
Abstract
Background
Molecular hydrogen (H2) functions as an extensive protector against oxidative stress, inflammation and allergic reaction in various biological models and clinical tests; however, its essential mechanisms remain unknown. H2directly reacts with the strong reactive nitrogen species peroxynitrite (ONOO-) as well as hydroxyl radicals (•OH), but not with nitric oxide radical (NO•). We hypothesized that one of the H2functions is caused by reducing cellular ONOO-, which is generated by the rapid reaction of NO• with superoxides (•O2-). To verify this hypothesis, we examined whether H2could restore cytotoxicity and transcriptional alterations induced by ONOO-derived from NO• in chondrocytes.
Methods
We treated cultured chondrocytes from porcine hindlimb cartilage or from rat meniscus fibrecartilage with a donor of NO•, S-nitroso-N-acetylpenicillamine (SNAP) in the presence or absence of H2. Chondrocyte viability was determined using a LIVE/DEAD Viability/Cytotoxicity Kit. Gene expressions of the matrix proteins of cartilage and the matrix metalloproteinases were analyzed by reverse transcriptase-coupled real-time PCR method.
Results
SNAP treatment increased the levels of nitrated proteins. H2decreased the levels of the nitrated proteins, and suppressed chondrocyte death. It is known that the matrix proteins of cartilage (including aggrecan and type II collagen) and matrix metalloproteinases (such as MMP3 and MMP13) are down- and up-regulated by ONOO-, respectively. H2restoratively increased the gene expressions of aggrecan and type II collagen in the presence of H2. Conversely, the gene expressions of MMP3 and MMP13 were restoratively down-regulated with H2. Thus, H2acted to restore transcriptional alterations induced by ONOO-.
Conclusions
These results imply that one of the functions of H2exhibits cytoprotective effects and transcriptional alterations through reducing ONOO-. Moreover, novel pharmacological strategies aimed at selective removal of ONOO-may represent a powerful method for preventive and therapeutic use of H2for joint diseases.
Similar content being viewed by others

Background
We have reported that molecular hydrogen (H2) has potential as a novel antioxidant in preventive and therapeutic applications [1]. Furthermore, H2exhibits not only anti-oxidative stress effects [2, 3], but also has various anti-inflammatory [4, 5] and anti-allergic effects [6]. Since the publication of the first article on the biological contribution of H2in 2007, more than 80 articles involved in H2have been published to establish the apparent activity of H2from various medical aspects [7–9].
H2reacted with strong reactive oxygen/nitrogen species including hydroxyl radical and peroxinitrite (ONOO-) in cell-free reactions and protected cultured cells depending upon the decrease of hydroxyl radicals (•OH) [1]. Subsequent and recent experiments including ours indicated that a small amount of hydrogen is also effective against various stimuli [8, 9]. When model animals consumed H2by drinking water with dissolved H2, a small amount of H2was extensively effective [10–12]; however, it may be difficult to explain that direct reduction of •OH by a very small amount of H2reveals all the functions of H2, because the saturated level of H2is only 0.8 mM and the dwelling time of •OH is very short in the body [11, 13]. In fact, drinking 0.04 or 0.08 mM H2was shown to be effective [14, 15]. Although we have recently shown that H2can be accumulated with hepatic glycogen, it is unlikely that the amount of H2is sufficient to exhibit all of its functions [15].
Moreover, H2regulated various gene expressions; however, there is no evidence that H2directly reacts with factors involved in transcriptional regulation including FGF21 [15], inflammatory cytokines [11], HMGB1 [16], and HO-1 [17]. It remains unclear whether such regulations are the cause or consequence of the effects against oxidative stress. Moreover, the primary molecular target of H2remains unknown.
ONOO-is produced by the rapid reaction of nitric monoxide (NO•)with superoxide anion radicals (•O2-) [18, 19]. We have shown that H2reduces ONOO-as well as •OH [1]. Different from •OH, ONOO-has a longer lifespan and the potential to regulate gene expression through nitration of target proteins [20, 21]. Thus, we hypothesized that one of the H2functions is caused by reducing cellular ONOO-.
Here, to verify this hypothesis, we examined protective and regulatory effects of H2on NO•-derived oxidative stress to chondrocytes. We found that H2protected chondrocytes from oxidative stress, and alternated gene expressions, contrary to the manner of transcriptional regulation by ONOO-. This study implies that at least one of the H2functions is responsible for the reduction of ONOO-.
