
Abstract
Decline in circulating sex steroid hormones accompanies several age-associated pathologies which may influence human healthspan. Mitochondria play important roles in biosynthesis of sex steroid hormones, and these hormones can also regulate mitochondrial function. Understanding the cross talk between mitochondria and sex steroid hormones may provide insights into the pathologies associated with aging. The aim of this review is to summarize the current knowledge regarding the interplay between mitochondria and sex steroid hormones during the aging process. The review describes the effect of mitochondria on sex steroid hormone production in the gonads, and then enumerates the contribution of sex steroid hormones on mitochondrial function in hormone responsive cells. Decline in sex steroid hormones and accumulation of mitochondrial damage may create a positive feedback loop that contributes to the progressive degeneration in tissue function during aging. The review further speculates whether regulation between mitochondrial function and sex steroid hormone action can potentially influence healthspan.
Similar content being viewed by others

Review
Introduction
Sex steroid hormones play important roles in maintaining normal reproductive and non-reproductive functions. Because aging is accompanied by decreasing levels of circulating sex steroid hormones (that is, estrogen, progesterone, and testosterone) [1, 2], many researchers and clinicians had previously hypothesized that the decline in these hormones promotes tissue degeneration and age-related pathologies. As a result, hormone replacement therapies have already been used to alleviate age-related symptoms in men and women. However, the efficacy of these treatments remains debatable. Hence, it is important to evaluate the contribution of sex steroid hormones on healthspan.
Mitochondria regulate sex steroid hormone biosynthesis, and sex steroid hormones are also implicated in controlling mitochondrial function. This co-regulation between mitochondria and sex steroid hormone signaling is important for maintaining proper physiology. Disrupting the crosstalk between mitochondrial function and sex steroid hormone action may alter cellular responses and accelerate aging-associated phenotypes. Understanding the relationship between mitochondria and sex steroid hormones may provide insights into the efficacy of using hormone replacement therapies to treat age-related diseases. This review summarizes the current knowledge regarding the crosstalk between mitochondria and sex steroid hormones during aging, and examines whether the interplay between mitochondrial function and sex steroid hormone action can potentially influence healthspan.
Sex steroid hormones and aging
Sex steroid hormone production declines during reproductive aging
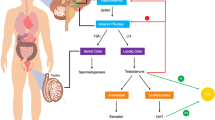
Reproductive aging is defined as the gradual decline in fertility during the aging process. It is accompanied by decreasing levels of circulating sex steroid hormones estrogen, progesterone, and testosterone [1, 2]. In women, the end of reproductive aging occurs when ovarian follicles drop below a threshold (approximately < 1,000 follicles), resulting in cessation of the menstrual cycle which is termed menopause [3]. The stock of non-renewing ovarian follicle reserves begins to decline from birth as a result of continuous maturation, ovulation, and atresia [4]. Ovarian follicles eventually become exhausted by the age of 51, which corresponds to the median age of menopause in the general population [5]. Because granulosa cells and theca cells of the ovarian follicles are the major sources of sex steroid biosynthesis in women, reproductive aging eventually leads to reduced estrogen and progesterone production [6], although the drop in progesterone levels surpasses the drop in estrogen levels during the menopausal transition [7].
In men, reproductive aging is accompanied by a decline in the ability of the testes to produce testosterone [8]. Leydig cells of the testes provide a significant source of testosterone in males. Reduced number of Leydig cells and decreased ability to respond to intracellular signaling during aging may contribute to low testosterone levels in aging men [9–11]. Testosterone production gradually decreases by approximately 1%–3% per year, beginning around the age of 35–40 years [12], and can eventually result in hypogonadism (<325 ng/dL of total serum testosterone) in older men. Partial androgen deficiency (sometimes referred as andropause) occurs in ~20% of men by age 60 and ~50% of men by age 80 [13].
Sex steroid hormone replacement may extend healthspan but not lifespan
The decline in sex steroid hormones with age is thought to reduce healthspan. In women, menopause correlates with several age-related diseases, such as hot flashes, bone loss, weight gain, vaginal atrophy, night sweats, sleep problems, mood changes, and skin atrophy [14, 15]. Estrogen replacement alleviates some of these menopausal symptoms, resulting in lowered frequency and severity of hot flashes, decreased bone loss and vertebral fractures, and increased bone mass [16]. Estrogen can also provide many beneficial effects to skin physiology and protects against skin aging [17]. In female mice, loss of sex steroid hormones by ovariectomy also presents numerous age-related diseases similar to those found in humans, such as accelerated bone loss, decreased uterine weight, increased fat mass, and impaired wound healing, which can all be alleviated by estrogen replacement [18–20].
Because estrogen increases the risk of endometrial cancers to women, progesterone supplements were added to estrogen replacement therapies to inhibit the proliferative effects of estrogen in the uterus. Estrogen plus progesterone replacement therapy provides many beneficial effects similar to those of estrogen alone treatment, but it also seems to increase the risk for other types of diseases, such as breast cancer [16]. Findings from the Women’s Health Initiative show a high risk-to-benefit ratio of using hormone replacement therapies. However, criticisms of the regimens used suggest that health status or age at initiation of treatment may contribute to severity of the side effects associated with hormone replacement therapy [21, 22]. Analysis of the current Kronos Early Estrogen Prevention Study (KEEPS) [23] and the Early versus Late Intervention Trial with Estradiol (ELITE) will hopefully address whether hormone replacement therapy is beneficial within a critical window of time [24, 25].
In men, hypogonadism at old age is correlated with age-related symptoms, such as impotence, decreased virility, decreased libido, and limited physical vigor [26]. However, it remains debatable whether these age-related phenotypes are mainly due to the decline in testosterone levels. In fact, while testosterone replacement can alleviate some of the aging characteristics associated with partial androgen deficiency [27–31], testosterone supplementation in aged men does not always seem to improve physical function [32]. Moreover, increased skeletal muscle mass due to testosterone does not seem to be specific to aged men [33]. In male mice, testosterone can increase skeletal muscle regeneration [34], but it may also accelerate aging phenotypes in other tissues, such as heart and kidneys [35]. Hence, the overall impact of testosterone on healthspan needs further investigation. The current Testosterone Trial will hopefully address the benefit and risk of testosterone replacement in hypogonadal men above the age of 65.
