa M, Takagi J, Matsuda Y, Shimada T, Hara-Nishimura I, Kohchi T: CRISPR/Cas9-mediated targeted mutagenesis in the liverwort Marchantia polymorpha L. Plant Cell Physiol. 2014, 55 (3): 475-481. 10.1093/pcp/pcu014.
Upadhyay SK, Kumar J, Alok A, Tuli R: RNA-Guided Genome Editing for Target Gene Mutations in Wheat. G3 (Bethesda). 2013, 3 (12): 2233-2238. 2013.
Wang Y, Cheng X, Shan Q, Zhang Y, Liu J, Gao C, Qiu JL: Simultaneous editing of three homoeoalleles in hexaploid bread wheat confers heritable resistance to powdery mildew. Nat Biotechnol. 2014, 32 (9): 947-951. 10.1038/nbt.2969.
Doyle EL, Booher NJ, Standage DS, Voytas DF, Brendel VP, Vandyk JK, Bogdanove AJ: TAL Effector-Nucleotide Targeter (TALE-NT) 2.0: tools for TAL effector design and target prediction. Nucleic Acids Res. 2012, 40: W117-W122. 10.1093/nar/gks608. Web Server issue
Zhou Q, Zhang G, Zhang Y, Xu S, Zhao R, Zhan Z, Li X, Ding Y, Yang S, Wang W: On the origin of new genes in Drosophila. Genome Res. 2008, 18 (9): 1446-1455. 10.1101/gr.076588.108.
Murphy DN, McLysaght A: De novo origin of protein-coding genes in murine rodents. PLoS One. 2012, 7 (11): e48650-10.1371/journal.pone.0048650.
Ding Y, Zhao L, Yang SA, Jiang Y, Chen YA, Zhao RP, Zhang Y, Zhang GJ, Dong Y, Yu HJ, Zhou Q, Wang W: A young Drosophila duplicate gene plays essential roles in spermatogenesis by regulating several Y- linked male fertility genes. PLoS Genet. 2010, 6 (12)):
Zhan Z, Ding Y, Zhao R, Zhang Y, Yu H, Zhou Q, Yang S, Xiang H, Wang W: Rapid functional divergence of a newly evolved polyubiquitin gene in Drosophila and its role in the trade-off between male fecundity and lifespan. Mol Biol Evol. 2012, 29 (5): 1407-1416. 10.1093/molbev/msr299.
Rogers RL, Bedford T, Lyons AM, Hartl DL: Adaptive impact of the chimeric gene Quetzalcoatl in Drosophila melanogaster. Proc Natl Acad Sci U S A. 2010, 107 (24): 10943-10948. 10.1073/pnas.1006503107.
Chen S, Ni X, Krinsky BH, Zhang YE, Vibranovski MD, White KP, Long M: Reshaping of global gene expression networks and sex-biased gene expression by integration of a young gene. EMBO J. 2012, 31 (12): 2798-2809. 10.1038/emboj.2012.108.
Wang W, Brunet FG, Nevo E, Long M: Origin of sphinx, a young chimeric RNA gene in Drosophila melanogaster. Proc Natl Acad Sci U S A. 2002, 99 (7): 4448-4453. 10.1073/pnas.072066399.
Dai H, Chen Y, Chen S, Mao Q, Kennedy D, Landback P, Eyre-Walker A, Du W, Long M: The evolution of courtship behaviors through the origination of a new gene in Drosophila. Proc Natl Acad Sci U S A. 2008, 105 (21): 7478-7483. 10.1073/pnas.0800693105.
Ross BD, Rosin L, Thomae AW, Hiatt MA, Vermaak D, de la Cruz AF, Imhof A, Mellone BG, Malik HS: Stepwise evolution of essential centromere function in a Drosophila neogene. Science. 2013, 340 (6137): 1211-1214. 10.1126/science.1234393.
Saleem S, Schwedes CC, Ellis LL, Grady ST, Adams RL, Johnson N, Whittington JR, Carney GE: Drosophila melanogaster p24 trafficking proteins have vital roles in development and reproduction. Mech Dev. 2012, 129 (5–8): 177-191.
Li D, Dong Y, Jiang Y, Jiang H, Cai J, Wang W: A de novo originated gene depresses budding yeast mating pathway and is repressed by the protein encoded by its antisense strand. Cell Res. 2010, 20 (4): 408-420. 10.1038/cr.2010.31.
Ragsdale EJ, Muller MR, Rodelsperger C, Sommer RJ: A developmental switch coupled to the evolution of plasticity acts through a sulfatase. Cell. 2013, 155 (4): 922-933. 10.1016/j.cell.2013.09.054.