Methods
Cartilage slice culture
A fresh hindlimb of a slaughtered male seven-month-old pig was purchased from Tokyo Shibaura Organ Co., Ltd. (Minato-ku, Tokyo, Japan). There were no possible contaminant diseases. Cartilage from the healthy porcine hindlimb (metatarsophalangeal joint) was cut into pieces for culture (2 mm width × 7 mm length × full thickness) as described previously [22]. Male Sprague-Dawley rats of 10 weeks of age were purchased from Nippon SLC (Hamamatsu, Shizuoka, Japan). Cartilage from the meniscus of a rat was also sliced into pieces (full width × full length × 0.5 mm thickness) for culture. Since the meniscus structure is not uniform and the peripheral part contains fewer chondrocytes, we used slices prepared from the middle part of the meniscus.
The slices were randomly divided into two experimental groups and incubated at 37°C in Dulbecco's modified Eagle's medium (DMEM)/Ham F-12 mixed medium (Gibco Invitrogen, Grand Island, NY, USA) supplemented with 10% fetal calf serum (FBS), penicillin (100 U/ml), and streptomycin (100 μg/ml).
The care and use of laboratory animals were in accordance with the NIH guidelines. This study was approved by the Animal Care and Use Committee of Nippon Medical School (Bunkyo-ku, Tokyo, Japan).
Hydrogen treatment
We prepared H2-dissolved culture medium as described previously [1]. In brief, we dissolved H2in the medium by bubbling H2gas to the saturated level. We also dissolved O2in a second medium by bubbling O2gas, and CO2in a third medium by bubbling CO2gas. We combined these media to give a medium consisting of 75% H2, 20% O2, 5% CO2(vol/vol/vol). We then cultured the cartilage slices in a closed culture flask filled with the medium. Control medium contained 75% N2instead of H2. The H2concentration was maintained for 24 hr as described [15].
Cell death assay
The cartilage slices were incubated for 12 - 80 hr in medium containing 0.3 - 3 mM S-nitroso-N-acetyl-D, L-penicillamine (SNAP) (Cayman Chemical, Ann Arbor, MI, USA) in the presence or absence of H2[22, 23]. Chondrocyte viability was determined using a LIVE/DEAD Viability/Cytotoxicity Kit (Molecular Probes, Eugene, OR, USA). Living, dying and dead cells were stained with green, yellow (combination of green and red) and red fluorescence, respectively, and visualized with a confocal scanning laser microscope (FLUOVIEW FV300; Olympus, Tokyo, Japan).
Immunohistochemical staining
Frozen sections of 6 μm-thick were fixed with 10% formalin and treated with 0.3% hydrogen peroxide in methanol to inhibit endogenous peroxidase activity. The sections were incubated with 10% Block Ace (DS Pharma Biomedical Co., Ltd., Suita, Osaka, Japan) in phosphate buffered saline (PBS) and then incubated with anti-nitrotyrosine monoclonal antibody (Calbiochem, San Diego, CA, USA; 1:100 dilution with 10% Block Ace in PBS) overnight at 4°C. Nitrotyrosine residues were visualized with DAB using horseradish peroxidase (HRP)-conjugated secondary antibody (Santa Cruz Biotechnology, Inc. Santa Cruz, CA, USA) and a HistoMark ORANGE kit (KPL, Gaithersburg, MD, USA). As a positive control for staining, we used sections from cartilage treated with 1 mM 3-morpholinosydnonimine (SIN-1) (Sigma-Aldrich, St. Louis, MO, USA), which generates both superoxide anion and nitric oxide that spontaneously produce peroxynitrite. The positive area was estimated using the Image J program (version 1.41; National Institutes of Health, Bethesda, MD, USA) from four sections for each group.
RNA isolation and RT-PCR
Total RNA was isolated from the cartilage using an RNeasy Mini kit (QIAGEN, Valencia, CA, USA). Complementary DNA synthesized by SuperScript II Reverse Transcriptase (Invitrogen, Carlsbad, CA, USA) was analyzed by quantitative PCR using the Thermal Cycler Dice Real Time System TP800 (TAKARA BIO Inc., Otsu, Shiga, Japan). All samples were normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) expression. Primer and probe sequences for each PCR are listed in Table 1.