While sex steroid hormones may potentially extend healthspan, the effect of sex steroid hormones on lifespan is less clear. For example, while increasing age at menopause in women is associated with an overall increased lifespan [36], menopausal age does not seem to predict lifespan in non-human primates, as chimpanzees rarely reach menopause whether in captivity or in the wild [37, 38]. Moreover, the length of postmenopausal years (that is, period of reduced sex steroid hormones) does not seem to be proportionally correlated with age at death [39]. In males, castration in rats and dogs increases lifespan [40, 41]. However, while some studies show that castration in humans (that is, eunuchs) extends lifespan [42, 43], another report mentions no change in lifespan [44]. Nevertheless, several studies have linked reduced sex steroid hormones to many age-related diseases. Hence, while loss of sex steroid hormones may not significantly influence longevity and lifespan, reduced sex steroid hormones can potentially shorten overall healthspan.
Mitochondria and sex steroid hormone biosynthesis
Sex steroid hormone biogenesis
Mitochondria play an essential role during the initial steps of sex steroid hormone biosynthesis, particularly by producing the sex steroid hormone precursor pregnenolone. Import of cholesterol from the outer to the inner mitochondrial membrane is a rate limiting step during the initial biosynthesis of sex steroid hormones [45]. Cholesterol transport involves interaction between the steroidogenic acute regulatory protein (StAR) and a multi-component molecular complex, which is composed of an 18 kDa translocator protein (TSPO), the voltage dependent anion channel, TSPO-associated protein 7, and protein kinase A subunit 1a [46]. Once imported, cholesterol is converted to pregnenolone by the cytochrome P450 side-chain cleavage (P450scc; or CYP11A1) enzyme, located on the inner membrane of the mitochondria, involving the oxidation of nicotinamide adenine dinucleotide phosphate (NADPH) [47]. Pregnenolone can then be exported out of the mitochondria and converted by specific microsomal P450 enzymes into the different sex steroid hormones [48].
In males, testosterone biogenesis mostly occurs in Leydig cells through the sequential conversion of pregnenolone to 17α-hydroxypregnenolone, dehydroepiandrosterone (DHEA), androstenedione, and testosterone [47]. In females, however, estrogen and progesterone biosynthesis occurs through the crosstalk between the granulosa and theca cells of the ovarian follicle. Pregnenolone in granulosa cells can diffuse to theca cells and be converted into androstenedione, which then re-diffuses back to granulosa cells and is eventually processed to estrogen. In some instances, androstenedione produced by the theca cells can also be immediately converted to testosterone in these cells. During the luteal phase of the menstrual cycle, progesterone is produced in luteinized granulosa cells by direct conversion from pregnenolone.
Mitochondrial damage may promote menopause and partial androgen deficiency
Mitochondria can produce reactive oxygen species (ROS) at multiple sites of the electron transport chain [49]. During mitochondrial respiration, the electron transport chain generates a flux of electrons, capable of establishing a proton gradient within the mitochondria. While this proton gradient is necessary for ATP production, electrons produced during oxidative phosphorylation need to be completely neutralized to water upon reaction with oxygen at complex IV (that is, cytochrome c oxidase). However, partial reduction of oxygen can occur upstream of complex IV, resulting in production of superoxides. These superoxides are normally eliminated by antioxidants (that is, superoxide dismutases, SOD) in the mitochondria. However, in certain conditions, some superoxides are thought to escape the mitochondrial antioxidant system and cause damage to mitochondrial proteins and DNA. Damaged mitochondria become less efficient in transferring electrons across the respiratory chain, rendering more electrons to be converted to superoxides. This positive feedback loop of mitochondrial superoxide (ROS) production and mitochondrial damage was proposed to cause aging, a theory known as the free radical theory of aging [50, 51]. Consistent with this theory, damage to mitochondrial DNA, lipids, and proteins increases with age, and is accompanied by decreased mitochondrial membrane potential and impaired mitochondrial function [52, 53].
In women, drop in sex steroid hormones during menopause is mainly attributed to loss of ovarian follicles. Continuous ovulation and follicular atresia, plus inability of follicles to naturally regenerate, eventually lead to reduced sex steroid hormone production. Aside from this progressive decline in follicle numbers, it is suggested that excessive oxidative damage in the ovaries, due to a decrease in the levels of antioxidants, may be a potential contributing factor for reproductive aging [54]. While it is yet to be shown whether mitochondrial ROS generation promotes mitochondrial damage in ovarian follicles, increased number of damaged mitochondria have been observed in ovarian follicles of aged women [55, 56] and the proportion of granulosa cells with ruptured mitochondrial membranes significantly increase with age [55]. While human studies only reveal correlations between mitochondrial damage and loss of ovarian follicles, mouse studies show that mitochondrial damage can contribute to reproductive aging. Mitochondrial DNA damage in a mouse model with defective mitochondrial DNA polymerase can reduce female fertility [57]. Mitochondrial dysfunction in a mouse model with high mitochondrial ROS generation also results in infertility, defective folliculogenesis, and impaired ovulation [58]. Hence, mitochondrial damage may potentially accelerate the decline in ovarian follicles and partly contribute to reproductive aging in females.
In men, drop of testosterone levels during aging is associated with decreasing supply of mitochondrial steroid precursors by Leydig cells [9, 10]. In male rats, testosterone levels have also been shown to decline with age [59]. This age-related decline in testosterone steroidogenesis is consistent with decreased mitochondrial expression of StAR and CYP11A1 in Leydig cells of old versus young rats [60]. While the cause of this decline in StAR and CYP11A1 expression is still unclear, it has been proposed that ROS may contribute to this effect. Increased mitochondrial ROS was observed in Leydig cells of old versus young rats [61]. Transient elevation of ROS production after luteinizing hormone treatment caused more DNA damage in aged versus young Leydig cells [62]. Moreover, ROS (that is, H2O2) can inhibit testosterone biosynthesis in the mouse Leydig cell line MA-10 and a primary rat Leydig cell line [63, 64]. But because the concentration used is relatively high (100 to 250 μM), the physiological relevance of this ROS-inhibition to steroidogenesis remains questionable. Nevertheless, while the biological impact of mitochondrial ROS on steroidogenesis needs further validation, the role of mitochondria in producing steroid precursors suggests that properly functioning mitochondria are important to maintain sex steroid hormone levels. Hence, there is a need to critically evaluate the contribution of mitochondrial damage on sex steroid hormone production during aging.