Chen S, Zhang YE, Long M: New genes in Drosophila quickly become essential. Science. 2010, 330 (6011): 1682-1685. 10.1126/science.1196380.
Zhao L, Saelao P, Jones CD, Begun DJ: Origin and spread of de novo genes in Drosophila melanogaster populations. Science. 2014, 343 (6172): 769-772. 10.1126/science.1248286.
Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, Lehoczky J, LeVine R, McEwan P, McKernan K, Meldrim J, Mesirov JP, Miranda C, Morris W, Naylor J, Raymond C, Rosetti M, Santos R, Sheridan A, Sougnez C: Initial sequencing and analysis of the human genome. Nature. 2001, 409 (6822): 860-921. 10.1038/35057062.
Ghildiyal M, Zamore PD: Small silencing RNAs: an expanding universe. Nat Rev Genet. 2009, 10 (2): 94-108. 10.1038/nrg2504.
Guttman M, Rinn JL: Modular regulatory principles of large non-coding RNAs. Nature. 2012, 482 (7385): 339-346. 10.1038/nature10887.
Alexander RP, Fang G, Rozowsky J, Snyder M, Gerstein MB: Annotating non-coding regions of the genome. Nat Rev Genet. 2010, 11 (8): 559-571. 10.1038/nrg2814.
Lindblad-Toh K, Garber M, Zuk O, Lin MF, Parker BJ, Washietl S, Kheradpour P, Ernst J, Jordan G, Mauceli E, Ward LD, Lowe CB, Holloway AK, Clamp M, Gnerre S, Alfoldi J, Beal K, Chang J, Clawson H, Cuff J, Di Palma F, Fitzgerald S, Flicek P, Guttman M, Hubisz MJ, Jaffe DB, Jungreis I, Kent WJ, Kostka D, Lara M: A high-resolution map of human evolutionary constraint using 29 mammals. Nature. 2011, 478 (7370): 476-482. 10.1038/nature10530.
Wu DD, Irwin DM, Zhang YP: De novo origin of human protein-coding genes. PLoS Genet. 2011, 7 (11): e1002379-10.1371/journal.pgen.1002379.
Qureshi IA, Mehler MF: Emerging roles of non-coding RNAs in brain evolution, development, plasticity and disease. Nat Rev Neurosci. 2012, 13 (8): 528-541. 10.1038/nrn3234.
Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG, Zhang F: Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014, 343 (6166): 84-87. 10.1126/science.1247005.
Zhou Y, Zhu S, Cai C, Yuan P, Li C, Huang Y, Wei W: High-throughput screening of a CRISPR/Cas9 library for functional genomics in human cells. Nature. 2014, 509 (7501): 487-491. 10.1038/nature13166.
Meyer RS, Purugganan MD: Evolution of crop species: genetics of domestication and diversification. Nat Rev Genet. 2013, 14 (12): 840-852. 10.1038/nrg3605.
Mannion AM: Domestication and the origins of agriculture: an appraisal. Prog Phys Geogr. 1999, 23 (1): 37-56. 10.1177/030913339902300102.
Diamond J: Evolution, consequences and future of plant and animal domestication. Nature. 2002, 418 (6898): 700-707. 10.1038/nature01019.
Brown TA, Jones MK, Powell W, Allaby RG: The complex origins of domesticated crops in the Fertile Crescent. Trends Ecol Evol. 2009, 24 (2): 103-109. 10.1016/j.tree.2008.09.008.
Doebley JF, Gaut BS, Smith BD: The molecular genetics of crop domestication. Cell. 2006, 127 (7): 1309-1321. 10.1016/j.cell.2006.12.006.
Xu X, Liu X, Ge S, Jensen JD, Hu F, Li X, Dong Y, Gutenkunst RN, Fang L, Huang L, Li J, He W, Zhang G, Zheng X, Zhang F, Li Y, Yu C, Kristiansen K, Zhang X, Wang J, Wright M, McCouch S, Nielsen R, Wang J, Wang W: Resequencing 50 accessions of cultivated and wild rice yields markers for identifying agronomically important genes. Nat Biotechnol. 2012, 30 (1): 105-111.
Li M, Tian S, Jin L, Zhou G, Li Y, Zhang Y, Wang T, Yeung CK, Chen L, Ma J, Zhang J, Jiang A, Li J, Zhou C, Zhang J, Liu Y, Sun X, Zhao H, Niu Z, Lou P, Xian L, Shen X, Liu S, Zhang S, Zhang M, Zhu L, Shuai S, Bai L, Tang G, Liu H: Genomic analyses identify distinct patterns of selection in domesticated pigs and Tibetan wild boars. Nat Genet. 2013, 45 (12): 1431-1438. 10.1038/ng.2811.