Immunoblot analysis
Specimens were homogenized with a micro-homogenizer in SDS (sodium dodecyl sulfate) buffer (1% SDS in PBS), and then centrifuged at 10,000 g for 10 min at 4°C to remove debris. Supernatants were subjected to SDS-PAGE (SDS-polyacrylamide gel electrophoresis) followed by electrotransfer onto a PVDF membrane. The blotted membranes were blocked with Block Ace (DS Pharma Biomedical Co., Ltd.) and incubated with anti-aggrecan polyclonal antibody (Abcam, Cambridge, UK; 1:1,000 dilution), anti-MMP13 polyclonal antibody (Santa Cruz Biotechnology, Inc. 1:1,000 dilution) or anti-actin monoclonal antibody (Sigma-Aldrich; 1:500 dilution) overnight at 4°C. Each band was visualized with horseradish peroxidase (HRP)-conjugated secondary antibody (Santa Cruz Biotechnology, Inc.) and an ECL plus Western blotting detection system (GE Healthcare, Piscataway, NJ, USA).
Statistical analysis
We performed statistical analysis using StatView software (SAS Institute) by applying an unpaired two-tailed Student's t-test and ANOVA followed by Fisher's exact test, as described previously [1]. Differences were considered significant at p < 0.05.
Results
H2protects chondrocytes of hyaline and fibrecartilage from cell death
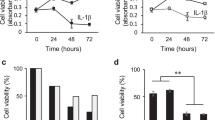
It is reported that cultured chondrocytes are sensitive to exposure to SNAP, a donor of NO• [22] and that H2exhibits no direct reaction with NO• in cultured cells as well as in a cell-free reaction. To verify the hypothesis that H2protects cells by reducing ONOO-, we examined the effect of H2on cell death induced by SNAP by using hyaline cartilage slices from a porcine hindlimb metatarsophalangeal joint as a target. Chondrocyte viability was determined using a LIVE/DEAD Viability/Cytotoxicity Kit, which provides quantitative analyses of the proportion of live and dead cells in a mixed population (Figure 1A). In living cells, membrane-permeated calcein AM is cleaved by esterases to yield cytoplasmic green fluorescence, and in dead cells membrane-impermeable ethidium homodimer-1 labels nucleic acids with red fluorescence. Dying cells, whose membrane structure has been disrupted but still have some esterase activity, were double stained as yellow. We counted green, red and yellow cells for statistical analysis (Additional file 1: Table S1). Cell viability was calculated as the percentage of green cell numbers against total cell numbers (Figure 1B). Significant protection of chondrocytes by H2was observed in the treatment with 3 mM SNAP for 12 hr (Figure 1A and 1B). More evident effects were obtained with longer SNAP treatment (Figure 1B).

Hydrogen protects chondrocytes of hyaline cartilage from cell death. (A) Porcine cartilage slices were incubated with 0, 1 or 3 mM SNAP in the presence or absence of hydrogen for 12 hr at 37°C. Cells were stained with a mixture of calcein AM (Live cell: green) and ethidium homodimer (Dead cell: red) as described in Materials and methods. Scale bar: 100 μm. (B) Chondrocyte viability was determined by counting green and red cells from three areas of each slice. Six slices were used for each experimental group. The slices were incubated with 0, 1 or 3 mM SNAP in the presence or absence of hydrogen for 12, 24 or 36 hr at 37°C. Data are the means ± SD (n= 6). *p< 0.05; **p< 0.01.
Next, we examined another type of cartilage, meniscus fibrecartilage, isolated from rats instead of swine specimens. Because it is easier to isolate swine than rat cartilage, we used swine cartilage for preliminary experiments; however, for further analysis, cartilage from rats is more suitable for RNA and protein analysis because genomic databases and antibodies are available. Treatment with 1 mM SNAP induced cell death in a time-dependent manner and H2suppressed chondrocyte death at each time point (Figure 2A and 2B, Additional file 2: Table S2). H2significantly protected chondrocytes from death with various concentrations of SNAP treatment for 48 hr (Figure 2C, Additional file 3: Table S3). These results indicate that H2protects chondrocytes by stimuli derived from NO•, although H2has no potential to react with NO•.