Sex steroid hormones and mitochondrial function
Sex steroid hormone nuclear receptors regulate mitochondrial gene expression
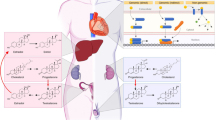
While mitochondria mediate sex steroid hormone production, sex steroid hormones can also regulate mitochondrial function. Sex steroid hormones estrogen, progesterone, and testosterone classically function by binding to their nuclear receptors: estrogen receptors (ERα and ERβ1-6), progesterone receptors (PR-A and PR-B), and androgen receptors (AR1 and AR2), respectively. These receptors along with specific nuclear receptor co-regulators can directly or indirectly bind to nuclear DNA regulatory elements and influence gene expression [65]. While the direct contributions of testosterone and progesterone on mitochondrial function are less studied, the effect of estrogen on mitochondrial function and biosynthesis is more evident. Estrogen, through its receptor, can directly modulate expression of genes important for mitochondrial function (Figure 1). Estrogen can directly up-regulate transcription of nuclear respiratory factor-1 (NRF1), a key transcription factor necessary for regulating expression of most of the mitochondrial respiratory chain complex proteins [66]. NRF1 promoter contains putative estrogen receptor response elements capable of binding both ERs [66, 67]. ERα can also interact with peroxisome proliferator-activated receptor gamma coactivator 1 (PGC-1), another important transcription factor that promotes transcription of NRFs and other mitochondrial proteins [68]. Regulation of NRFs and PGC-1 by estrogen is thought to play an important role in modulating overall mitochondrial biogenesis and function [69].

Molecular targets of estrogen in regulating mitochondrial function. 1. Estrogen binds to nuclear estrogen receptors (ERα and ERβ) to directly regulate tissue-specific expression of genes necessary for cellular function. 2. Estrogen binds to nuclear ERs and regulates expression of mitochondrial genes by promoting gene expression of transcription factor NRF1 and enhancing transcriptional activity of PGC1α. 3. Estrogen binds to membrane ERs and activates signaling cascades that protect mitochondria from oxidative damage. 4. Estrogen binds to mitochondrial ERs and limits ROS generation in the mitochondria. 5. Estrogen binds to mitochondrial ERs and regulates transcription of mitochondrial-encoded mitochondrial genes.
Sex steroid hormone membrane receptors protect against mitochondrial oxidative damage
Aside from the function of sex steroid hormones in the nucleus, sex steroid hormones can also have rapid, nongenomic actions by activating plasma membrane-associated receptors, leading to intracellular protein kinase-mediated phosphorylation signaling cascades [70]. While studies regarding the contribution of plasma membrane-associated receptors on mitochondrial function are very limited, some reports suggest a role for this signaling in mitochondria. Estrogen, by acting on plasma membrane-associated ER, is thought to trigger a phosphorylation cascade and limit mitochondrial oxidative damage (Figure 1). One of the early studies suggesting this phenomenon was when a BSA-conjugated form of estrogen, which is membrane-impermeable, was able to undergo rapid internalization and translocation into mitochondria but not the nucleus [71, 72]. Estrogen, acting through intracellular phosphorylation cascades, may protect mitochondria from oxidative damage. One study shows that estrogen can activate the MAP kinase and NF-kB pathways to reduce cellular levels of hydrogen peroxides by stimulating the nuclear transcription of mitochondrial antioxidant enzyme Sod2[73]. However, it remains to be confirmed whether this estrogen-mediated Sod2 mRNA regulation is necessary to protect against mitochondrial oxidative damage. Another study also shows that estrogen can activate ERK, permit its translocation to the mitochondria, and enhance cytochrome c oxidase complex IV activity [74]. Whether these estrogen actions are due to plasma membrane-associated receptors and whether they play a significant role in protecting mitochondria against oxidative damage needs further investigation.
Mitochondria-associated sex steroid hormone receptors protect against mitochondrial oxidative damage
Sex steroid hormone receptors, particularly ERs, have been observed to localize in mitochondria and contribute to mitochondrial function (Figure 1). Despite the controversy regarding mitochondrial localization of ERs [75, 76], accumulating evidence suggest that both ERα and ERβ are indeed present in the mitochondria, depending on cell type [77–80]. ERβ seems to be the ER that is more frequently present in mitochondria of most cell types [81–85]. Knockdown of the ERβ1 isoform, which predominantly localizes to mitochondria, eliminates estrogen-dependent protection against peroxide-induced mitochondrial membrane depolarization [86]. Using a targeting vector containing mitochondrial or nuclear localization sequences, ER can be efficiently targeted to mitochondria or nucleus [84]. Expression of a mitochondrial-targeted, but not a nuclear-targeted ER, confers estrogen-dependent inhibition of UV-induced mitochondrial depolarization in a breast cancer cell line MCF-7 by enhancing mitochondrial SOD2 protein activity, independent of its transcriptional regulation [84]. However, the mechanism of ER in regulating SOD2 protein activity remains unclear and needs further confirmation.
Sex steroid hormone receptors in the mitochondria can regulate transcription of mitochondrial encoded genes [87]. Mitochondrial DNA contains hormone response elements, which allow binding of steroid hormone receptors. Indeed, ER in the mitochondria can bind to estrogen response elements (ERE) located in mitochondrial DNA (Figure 1) [88]. This estrogen-mediated ER binding to mitochondrial DNA is thought to increase expression of mitochondrial-encoded mitochondrial genes associated with the electron transport chain [89]. Aside from regulating transcription of mitochondrial encoded genes, sex steroid hormone receptors have been suggested to bind to mitochondrial proteins. For example, ERβ has been shown to co-immunoprecipitate with the mitochondrial protein ATP synthase [90]. However, whether this interaction is functional remains to be addressed.
Hormone replacements improve mitochondrial function in healthy cells
Estrogen can inhibit mitochondrial ROS generation in primary cells, such as endothelial cells, cardiomyocytes, and epithelial lens cells [86, 91–93]. Animal experiments also demonstrate that estrogen can reduce mitochondrial ROS production and enhance mitochondrial respiration in normal brains of male and female rats [94, 95]. Estrogen seems to limit mitochondrial ROS production in cell types, which are predominantly expressing ERβ, but a few studies have also supported a protective role of ERα [96].
While estrogen replacement seems to provide overall beneficial effects on mitochondrial function, timing of treatment and type of cell may be important for this phenomenon. Estrogen can decrease mitochondrial ROS production in non-cancer primary cells, but it can also increase ROS production in damaged estrogen-responsive cancer cells [97–99]. This suggests that while estrogen can protect normal cells from oxidative stress, it exacerbates oxidative stress in damaged cells, that is, cancer cells. While it remains unclear what mechanisms regulate this contradictory effect of estrogen on mitochondrial ROS production, the contribution of damaged versus healthy cell on estrogen action is consistent with the critical window and healthy cell hypothesis of estrogen replacement therapy [22, 100]. This hypothesis proposes that estrogen replacement is only beneficial if performed at the appropriate time of a woman’s life, before she accumulates a certain threshold of cellular damage. Indeed, estrogen replacement therapies seem to be more beneficial in younger versus older women [25]. This is one of the rationales for the current KEEPS and ELITE studies on estrogen replacement therapies [23]. Results from these studies will hopefully address some of these questions.