Xia Q, Guo Y, Zhang Z, Li D, Xuan Z, Li Z, Dai F, Li Y, Cheng D, Li R, Cheng T, Jiang T, Becquet C, Xu X, Liu C, Zha X, Fan W, Lin Y, Shen Y, Jiang L, Jensen J, Hellmann I, Tang S, Zhao P, Xu H, Yu C, Zhang G, Li J, Cao J, Liu S: Complete resequencing of 40 genomes reveals domestication events and genes in silkworm (Bombyx). Science. 2009, 326 (5951): 433-436. 10.1126/science.1176620.
Shan Q, Wang Y, Li J, Zhang Y, Chen K, Liang Z, Zhang K, Liu J, Xi JJ, Qiu JL, Gao C: Targeted genome modification of crop plants using a CRISPR-Cas system. Nat Biotechnol. 2013, 31 (8): 686-688. 10.1038/nbt.2650.
Li T, Liu B, Spalding MH, Weeks DP, Yang B: High-efficiency TALEN-based gene editing produces disease-resistant rice. Nat Biotechnol. 2012, 30 (5): 390-392. 10.1038/nbt.2199.
Liang Z, Zhang K, Chen K, Gao C: Targeted mutagenesis in Zea mays using TALENs and the CRISPR/Cas system. J Genet genomics =Yi chuan xue bao. 2014, 41 (2): 63-68. 10.1016/j.jgg.2013.12.001.
Lillico SG, Proudfoot C, Carlson DF, Stverakova D, Neil C, Blain C, King TJ, Ritchie WA, Tan W, Mileham AJ, McLaren DG, Fahrenkrug SC, Whitelaw CB: Live pigs produced from genome edited zygotes. Sci Rep. 2013, 3: 2847.
Carlson DF, Tan WF, Lillico SG, Stverakova D, Proudfoot C, Christian M, Voytas DF, Long CR, Whitelaw CBA, Fahrenkrug SC: Efficient TALEN-mediated gene knockout in livestock. Proc Natl Acad Sci U S A. 2012, 109 (43): 17382-17387. 10.1073/pnas.1211446109.
Tan WF, Carlson DF, Lancto CA, Garbe JR, Webster DA, Hackett PB, Fahrenkrug SC: Efficient nonmeiotic allele introgression in livestock using custom endonucleases. Proc Natl Acad Sci U S A. 2013, 110 (41): 16526-16531. 10.1073/pnas.1310478110.
Stapley J, Reger J, Feulner PG, Smadja C, Galindo J, Ekblom R, Bennison C, Ball AD, Beckerman AP, Slate J: Adaptation genomics: the next generation. Trends Ecol Evol. 2010, 25 (12): 705-712. 10.1016/j.tree.2010.09.002.
Heliconius Genome C: Butterfly genome reveals promiscuous exchange of mimicry adaptations among species. Nature. 2012, 487 (7405): 94-98.
Zhang G, Fang X, Guo X, Li L, Luo R, Xu F, Yang P, Zhang L, Wang X, Qi H, Xiong Z, Que H, Xie Y, Holland PW, Paps J, Zhu Y, Wu F, Chen Y, Wang J, Peng C, Meng J, Yang L, Liu J, Wen B, Zhang N, Huang Z, Zhu Q, Feng Y, Mount A, Hedgecock D: The oyster genome reveals stress adaptation and complexity of shell formation. Nature. 2012, 490 (7418): 49-54. 10.1038/nature11413.
Jeukens J, Bittner D, Knudsen R, Bernatchez L: Candidate genes and adaptive radiation: insights from transcriptional adaptation to the limnetic niche among coregonine fishes (Coregonus spp., Salmonidae). Mol Biol Evol. 2009, 26 (1): 155-166.
Brachi B, Faure N, Horton M, Flahauw E, Vazquez A, Nordborg M, Bergelson J, Cuguen J, Roux F: Linkage and association mapping of Arabidopsis thaliana flowering time in nature. PLoS Genet. 2010, 6 (5): e1000940-10.1371/journal.pgen.1000940.
Hohenlohe PA, Bassham S, Etter PD, Stiffler N, Johnson EA, Cresko WA: Population genomics of parallel adaptation in threespine stickleback using sequenced RAD tas. PLoS Genet. 2010, 6 (2): e1000862-10.1371/journal.pgen.1000862.
Burke MK, Dunham JP, Shahrestani P, Thornton KR, Rose MR, Long AD: Genome-wide analysis of a long-term evolution experiment with Drosophila. Nature. 2010, 467 (7315): 587-U111. 10.1038/nature09352.
Turner TL, Bourne EC, Von Wettberg EJ, Hu TT, Nuzhdin SV: Population resequencing reveals local adaptation of Arabidopsis lyrata to serpentine soils. Nat Genet. 2010, 42 (3): 260-U242. 10.1038/ng.515.