Hydrogen protects chondrocytes of fibrocartilages from cell death. (A) Meniscus fibrocartilages from SD rats was incubated with 1 mM SNAP in the presence or absence of hydrogen for 0, 6, 20, 48, or 80 hr at 37°C. Cells were stained with calcein AM (Live cell: green) and ethidium homodimer (Dead cell: red) as described in Materials and methods. Scale bar: 40 μm. (B) Chondrocyte viability was determined by counting green and red cells from three regions of each slice. Six slices were used for each experimental group. The slices were incubated with 1 mM SNAP in the presence or absence of hydrogen for the indicated periods at 37°C. Data are the means ± SD (n= 6). *p< 0.05; ***p< 0.001. (C) The slices were incubated with 0, 0.3, 1, or 3 mM SNAP in the presence or absence of hydrogen for 48 hr at 37°C. Data are the means ± SD (n= 6). *p< 0.05; ***p< 0.001.
H2decreases nitrotyrosine in chondrocytes and matrix of cartilage induced by SNAP
ONOO-is a strong modifier of nitration in proteins. To confirm that H2decreased ONOO-derived from NO•, we examined levels of nitrotyrosine residues in cartilage immunohistologically. In fact, NO• increased the levels of nitrotyrosine, and H2restored its increase (Figure 3). Thus, H2should decrease ONOO-derived from NO•.

Hydrogen decreases nitrotyrosine in chondrocytes and cartilage matrix. (A) Meniscus fibrocartilage from SD rats was incubated with 1 mM SNAP in the presence or absence of hydrogen for 3 hr at 37°C. Frozen sections were stained with anti-nitorotyrosine antibody and visualized with DAB as described in Methods. The inset shows chondrocytes. Cartilage incubated with 1 mM SIN-1 and without SNAP was used for positive and negative staining controls, respectively. Scale bar: 40 μm. (B) Levels of nitrotyrosine in cartilage were estimated from anti-nitrotyrosine immunostaining using an image analysis program, Image J program. Data are the mean ± SD (n = 4). *p < 0.05; control versus hydrogen in 1 mM SNAP-treated groups.
H2restores down-regulation of matrix expression and up-regulation of matrix-metallo protease expression induced by SNAP
It was also reported that ONOO-down-regulates gene expressions of the cartilage matrix proteins including aggrecan and type II collagen [24]. Conversely, levels of matrix-metallo protease are known to be up-regulated by ONOO-[24]. We then investigated the effect of H2on the expression of chondrocyte-specific matrix genes. Isolated meniscus fibrecartilage was incubated in DMEM/F-12 supplemented with 10% FBS with or without 1 mM SNAP in the presence or absence of H2. The levels of mRNA for the matrix proteins of type II collagen and aggrecan core protein were quantified with real-time PCR coupled with reverse transcription (Figure 4A and 4B). Indeed, SNAP down-regulated aggrecan and collagen II gene expressions as expected. The decreased gene expressions of the matrix proteins were significantly restored by H2-dissolved culture medium, suggesting that the decreased ONOO-restored the gene expression.

Hydrogen alters mRNA and protein expressions of matrix proteins and matrix-metalloproteases (MMPs). Meniscus fibrocartilage from rats was incubated with 1 mM SNAP in the presence or absence of hydrogen for 4 hr or 20 hr at 37°C. Total RNA was extracted from 4 hr-incubated cartilage and the expression levels of aggrecan (A), type II collagen (B), GAPDH (C), MMP3 (D) and MMP13 (E) were analyzed by real-time PCR coupled with reverse transcription. Data are the mean ± SD (n = 4). *p< 0.05; **p< 0.01. (F) Total protein was extracted from 20 hr-incubated cartilage and the expression levels of aggrecan, MMP13 and actin were analyzed by immunoblotting.
The possibility cannot be ruled out that oxidative damage derived from SNAP may reduce any gene expressions in a non-specific manner. We examined therefore the effect of H2on catabolic enzyme genes induced by SNAP, because levels of matrix-metallo protease are known to be contradictorily up-regulated by ONOO-[24]. The levels of MMP-3 and MMP-13 mRNA were measured with quantitative real-time PCR after treatment with SNAP with or without H2(Figure 4D and 4E). Moreover, the alterations of the aggrecan and MMP-13 proteins corresponded to their mRNA levels (Figure 4F). Thus, SNAP up-regulated MMP-3 and MMP-13 gene expressions as expected, whereas H2significantly suppressed MMP gene expressions, suggesting that H2restored the increased expressions by decreasing ONOO-.