The effect of testosterone replacement on mitochondrial function is less well understood. Low levels of testosterone in males seem to be associated with reduced expression of mitochondrial respiratory genes and activity [101]. Orchiectomy in young male mice decreases expression of genes associated with energy metabolism, oxidative phosphorylation, and ubiquinone pathways [102]. AR overexpression in myocytes increases mitochondrial enzyme activities and oxygen consumption [103, 104]. Testosterone therapy potentiates the effect of low-intensity physical training in old male mice by increasing mitochondrial biogenesis, improving mitochondrial quality, and increasing spontaneous physical activity, respiration, muscle mass, and grip strength [105]. However, testosterone has also been shown to reduce mitochondrial function [106]. Whether or not testosterone play a major role in mitochondrial function needs further investigation.
The positive feedback loop of mitochondrial damage
Mitochondria play important roles in the biosynthesis of sex steroid hormones by housing important enzymes for steroidogenesis and by maintaining cells that produce these hormones (Figure 2). Conversely, sex steroid hormones help to maintain mitochondrial function and limit aging phenotypes. According to the free radical theory of aging, mitochondrial ROS produced during cellular respiration can result in mitochondrial damage, which then initiates a positive feedback loop of increased mitochondrial ROS generation and mitochondrial damage with age. Accumulation of damaged mitochondria can compromise steroidogenesis by damaging gonadal cells, resulting in decreased sex steroid hormone levels. Decline in sex steroid hormones exacerbates mitochondrial dysfunction and further promotes mitochondrial damage. This cycle of mitochondrial damage and decline in sex steroid hormone levels can potentially contribute to the progressive degeneration in tissue function during aging.

Mitochondria and sex steroid hormones during aging. Mitochondria produce sex steroid hormones in the gonads through initial conversion of cholesterol to pregnenolone. Sex steroid hormones improve and/or maintain mitochondrial function in hormone responsive cells by regulating: 1. gene expression of nuclear-encoded mitochondrial proteins, 2. gene expression of mitochondrial-encoded mitochondrial proteins, and/or 3. activity of mitochondrial proteins. Increased oxidative stress during aging damages gonadal cells and/or impair steroidogenesis. Decline in sex steroid hormone biosynthesis during aging compromises mitochondrial function in hormone responsive tissues and contribute to age-related pathologies.
While the free radical theory provides a potential model for the cause of aging, several studies have questioned whether accumulation of mitochondrial oxidative damage actually drives the aging process [107, 108]. Some studies support the theory [109, 110], but others suggest that increased mitochondrial ROS production does not decrease lifespan [111], but can even potentially extend lifespan [112]. It is possible that depending on the amount and context, mitochondrial ROS production can provide beneficial or detrimental effects. Excessive mitochondrial ROS may result in cellular damage and aging, but low mitochondrial ROS production may provide a hormetic effect that extends lifespan [113]. Further research needs to be performed to confirm the validity of the theory.
It is also important to note, that while this review focused on the effects of sex steroid hormones on mitochondrial function, sex steroid hormones can also regulate other cell function independent of mitochondria (Figure 1). Moreover, while estrogen can directly and indirectly regulate mitochondrial biogenesis and function, it remains to be confirmed whether testosterone can significantly contribute to mitochondrial function.
Conclusions
Current studies show that mitochondria are important for the initial step of steroidogenesis, and sex steroid hormones (that is, estrogens) are capable of regulating mitochondrial biogenesis and function. Dysregulation of mitochondrial function and sex steroid hormone action may compromise cellular integrity and lead to progressive decline in tissue function. While the positive feedback loop of mitochondrial damage has been suggested to partly contribute to aging and age-related phenotypes, several questions still need to be addressed to validate this theory. Nevertheless, research on this topic may potentially provide insights into extending healthspan in humans.
Abbreviations
- KEEPS:
-
Kronos Early Estrogen Prevention Study
- ELITE:
-
Early versus Late Intervention Trial with Estradiol
- StAR:
-
Steroidogenic acute regulatory protein
- TSPO:
-
Translocator protein
- P450scc:
-
Cytochrome P450 side-chain cleavage
- CYP11A1:
-
Cytochrome P450, family 11, subfamily A, polypeptide 1
- NADPH:
-
Nicotinamide adenine dinucleotide phosphate
- DHEA:
-
Dehydroepiandrosterone
- ROS:
-
Reactive oxygen species
- SOD:
-
Superoxide dismutase
- ER:
-
Estrogen receptor
- PR:
-
Progesterone receptor
- AR:
-
Androgen receptor
- NRF1:
-
Nuclear respiratory factor-1
- PGC-1:
-
Peroxisome proliferator-activated receptor gamma coactivator 1
- ERE:
-
Estrogen response elements.
References
Sherman BM, West JH, Korenman SG: The menopausal transition: analysis of LH, FSH, estradiol, and progesterone concentrations during menstrual cycles of older women. J Clin Endocrinol Metab. 1976, 42: 629-636.
Ferrini RL, Barrett-Connor E: Sex hormones and age: a cross-sectional study of testosterone and estradiol and their bioavailable fractions in community-dwelling men. Am J Epidemiol. 1998, 147: 750-754.
Richardson SJ, Senikas V, Nelson JF: Follicular depletion during the menopausal transition: evidence for accelerated loss and ultimate exhaustion. J Clin Endocrinol Metab. 1987, 65: 1231-1237.
McGee EA, Hsueh AJ: Initial and cyclic recruitment of ovarian follicles. Endocr Rev. 2000, 21: 200-214.
Wallace WH, Kelsey TW: Human ovarian reserve from conception to the menopause. PLoS One. 2010, 5: e8772-
Young JM, McNeilly AS: Theca: the forgotten cell of the ovarian follicle. Reproduction. 2010, 140: 489-504.
Finch CE: The menopause and aging, a comparative perspective. J Steroid Biochem Mol Biol. in press
Veldhuis JD: Aging and hormones of the hypothalamo-pituitary axis: gonadotropic axis in men and somatotropic axes in men and women. Ageing Res Rev. 2008, 7: 189-208.