Jao LE, Wente SR, Chen W: Efficient multiplex biallelic zebrafish genome editing using a CRISPR nuclease system. Proc Natl Acad Sci U S A. 2013, 110 (34): 13904-13909. 10.1073/pnas.1308335110.
Sanjana NE, Cong L, Zhou Y, Cunniff MM, Feng G, Zhang F: A transcription activator-like effector toolbox for genome engineering. Nat Protocols. 2012, 7 (1): 171-192. 10.1038/nprot.2011.431.
Heigwer F, Kerr G, Boutros M: E-CRISP: fast CRISPR target site identification. Nat Methods. 2014, 11 (2): 122-123. 10.1038/nmeth.2812.
Xie K, Zhang J, Yang Y: Genome-wide prediction of highly specific guide RNA spacers for CRISPR-Cas9-mediated genome editing in model plants and major crops. Mol Plant. 2014, 7 (5): 923-926. 10.1093/mp/ssu009.
Xie S, Shen B, Zhang C, Huang X, Zhang Y: sgRNAcas9: a software package for designing CRISPR sgRNA and evaluating potential off-target cleavage sites. PloS one. 2014, 9 (6): e100448-10.1371/journal.pone.0100448.
TALEN & CRISPR web tools and company service wiki. [https://github.com/gigascience/paper-chen2014/wiki]
Pattanayak V, Lin S, Guilinger JP, Ma E, Doudna JA, Liu DR: High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat Biotechnol. 2013, 31 (9): 839-843. 10.1038/nbt.2673.
Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, Scott DA, Inoue A, Matoba S, Zhang Y, Zhang F: Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013, 154 (6): 1380-1389. 10.1016/j.cell.2013.08.021.
Shen B, Zhang W, Zhang J, Zhou J, Wang J, Chen L, Wang L, Hodgkins A, Iyer V, Huang X, Skarnes WC: Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nat Methods. 2014, 11 (4): 399-402. 10.1038/nmeth.2857.
Tsai SQ, Wyvekens N, Khayter C, Foden JA, Thapar V, Reyon D, Goodwin MJ, Aryee MJ, Joung JK: Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat Biotechnol. 2014, 32 (6): 569-576. 10.1038/nbt.2908.
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E: A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012, 337 (6096): 816-821. 10.1126/science.1225829.
Jiang WY, Bikard D, Cox D, Zhang F, Marraffini LA: RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol. 2013, 31 (3): 233-239. 10.1038/nbt.2508.
Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li YQ, Fine EJ, Wu XB, Shalem O, Cradick TJ, Marraffini LA, Bao G, Zhang F: DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013, 31 (9): 827-10.1038/nbt.2647. −+
Fu YF, Sander JD, Reyon D, Cascio VM, Joung JK: Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol. 2014, 32 (3): 279-284. 10.1038/nbt.2808.
Acknowledgements
This work was supported by a 973 program (2013CB835200) to W.W, a key grant of West Light Foundation of the Chinese Academy of Sciences to H.X.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contribution
GJZ, WW raised the idea and designed the main structure of the manuscript. LC and LYT wrote the manuscript. GJZ and H.X. made the major revisions. QYL, LJJ predict the candidate editable region of genomes. All of the authors read the manuscript. All authors read and approved the final manuscript.
Lei Chen, Linyi Tang contributed equally to this work.
Electronic supplementary material
13742_2014_72_MOESM1_ESM.pdf
Additional file 1: Table S1: Online tools for TALEN and CRISPR/Cas9. Collected online tools for TALEN and CRISPR/Cas9 are presented in this table. Updates can be accessed in GitHub [107]. Table S2. Commercial service for TALEN and CRISPR/Cas9. Collected commercial service for TALEN and CRISPR/Cas9 are presented in this table. Updates could can accessed in GitHub [107]. Table S3. Representative applications of genome editing. A summary of the representative applications in different organisms. (PDF 215 KB)
13742_2014_72_MOESM2_ESM.txt
Additional file 2: CRISPR/ Cas9 and TALEN prediction details of coding and promoter regions for 26 organisms. CRISPR/Cas9 and TALEN prediction details of coding and promoter regions for 26 organisms have been presented. Column 1 presents the gene IDs, Column 2 and Column3 presents whether promoter regions would be targeted by CRISPR/Cas9 and TALEN, Column 4 and Column 5 presents whether coding regions would be targeted by CRISPR/Cas9 and TALEN. (TXT 12 MB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Chen, L., Tang, L., Xiang, H. et al. Advances in genome editing technology and its promising application in evolutionary and ecological studies. GigaSci 3, 24 (2014). https://doi.org/10.1186/2047-217X-3-24
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2047-217X-3-24