Discussion
Joint diseases including osteoarthritis (OA) and rheumatoid arthritis (RA) are the most common disabling diseases, especially among elderly people. Arthritis is a degenerative disease involving abnormalities in chondrocytes, articular cartilage and other joint tissue, and is mediated by a number of underlying biochemical and physical stimuli [25, 26]. Recent studies revealed that oxidative stress plays a leading role in the initiation and progression of the disease process [27, 28]. As a joint disease model of aged patients, we stimulated chondrocytes with oxidative stress derived from NO•. The cartilage consists mostly of the extracellular matrix, which is synthesized by chondrocytes [28, 29]. The extracellular matrix is composed of collagens and proteoglycans that are responsible for the important compressive and tensile properties of cartilage [28].
The major oxidative stress generated by chondrocytes is one of the most powerful oxidants ONOO-, which was produced by the rapid reaction of NO• with •O2-[18, 19]. At an earlier stage, NO• has been considered as the primary inducer of chondrocyte death [30]; however, it has been revealed that the oxidative strength of NO• is not sufficient to initiate cell death [31, 32]. A series of experiments have indicated that the major cytotoxicity attributed to NO• is rather due to ONOO-[20, 33]. Increased ONOO-formation has been observed in cartilage and subchondral bone in rodent models [34–36] and in cartilage in OA and RA patients [37–39]. ONOO-induces cell death and regulates the decreased expression of collagens and proteoglycans and increased matrix metallo proteinases in chondrocytes, resulting in matrix degradation [24, 40]. Thus, chondrocyte is a suitable target for investing the effect of H2regarding ONOO-in this study.
In this study, we show that H2protected chondrocytes from death induced by SNAP. SNAP is a donor of NO•; however, NO• has no strong toxicity itself and H2has no potential to reduce NO•. Our previous study demonstrated that H2reduces ONOO-in a cell-free system [1]. Thus, we speculate that H2would protect SNAP-treated chondrocytes by decreasing ONOO-. More importantly, it has been reported that drinking hydrogen water suppress the nitration of kidney proteins, although H2received from hydrogen water remained for only short period in the organ (less than 5 min) [11]. In this study, we have shown that H2in medium suppress the nitration of the chondrocyte proteins (Figure 3). Thus, it is possible that even a very small amount of H2exhibits anti-oxidative effects by reducing ONOO-in many situations.
Several laboratories including ours have reported that H2altered gene expressions involved in inflammation or energy metabolism when animals drank hydrogen water [15, 17]; however, it is an open question why H2alters gene expressions, because there is no evidence that H2directly influences gene expressions. On the other hand, ONOO-has the potential to regulate gene expressions through the nitration of factors involved in transcriptional regulation [20]. As mentioned above, drinking hydrogen water suppresses the nitration of proteins; thus, it is possible that the very small amount of H2consumed by drinking hydrogen water influences nitration in in vivoexperiments and results in regulatory as well as anti-oxidative effects [11]. These results agree with the present finding that H2suppressed the nitration of proteins.
Taken together, this study implies that one of the H2functions, including transcriptional alterations, is caused through reducing ONOO-derived from NO•.
Novel pharmacological strategies aimed at selective removal of ONOO-may represent a powerful method for preventive and therapeutic use of H2for joint diseases. Cartilage has no blood vessels and nutrients are supplied through fluid. Since H2has a great advantage to rapidly diffuse into tissues even without blood flow [41, 42], it may be useful to prevent joint diseases by reducing oxidative stress and by suppressing the decrease in matrix proteins and inhibiting degradation by proteinases.
Conclusions
This study implies that one of the H2functions, including transcriptional alterations, is caused through reducing ONOO-derived from NO•. Novel pharmacological strategies aimed at selective removal of ONOO-may represent a powerful method for preventive and therapeutic use of H2for joint diseases.
Abbreviations
- SNAP:
-
S-nitroso-N-acetylpenicillamine
- MMP:
-
matrix metalloproteinase
- GAPDH:
-
glyceraldehyde 3-phosphate dehydrogenase
- OA:
-
osteoarthritis
- RA:
-
rheumatoid arthritis.