Neaves WB, Johnson L, Porter JC, Parker CR, Petty CS: Leydig cell numbers, daily sperm production, and serum gonadotropin levels in aging men. J Clin Endocrinol Metab. 1984, 59: 756-763.
Takahashi J, Higashi Y, LaNasa JA, Yoshida K, Winters SJ, Oshima H, Troen P: Studies of the human testis: XVIII: simultaneous measurement of nine intratesticular steroids: evidence for reduced mitochondrial function in testis of elderly men. J Clin Endocrinol Metab. 1983, 56: 1178-1187.
Mulligan T, Iranmanesh A, Veldhuis JD: Pulsatile iv infusion of recombinant human LH in leuprolide-suppressed men unmasks impoverished Leydig-cell secretory responsiveness to midphysiological LH drive in the aging male. J Clin Endocrinol Metab. 2001, 86: 5547-5553.
Feldman HA, Longcope C, Derby CA, Johannes CB, Araujo AB, Coviello AD, Bremner WJ, McKinlay JB: Age trends in the level of serum testosterone and other hormones in middle-aged men: longitudinal results from the Massachusetts male aging study. J Clin Endocrinol Metab. 2002, 87: 589-598.
Harman SM, Metter EJ, Tobin JD, Pearson J, Blackman MR: Longitudinal effects of aging on serum total and free testosterone levels in healthy men: Baltimore longitudinal study of aging. J Clin Endocrinol Metab. 2001, 86: 724-731.
Butler L, Santoro N: The reproductive endocrinology of the menopausal transition. Steroids. 2011, 76: 627-635.
Emmerson E, Hardman MJ: The role of estrogen deficiency in skin ageing and wound healing. Biogerontology. 2012, 13: 3-20.
Endocrine S, Santen RJ, Allred DC, Ardoin SP, Archer DF, Boyd N, Braunstein GD, Burger HG, Colditz GA, Davis SR, Gambacciani M, Gower BA, Henderson VW, Jarjour WN, Karas RH, Kleerekoper M, Lobo RA, Manson JE, Marsden J, Martin KA, Martin L, Pinkerton JV, Rubinow DR, Teede H, Thiboutot DM, Utian WH: Postmenopausal hormone therapy: an endocrine society scientific statement. J Clin Endocrinol Metab. 2010, 95: s1-s66.
Stevenson S, Thornton J: Effect of estrogens on skin aging and the potential role of SERMs. Clin Interv Aging. 2007, 2: 283-297.
Määttä JA, Büki KG, Gu G, Alanne MH, Vääräniemi J, Liljenbäck H, Poutanen M, Härkönen P, Väänänen K: Inactivation of estrogen receptor α in bone-forming cells induces bone loss in female mice. FASEB J. 2013, 27: 478-488.
Campbell L, Emmerson E, Davies F, Gilliver SC, Krust A, Chambon P, Ashcroft GS, Hardman MJ: Estrogen promotes cutaneous wound healing via estrogen receptor beta independent of its antiinflammatory activities. J Exp Med. 2010, 207: 1825-1833.
Lindberg MK, Weihua Z, Andersson N, Movérare S, Gao H, Vidal O, Erlandsson M, Windahl S, Andersson G, Lubahn DB, Carlsten H, Dahlman-Wright K, Gustafsson JA, Ohlsson C: Estrogen receptor specificity for the effects of estrogen in ovariectomized mice. J Endocrinol. 2002, 174: 167-178.
Grodstein F, Manson JE, Stampfer MJ: Hormone therapy and coronary heart disease: the role of time since menopause and age at hormone initiation. J Womens Health (Larchmt). 2006, 15: 35-44.
Henderson VW, Brinton RD: Menopause and mitochondria: windows into estrogen effects on Alzheimer’s disease risk and therapy. Prog Brain Res. 2010, 182: 77-96.
Wharton W, Gleason CE, Miller VM, Asthana S: Rationale and design of the Kronos Early Estrogen Prevention Study (KEEPS) and the KEEPS cognitive and affective sub study (KEEPS Cog). Brain Res. 2013, 1514: 12-17.
Velarde MC: Pleiotropic actions of estrogen: a mitochondrial matter. Physiol Genomics. 2013, 45: 106-109.
Velarde MC: Reply to Turner and Kerber. Physiol Genomics. 2013, 45: 448-
Wu FC, Tajar A, Beynon JM, Pye SR, Silman AJ, Finn JD, O’Neill TW, Bartfai G, Casanueva FF, Forti G, Giwercman A, Han TS, Kula K, Lean ME, Pendleton N, Punab M, Boonen S, Vanderschueren D, Labrie F, Huhtaniemi IT, EMAS Group: Identification of late-onset hypogonadism in middle-aged and elderly men. N Engl J Med. 2010, 363: 123-135.
Cunningham GR: Andropause or male menopause? Rationale for testosterone replacement therapy in older men with low testosterone levels. Endocr Pract. 2013, 6: 1-18.
Ulloor J, Zhang A, Brooks B, Nguyen AH, Eder R, LeBrasseur N, Elmi A, Appleman E, Hede-Brierley L, Bhasin G, Bhatia A, Lazzari A, Davis S, Ni P, Collins L, Bhasin S: Clinical meaningfulness of the changes in muscle performance and physical function associated with testosterone administration in older men with mobility limitation. J Gerontol A Biol Sci Med Sci. 2011, 66: 1090-1099.
Srinivas-Shankar U, Roberts SA, Connolly MJ, O’Connell MD, Adams JE, Oldham JA, Wu FC: Effects of testosterone on muscle strength, physical function, body composition, and quality of life in intermediate-frail and frail elderly men: a randomized, double-blind, placebo-controlled study. J Clin Endocrinol Metab. 2010, 95: 639-650.
Snyder PJ, Peachey H, Hannoush P, Berlin JA, Loh L, Lenrow DA, Holmes JH, Dlewati A, Santanna J, Rosen CJ, Strom BL: Effect of testosterone treatment on body composition and muscle strength in men over 65 years of age. J Clin Endocrinol Metab. 1999, 84: 2647-2653.
Ferrando AA, Sheffield-Moore M, Yeckel CW, Gilkison C, Jiang J, Achacosa A, Lieberman SA, Tipton K, Wolfe RR, Urban RJ: Testosterone administration to older men improves muscle function: molecular and physiological mechanisms. Am J Physiol Endocrinol Metab. 2002, 282: E601-607.
Kenny AM, Kleppinger A, Annis K, Rathier M, Browner B, Judge JO, McGee D: Effects of transdermal testosterone on bone and muscle in older men with low bioavailable testosterone levels, low bone mass, and physical frailty. J Am Geriatr Soc. 2010, 58: 1134-1143.
Bhasin S, Storer TW, Berman N, Callegari C, Clevenger B, Phillips J, Bunnell TJ, Tricker R, Shirazi A, Casaburi R: The effects of supraphysiologic doses of testosterone on muscle size and strength in normal men. N Engl J Med. 1996, 335: 1-7.
Serra C, Tangherlini F, Rudy S, Lee D, Toraldo G, Sandor NL, Zhang A, Jasuja R, Bhasin S: Testosterone improves the regeneration of old and young mouse skeletal muscle. J Gerontol A Biol Sci Med Sci. 2013, 68: 17-26.
Hewitson TD, Zhao C, Wigg B, Lee SW, Simpson ER, Boon WC, Samuel CS: Relaxin and castration in male mice protect from, but testosterone exacerbates, age-related cardiac and renal fibrosis, whereas estrogens are an independent determinant of organ size. Endocrinology. 2012, 153: 188-199.
Ossewaarde ME, Bots ML, Verbeek AL, Peeters PH, van der Graaf Y, Grobbee DE, van der Schouw YT: Age at menopause, cause-specific mortality and total life expectancy. Epidemiology. 2005, 16: 556-562.
Lacreuse A, Chennareddi L, Gould KG, Hawkes K, Wijayawardana SR, Chen J, Easley KA, Herndon JG: Menstrual cycles continue into advanced old age in the common chimpanzee (Pan troglodytes). Biol Reprod. 2008, 79: 407-412.
Emery Thompson M, Jones JH, Pusey AE, Brewer-Marsden S, Goodall J, Marsden D, Matsuzawa T, Nishida T, Reynolds V, Sugiyama Y, Wrangham RW: Aging and fertility patterns in wild chimpanzees provide insights into the evolution of menopause. Curr Biol. 2007, 17: 2150-2156.
Snowdon DA: Early natural menopause and the duration of postmenopausal life. Findings from a mathematical model of life expectancy. J Am Geriatr Soc. 1990, 38: 402-408.
Drori D, Folman Y: Environmental effects on longevity in the male rat: exercise, mating, castration and restricted feeding. Exp Gerontol. 1976, 11: 25-32.
Michell AR: Longevity of British breeds of dog and its relationships with sex, size, cardiovascular variables and disease. Vet Rec. 1999, 145: 625-629.
Min KJ, Lee CK, Park HN: The lifespan of Korean eunuchs. Curr Biol. 2012, 22: R792-793.
Hamilton JB, Mestler GE: Mortality and survival: comparison of eunuchs with intact men and women in a mentally retarded population. J Gerontol. 1969, 24: 395-411.
Nieschlag E, Nieschlag S, Behre HM: Lifespan and testosterone. Nature. 1993, 366: 215-
Papadopoulos V, Miller WL: Role of mitochondria in steroidogenesis. Best Pract Res Clin Endocrinol Metab. 2012, 26: 771-790.
Miller WL: Steroid hormone synthesis in mitochondria. Mol Cell Endocrinol. 2013, 379: 62-73.
Miller WL, Auchus RJ: The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr Rev. 2011, 32: 81-151.
Miller WL: Minireview: regulation of steroidogenesis by electron transfer. Endocrinology. 2005, 146: 2544-2550.
Quinlan CL, Perevoshchikova IV, Hey-Mogensen M, Orr AL, Brand MD: Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 2013, 1: 304-312.
Harman D: Free radical theory of aging: the ‘free radical’ diseases. Age. 1984, 7: 111-131.
Harman D: Origin and evolution of the free radical theory of aging: a brief personal history, 1954 to 2009. Biogerontology. 2009, 10: 773-781.
Shigenaga MK, Hagen TM, Ames BN: Oxidative damage and mitochondrial decay in aging. Proc Natl Acad Sci USA. 1994, 91: 10771-10778.
Hagen TM, Yowe DL, Bartholomew JC, Wehr CM, Do KL, Park JY, Ames BN: Mitochondrial decay in hepatocytes from old rats: membrane potential declines, heterogeneity and oxidants increase. Proc Natl Acad Sci USA. 1997, 94: 3064-3069.
Tatone C, Carbone MC, Falone S, Aimola P, Giardinelli A, Caserta D, Marci R, Pandolfi A, Ragnelli AM, Amicarelli F: Age-dependent changes in the expression of superoxide dismutases and catalase are associated with ultrastructural modifications in human granulosa cells. Mol Hum Reprod. 2006, 12: 655-660.
de Bruin JP, Dorland M, Spek ER, Posthuma G, van Haaften M, Looman CW, te Velde ER: Age-related changes in the ultrastructure of the resting follicle pool in human ovaries. Biol Reprod. 2004, 70: 419-424.
Seifer DB, DeJesus V, Hubbard K: Mitochondrial deletions in luteinized granulosa cells as a function of age in women undergoing in vitro fertilization. Fertil Steril. 2002, 78: 1046-1048.
Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly-Y M, Gidlöf S, Oldfors A, Wibom R, Törnell J, Jacobs HT, Larsson NG: Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004, 429: 417-423.
Lu B, Poirier C, Gaspar T, Gratzke C, Harrison W, Busija D, Matzuk MM, Andersson KE, Overbeek PA, Bishop CE: A mutation in the inner mitochondrial membrane peptidase 2-like gene (Immp2l) affects mitochondrial function and impairs fertility in mice. Biol Reprod. 2008, 78: 601-610.
Zirkin BR, Tenover JL: Aging and declining testosterone: past, present, and hopes for the future. J Androl. 2012, 33: 1111-1118.
Luo L, Chen H, Zirkin BR: Temporal relationships among testosterone production, steroidogenic acute regulatory protein (StAR), and P450 side-chain cleavage enzyme (P450scc) during Leydig cell aging. J Androl. 2005, 26: 25-31.
Chen H, Cangello D, Benson S, Folmer J, Zhu H, Trush MA, Zirkin BR: Age-related increase in mitochondrial superoxide generation in the testosterone-producing cells of Brown Norway rat testes: relationship to reduced steroidogenic function?. Exp Gerontol. 2001, 36: 1361-1373.
Beattie MC, Chen H, Fan J, Papadopoulos V, Miller P, Zirkin BR: Aging and luteinizing hormone effects on reactive oxygen species production and DNA damage in rat Leydig cells. Biol Reprod. 2013, 88: 100-
Diemer T, Allen JA, Hales KH, Hales DB: Reactive oxygen disrupts mitochondria in MA-10 tumor Leydig cells and inhibits steroidogenic acute regulatory (StAR) protein and steroidogenesis. Endocrinology. 2003, 144: 2882-2891.