References
Ohsawa I, Ishikawa M, Takahashi K, Watanabe M, Nishimaki K, Yamagata K, Katsura K, Katayama Y, Asoh S, Ohta S: Hydrogen acts as a therapeutic antioxidant by selectively reducing cytotoxic oxygen radicals. Nat Med. 2007, 13: 688-694. 10.1038/nm1577.
Fukuda K, Asoh S, Ishikawa M, Yamamoto Y, Ohsawa I, Ohta S: Inhalation of hydrogen gas suppresses hepatic injury caused by ischemia/reperfusion through reducing oxidative stress. Biochem Biophys Res Commun. 2007, 361: 670-674. 10.1016/j.bbrc.2007.07.088.
Ohsawa I, Nishimaki K, Yamagata K, Ishikawa M, Ohta S: Consumption of hydrogen water prevents atherosclerosis in apolipoprotein E knockout mice. Biochem Biophys Res Commun. 2008, 377: 1195-1198. 10.1016/j.bbrc.2008.10.156.
Kajiya M, Silva MJ, Sato K, Ouhara K, Kawai T: Hydrogen mediates suppression of colon inflammation induced by dextran sodium sulfate. Biochem Biophys Res Commun. 2009, 386: 11-15. 10.1016/j.bbrc.2009.05.117.
Kajiya M, Sato K, Silva MJ, Ouhara K, Do PM, Shanmugam KT, Kawai T: Hydrogen from intestinal bacteria is protective for Concanavalin A-induced hepatitis. Biochem Biophys Res Commun. 2009, 386: 316-321. 10.1016/j.bbrc.2009.06.024.
Itoh T, Fujita Y, Ito M, Masuda A, Ohno K, Ichihara M, Kojima T, Nozawa Y: Molecular hydrogen suppresses FcepsilonRI-mediated signal transduction and prevents degranulation of mast cells. Biochem Biophys Res Commun. 2009, 389: 651-656. 10.1016/j.bbrc.2009.09.047.
Huang CS, Kawamura T, Toyoda Y, Nakao A: Recent advances in hydrogen research as a therapeutic medical gas. Free Radic Res. 2010, 44: 971-982. 10.3109/10715762.2010.500328.
Ohta S: Molecular hydrogen is a novel antioxidant to efficiently reduce oxidative stress with potential for the improvement of mitochondrial diseases. Biochim Biophys Acta. 2011,
Ohta S: Recent progress toward hydrogen medicine: Potential of molecular hydrogen for preventive and therapeutic applications. Curr Pharm Des. 2011,
Nagata K, Nakashima-Kamimura N, Mikami T, Ohsawa I, Ohta S: Consumption of molecular hydrogen prevents the stress-induced impairments in hippocampus-dependent learning tasks during chronic physical restraint in mice. Neuropsychopharmacology. 2009, 34: 501-508. 10.1038/npp.2008.95.
Cardinal JS, Zhan J, Wang Y, Sugimoto R, Tsung A, McCurry KR, Billiar TR, Nakao A: Oral hydrogen water prevents chronic allograft nephropathy in rats. Kidney Int. 2010, 77: 101-109. 10.1038/ki.2009.421.
Fu Y, Ito M, Fujita Y, Ichihara M, Masuda A, Suzuki Y, Maesawa S, Kajita Y, Hirayama M, Ohsawa I, Ohta S, Ohno K: Molecular hydrogen is protective against 6-hydroxydopamine-induced nigrostriatal degeneration in a rat model of Parkinson's disease. Neurosci Lett. 2009, 453: 81-85. 10.1016/j.neulet.2009.02.016.
Nakashima-Kamimura N, Mori T, Ohsawa I, Asoh S, Ohta S: Molecular hydrogen alleviates nephrotoxicity induced by an anti-cancer drug cisplatin without compromising anti-tumor activity in mice. Cancer Chemother Pharmacol. 2009, 64: 753-761. 10.1007/s00280-008-0924-2.
Fujita K, Seike T, Yutsudo N, Ohno M, Yamada H, Yamaguchi H, Sakumi K, Yamakawa Y, Kido MA, Takaki A, Katafuchi T, Tanaka Y, Nakabeppu Y, Noda M: Hydrogen in drinking water reduces dopaminergic neuronal loss in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson's disease. PLoS One. 2009, 4: e7247-10.1371/journal.pone.0007247.