Li WR, Chen L, Chang ZJ, Xin H, Liu T, Zhang YQ, Li GY, Zhou F, Gong YQ, Gao ZZ, Xin ZC: Autophagic deficiency is related to steroidogenic decline in aged rat Leydig cells. Asian J Androl. 2011, 13: 881-888.
Lonard DM, O’Malley BW: Nuclear receptor coregulators: modulators of pathology and therapeutic targets. Nat Rev Endocrinol. 2012, 8: 598-604.
Mattingly KA, Ivanova MM, Riggs KA, Wickramasinghe NS, Barch MJ, Klinge CM: Estradiol stimulates transcription of nuclear respiratory factor-1 and increases mitochondrial biogenesis. Mol Endocrinol. 2008, 22: 609-622.
Ivanova MM, Luken KH, Zimmer AS, Lenzo FL, Smith RJ, Arteel MW, Kollenberg TJ, Mattingly KA, Klinge CM: Tamoxifen increases nuclear respiratory factor 1 transcription by activating estrogen receptor beta and AP-1 recruitment to adjacent promoter binding sites. FASEB J. 2011, 25: 1402-1416.
Tcherepanova I, Puigserver P, Norris JD, Spiegelman BM, McDonnell DP: Modulation of estrogen receptor-alpha transcriptional activity by the coactivator PGC-1. J Biol Chem. 2000, 275: 16302-16308.
Chen JQ, Cammarata PR, Baines CP, Yager JD: Regulation of mitochondrial respiratory chain biogenesis by estrogens/estrogen receptors and physiological, pathological and pharmacological implications. Biochim Biophys Acta. 2009, 1793: 1540-1570.
Hammes SR, Levin ER: Minireview: recent advances in extranuclear steroid receptor actions. Endocrinology. 2011, 152: 4489-4495.
Moats RK, Ramirez VD: Rapid uptake and binding of estradiol-17beta-6-(O-carboxymethyl) oxime:125I-labeled BSA by female rat liver. Biol Reprod. 1998, 58: 531-538.
Moats RK, Ramirez VD: Electron microscopic visualization of membrane-mediated uptake and translocation of estrogen-BSA:colloidal gold by hep G2 cells. J Endocrinol. 2000, 166: 631-647.
Borrás C, Gambini J, Gómez-Cabrera MC, Sastre J, Pallardó FV, Mann GE, Viña J: 17beta-oestradiol up-regulates longevity-related, antioxidant enzyme expression via the ERK1 and ERK2(MAPK)/NFkappaB cascade. Aging Cell. 2005, 4: 113-118.
Ronda AC, Vasconsuelo A, Boland R: 17β-Estradiol protects mitochondrial functions through extracellular-signal-regulated kinase in C2C12 muscle cells. Cell Physiol Biochem. 2013, 32: 1011-1023.
Yang SH, Prokai L, Simpkins JW: Correspondence regarding Schwend and Gustafsson, ‘False positives in MALDI-TOF detection of ERbeta in mitochondria’. Biochem Biophys Res Commun. 2006, 345: 917-918.
Schwend T, Gustafsson JA: False positives in MALDI-TOF detection of ERbetain mitochondria. Biochem Biophys Res Commun. 2006, 343: 707-711.
Razmara A, Sunday L, Stirone C, Wang XB, Krause DN, Duckles SP, Procaccio V: Mitochondrial effects of estrogen are mediated by estrogen receptor alpha in brain endothelial cells. J Pharmacol Exp Ther. 2008, 325: 782-790.
Yang SH, Liu R, Perez EJ, Wen Y, Stevens SM, Valencia T, Brun-Zinkernagel AM, Prokai L, Will Y, Dykens J, Koulen P, Simpkins JW: Mitochondrial localization of estrogen receptor beta. Proc Natl Acad Sci USA. 2004, 101: 4130-4135.
Milner TA, Ayoola K, Drake CT, Herrick SP, Tabori NE, McEwen BS, Warrier S, Alves SE: Ultrastructural localization of estrogen receptor beta immunoreactivity in the rat hippocampal formation. J Comp Neurol. 2005, 491: 81-95.
Milanesi L, Russo de Boland A, Boland R: Expression and localization of estrogen receptor alpha in the C2C12 murine skeletal muscle cell line. J Cell Biochem. 2008, 104: 1254-1273.
Monje P, Boland R: Subcellular distribution of native estrogen receptor alpha and beta isoforms in rabbit uterus and ovary. J Cell Biochem. 2001, 82: 467-479.
Cammarata PR, Chu S, Moor A, Wang Z, Yang SH, Simpkins JW: Subcellular distribution of native estrogen receptor alpha and beta subtypes in cultured human lens epithelial cells. Exp Eye Res. 2004, 78: 861-871.
Vasconsuelo A, Milanesi L, Boland R: Actions of 17β-estradiol and testosterone in the mitochondria and their implications in aging. Ageing Res Rev. 2013, 12: 907-917.
Pedram A, Razandi M, Wallace DC, Levin ER: Functional estrogen receptors in the mitochondria of breast cancer cells. Mol Biol Cell. 2006, 17: 2125-2137.
Chen JQ, Delannoy M, Cooke C, Yager JD: Mitochondrial localization of ERalpha and ERbeta in human MCF7 cells. Am J Physiol Endocrinol Metab. 2004, 286: E1011-1022.
Flynn JM, Dimitrijevich SD, Younes M, Skliris G, Murphy LC, Cammarata PR: Role of wild-type estrogen receptor-beta in mitochondrial cytoprotection of cultured normal male and female human lens epithelial cells. Am J Physiol Endocrinol Metab. 2008, 295: E637-647.
Chen JQ, Yager JD: Estrogen’s effects on mitochondrial gene expression: mechanisms and potential contributions to estrogen carcinogenesis. Ann N Y Acad Sci. 2004, 1028: 258-272.
Chen JQ, Eshete M, Alworth WL, Yager JD: Binding of MCF-7 cell mitochondrial proteins and recombinant human estrogen receptors alpha and beta to human mitochondrial DNA estrogen response elements. J Cell Biochem. 2004, 93: 358-973.
Arnold S, Victor MB, Beyer C: Estrogen and the regulation of mitochondrial structure and function in the brain. J Steroid Biochem Mol Biol. 2012, 131: 2-9.
Alvarez-Delgado C, Mendoza-Rodríguez CA, Picazo O, Cerbón M: Different expression of alpha and beta mitochondrial estrogen receptors in the aging rat brain: interaction with respiratory complex V. Exp Gerontol. 2010, 45: 580-585.
Stirone C, Duckles SP, Krause DN, Procaccio V: Estrogen increases mitochondrial efficiency and reduces oxidative stress in cerebral blood vessels. Mol Pharmacol. 2005, 68: 959-965.
Song JY, Kim MJ, Jo HH, Hwang SJ, Chae B, Chung JE, Kwon DJ, Lew YO, Lim YT, Kim JH, Kim JH, Kim MR: Antioxidant effect of estrogen on bovine aortic endothelial cells. J Steroid Biochem Mol Biol. 2009, 117: 74-80.
Kim JK, Pedram A, Razandi M, Levin ER: Estrogen prevents cardiomyocyte apoptosis through inhibition of reactive oxygen species and differential regulation of p38 kinase isoforms. J Biol Chem. 2006, 281: 6760-6767.
Razmara A, Duckles SP, Krause DN, Procaccio V: Estrogen suppresses brain mitochondrial oxidative stress in female and male rats. Brain Res. 2007, 1176: 71-81.
Irwin RW, Yao J, Hamilton RT, Cadenas E, Brinton RD, Nilsen J: Progesterone and estrogen regulate oxidative metabolism in brain mitochondria. Endocrinology. 2008, 149: 3167-3175.
Gollapudi L, Oblinger MM: Stable transfection of PC12 cells with estrogen receptor (ERalpha): protective effects of estrogen on cell survival after serum deprivation. J Neurosci Res. 1999, 56: 99-108.
Sastre-Serra J, Valle A, Company MM, Garau I, Oliver J, Roca P: Estrogen down-regulates uncoupling proteins and increases oxidative stress in breast cancer. Free Radic Biol Med. 2010, 48: 506-512.
Felty Q, Singh KP, Roy D: Estrogen-induced G1/S transition of G0-arrested estrogen-dependent breast cancer cells is regulated by mitochondrial oxidant signaling. Oncogene. 2005, 24: 4883-4893.
Roy D, Cai Q, Felty Q, Narayan S: Estrogen-induced generation of reactive oxygen and nitrogen species, gene damage, and estrogen-dependent cancers. J Toxicol Environ Health B Crit Rev. 2007, 10: 235-257.
López-Grueso R, Gambini J, Abdelaziz KM, Monleón D, Díaz A, El Alami M, Bonet-Costa V, Borrás C, Viña J: Early, but not late onset estrogen replacement therapy prevents oxidative stress and metabolic alterations caused by ovariectomy. Antioxid Redox Signal. 2014, 20: 236-246.
Pitteloud N, Mootha VK, Dwyer AA, Hardin M, Lee H, Eriksson KF, Tripathy D, Yialamas M, Groop L, Elahi D, Hayes FJ: Relationship between testosterone levels, insulin sensitivity, and mitochondrial function in men. Diabetes Care. 2005, 28: 1636-1642.
Ibebunjo C, Eash JK, Li C, Ma Q, Glass DJ: Voluntary running, skeletal muscle gene expression, and signaling inversely regulated by orchidectomy and testosterone replacement. Am J Physiol Endocrinol Metab. 2010, 300: E327-340.
Fernando SM, Rao P, Niel L, Chatterjee D, Stagljar M, Monks DA: Myocyte androgen receptors increase metabolic rate and improve body composition by reducing fat mass. Endocrinology. 2010, 151: 3125-3132.
Musa M, Fernando SM, Chatterjee D, Monks DA: Subcellular effects of myocyte-specific androgen receptor overexpression in mice. J Endocrinol. 2011, 210: 93-104.
Guo W, Wong S, Li M, Liang W, Liesa M, Serra C, Jasuja R, Bartke A, Kirkland JL, Shirihai O, Bhasin S: Testosterone plus low-intensity physical training in late life improves functional performance, skeletal muscle mitochondrial biogenesis, and mitochondrial quality control in male mice. PLoS One. 2012, 7: e51180-
Pansarasa O, D’Antona G, Gualea MR, Marzani B, Pellegrino MA, Marzatico F: ‘Oxidative stress’: effects of mild endurance training and testosterone treatment on rat gastrocnemius muscle. Eur J Appl Physiol. 2002, 87: 550-555.
Gladyshev VN: The free radical theory of aging is dead: long live the damage theory!. Antioxid Redox Signal. in press
Jacobs HT: The mitochondrial theory of aging: dead or alive?. Aging Cell. 2003, 2: 11-17.
Melov S, Doctrow SR, Schneider JA, Haberson J, Patel M, Coskun PE, Huffman K, Wallace DC, Malfroy B: Lifespan extension and rescue of spongiform encephalopathy in superoxide dismutase 2 nullizygous mice treated with superoxide dismutase-catalase mimetics. J Neurosci. 2001, 21: 8348-8353.
Kirby K, Hu J, Hilliker AJ, Phillips JP: RNA interference-mediated silencing of Sod2 in Drosophila leads to early adult-onset mortality and elevated endogenous oxidative stress. Proc Natl Acad Sci USA. 2002, 99: 16162-16167.
Van Raamsdonk JM, Hekimi S: Superoxide dismutase is dispensable for normal animal lifespan. Proc Natl Acad Sci USA. 2012, 109: 5785-5790.
Van Raamsdonk JM, Hekimi S: Deletion of the mitochondrial superoxide dismutase sod-2 extends lifespan in Caenorhabditis elegans. PLoS Genet. 2009, 5: e1000361-
Schmeisser S, Schmeisser K, Weimer S, Groth M, Priebe S, Fazius E, Kuhlow D, Pick D, Einax JW, Guthke R, Platzer M, Zarse K, Ristow M: Mitochondrial hormesis links low-dose arsenite exposure to lifespan extension. Aging Cell. 2013, 12: 508-517.
Acknowledgments
I thank Pierre-Yves Desprez, Samuel Curran, James M Flynn, Remi-Martin Laberge, and Christopher A Zambataro for critically reading the manuscript; and Isaac Daviet for his technical contributions in constructing Figure 2. Part of this work was supported by National Institute of Health, K99-AG041221 (Velarde).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author declares that he has no competing interests.
Authors’ contributions
MV drafted all the content of the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Velarde, M.C. Mitochondrial and sex steroid hormone crosstalk during aging. Longev Healthspan 3, 2 (2014). https://doi.org/10.1186/2046-2395-3-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2046-2395-3-2