Kamimura N, Nishimaki K, Ohsawa I, Ohta S: Molecular hydrogen improves obesity and diabetes by inducing hepatic FGF21 and stimulating energy metabolism in db/db mice. Obesity. 2011,
Xie K, Yu Y, Pei Y, Hou L, Chen S, Xiong L, Wang G: Protective effects of hydrogen gas on murine polymicrobial sepsis via reducing oxidative stress and HMGB1 release. Shock. 2010, 34: 90-97.
Huang CS, Kawamura T, Lee S, Tochigi N, Shigemura N, Buchholz BM, Kloke JD, Billiar TR, Toyoda Y, Nakao A: Hydrogen inhalation ameliorates ventilator-induced lung injury. Crit Care. 2010, 14: R234-10.1186/cc9389.
Huie RE, Padmaja S: The reaction of no with superoxide. Free Radic Res Commun. 1993, 18: 195-199. 10.3109/10715769309145868.
Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA: Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci USA. 1990, 87: 1620-1624. 10.1073/pnas.87.4.1620.
Pacher P, Beckman JS, Liaudet L: Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007, 87: 315-424. 10.1152/physrev.00029.2006.
Klotz LO, Schroeder P, Sies H: Peroxynitrite signaling: receptor tyrosine kinases and activation of stress-responsive pathways. Free Radic Biol Med. 2002, 33: 737-743. 10.1016/S0891-5849(02)00892-4.
Ozaki D, Sudo K, Asoh S, Yamagata K, Ito H, Ohta S: Transduction of anti-apoptotic proteins into chondrocytes in cartilage slice culture. Biochem Biophys Res Commun. 2004, 313: 522-527. 10.1016/j.bbrc.2003.11.144.
Sudo K, Asoh S, Ohsawa I, Ozaki D, Yamagata K, Ito H, Ohta S: The anti-cell death FNK protein protects cells from death induced by freezing and thawing. Biochem Biophys Res Commun. 2005, 330: 850-856. 10.1016/j.bbrc.2005.03.059.
Henrotin YE, Bruckner P, Pujol JP: The role of reactive oxygen species in homeostasis and degradation of cartilage. Osteoarthritis Cartilage. 2003, 11: 747-755. 10.1016/S1063-4584(03)00150-X.
Roach HI, Aigner T, Soder S, Haag J, Welkerling H: Pathobiology of osteoarthritis: pathomechanisms and potential therapeutic targets. Curr Drug Targets. 2007, 8: 271-282. 10.2174/138945007779940160.
Ge Z, Hu Y, Heng BC, Yang Z, Ouyang H, Lee EH, Cao T: Osteoarthritis and therapy. Arthritis Rheum. 2006, 55: 493-500. 10.1002/art.21994.
Halliwell B: Oxygen radicals, nitric oxide and human inflammatory joint disease. Ann Rheum Dis. 1995, 54: 505-510. 10.1136/ard.54.6.505.
Henrotin Y, Kurz B, Aigner T: Oxygen and reactive oxygen species in cartilage degradation: friends or foes?. Osteoarthritis Cartilage. 2005, 13: 643-654. 10.1016/j.joca.2005.04.002.
Muir H: The chondrocyte, architect of cartilage. Biomechanics, structure, function and molecular biology of cartilage matrix macromolecules. Bioessays. 1995, 17: 1039-1048. 10.1002/bies.950171208.
Blanco FJ, Ochs RL, Schwarz H, Lotz M: Chondrocyte apoptosis induced by nitric oxide. Am J Pathol. 1995, 146: 75-85.
Del Carlo M, Loeser RF: Nitric oxide-mediated chondrocyte cell death requires the generation of additional reactive oxygen species. Arthritis Rheum. 2002, 46: 394-403. 10.1002/art.10056.
Clements KM, Burton-Wurster N, Lust G: The spread of cell death from impact damaged cartilage: lack of evidence for the role of nitric oxide and caspases. Osteoarthritis Cartilage. 2004, 12: 577-585. 10.1016/j.joca.2004.04.006.
Whiteman M, Armstrong JS, Cheung NS, Siau JL, Rose P, Schantz JT, Jones DP, Halliwell B: Peroxynitrite mediates calcium-dependent mitochondrial dysfunction and cell death via activation of calpains. FASEB J. 2004, 18: 1395-1397.
Cuzzocrea S, Chatterjee PK, Mazzon E, McDonald MC, Dugo L, Di Paola R, Serraino I, Britti D, Caputi AP, Thiemermann C: Beneficial effects of GW274150, a novel, potent and selective inhibitor of iNOS activity, in a rodent model of collagen-induced arthritis. Eur J Pharmacol. 2002, 453: 119-129. 10.1016/S0014-2999(02)02338-5.
Szabo C, Virag L, Cuzzocrea S, Scott GS, Hake P, O'Connor MP, Zingarelli B, Salzman A, Kun E: Protection against peroxynitrite-induced fibroblast injury and arthritis development by inhibition of poly(ADP-ribose) synthase. Proc Natl Acad Sci USA. 1998, 95: 3867-3872. 10.1073/pnas.95.7.3867.
Yonekura Y, Koshiishi I, Yamada K, Mori A, Uchida S, Nakamura T, Utsumi H: Association between the expression of inducible nitric oxide synthase by chondrocytes and its nitric oxide-generating activity in adjuvant arthritis in rats. Nitric Oxide. 2003, 8: 164-169. 10.1016/S1089-8603(03)00025-9.
Loeser RF, Carlson CS, Del Carlo M, Cole A: Detection of nitrotyrosine in aging and osteoarthritic cartilage: Correlation of oxidative damage with the presence of interleukin-1beta and with chondrocyte resistance to insulin-like growth factor 1. Arthritis Rheum. 2002, 46: 2349-2357. 10.1002/art.10496.
Kaur H, Halliwell B: Evidence for nitric oxide-mediated oxidative damage in chronic inflammation. Nitrotyrosine in serum and synovial fluid from rheumatoid patients. FEBS Lett. 1994, 350: 9-12. 10.1016/0014-5793(94)00722-5.
Sandhu JK, Robertson S, Birnboim HC, Goldstein R: Distribution of protein nitrotyrosine in synovial tissues of patients with rheumatoid arthritis and osteoarthritis. J Rheumatol. 2003, 30: 1173-1181.
Abramson SB: Osteoarthritis and nitric oxide. Osteoarthritis Cartilage. 2008, 16 (Suppl 2): S15-20.
Hayashida K, Sano M, Ohsawa I, Shinmura K, Tamaki K, Kimura K, Endo J, Katayama T, Kawamura A, Kohsaka S, Makino S, Ohta S, Ogawa S, Fukuda K: Inhalation of hydrogen gas reduces infarct size in the rat model of myocardial ischemia-reperfusion injury. Biochem Biophys Res Commun. 2008, 373: 30-35. 10.1016/j.bbrc.2008.05.165.
Oharazawa H, Igarashi T, Yokota T, Fujii H, Suzuki H, Machide M, Takahashi H, Ohta S, Ohsawa I: Protection of the retina by rapid diffusion of hydrogen: administration of hydrogen-loaded eye drops in retinal ischemia-reperfusion injury. Invest Ophthalmol Vis Sci. 2010, 51: 487-492. 10.1167/iovs.09-4089.
Acknowledgements
This work was supported by a grant from the Ministry of Education, Culture, Sports, Science and Technology of Japan and the Ministry of Health, Labour and Welfare.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
SO and ST conceived the experiments. SO and NK designed the actual experiments. TH, TY and NK performed the experiments and data analysis. NK and SO interpreted the data and wrote the paper. All authors have been involved in drafting the manuscript it critically for important intellectual content; and have given final approval of the version to be published.
Electronic supplementary material
13618_2011_20_MOESM3_ESM.PDF
Additional file 3: Table S3 - Live, dying, and dead cell numbers of fibrocartilages treated with various concentration of SNAP for 48 hr. (PDF 67 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Hanaoka, T., Kamimura, N., Yokota, T. et al. Molecular hydrogen protects chondrocytes from oxidative stress and indirectly alters gene expressions through reducing peroxynitrite derived from nitric oxide. Med Gas Res 1, 18 (2011). https://doi.org/10.1186/2045-9912-1-18
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2045-9912-1-18