
Abstract
The Blood–brain barrier (BBB), present at the level of the endothelium of cerebral blood vessels, selectively restricts the blood-to-brain paracellular diffusion of compounds; it is mandatory for cerebral homeostasis and proper neuronal function. The barrier properties of these specialized endothelial cells notably depend on tight junctions (TJs) between adjacent cells: TJs are dynamic structures consisting of a number of transmembrane and membrane-associated cytoplasmic proteins, which are assembled in a multimolecular complex and acting as a platform for intracellular signaling. Although the structural composition of these complexes has been well described in the recent years, our knowledge about their functional regulation still remains fragmentary. Importantly, pericytes, embedded in the vascular basement membrane, and perivascular microglial cells, astrocytes and neurons contribute to the regulation of endothelial TJs and BBB function, altogether constituting the so-called neurovascular unit.
The present review summarizes our current understanding of the structure and functional regulation of endothelial TJs at the BBB. Accumulating evidence points to a correlation between BBB dysfunction, alteration of TJ complexes and progression of a variety of CNS diseases, such as stroke, multiple sclerosis and brain tumors, as well as neurodegenerative diseases like Parkinson’s and Alzheimer’s diseases. Understanding how TJ integrity is controlled may thus help improve drug delivery across the BBB and the design of therapeutic strategies for neurological disorders.
Similar content being viewed by others

Review
Background
The BBB maintains the homeostasis of the central nervous system (CNS) by (i) strictly limiting the passive diffusion of polar substances from the blood to the brain, (ii) mediating the transport of nutrients to the brain parenchyma as well as the efflux from the brain of toxic metabolites and xenobiotics, (iii) regulating the migration of circulating immune cells [1–3]. Formed by specialized vascular endothelial cells, the BBB is tightly controlled by pericytes, embedded in the vascular basement membrane, perivascular microglial cells, astrocytes and neurons which altogether constitute the neurovascular unit (NVU), a concept highlighting the functional cell-cell interactions supporting BBB function.
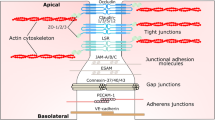
BBB endothelial cells display a unique phenotype characterized by the presence of TJs and the expression of specific polarized transport systems. TJs constitute the most apical intercellular junctional complex in polarized epithelium and endothelium, with three key biological functions: a barrier to paracellular diffusion of blood-borne polar substances [4], a fence preventing the lateral diffusion of lipids and integral membrane proteins, thus maintaining cell polarization [5–7] and an intracellular signaling platform which will be described below.
Brain endothelial TJ strands, like epithelial TJs, are composed of integral membrane proteins (occludin, claudins and junctional adhesion molecules (JAMs)) involved in intercellular contacts and interactions with cytoplasmic scaffolding proteins such as zonula occludens (ZO) proteins, actin cytoskeleton and associated proteins, such as protein kinases, small GTPases [8] and heterotrimeric G-proteins [9].
Excellent reviews have recently been published on the architecture of TJ complexes in epithelial and brain endothelial cells [10, 11]. Here we will briefly recall the main features of the structural organization of TJs at the BBB and will focus on transcriptional regulation, post-translational modifications and subcellular localization of TJ proteins and their consequences for BBB integrity with exposure to various environmental stimuli and during CNS disorders.
Components of TJs in brain endothelial cells
As in polarized epithelial cells where TJs have been mostly studied, the TJ backbone in brain endothelial cells consists of transmembrane proteins (occludin, claudins and JAMs) which recruit a number of membrane-associated cytoplasmic proteins.
Transmembrane proteins as the BBB TJ backbone
Occludin (60kDa), a tetraspan integral membrane protein, was the first TJ-specific protein identified [12, 13] in epithelial cells and shown to be functionally important for barrier function [14]. It is a member of the family of TJ-associated marvel proteins (TAMP) with tricellulin (marvelD2) [15] and marvelD3 [16, 17]. Both the MARVEL transmembrane domain of occludin, encompassing the four transmembrane helices, and its coiled coil cytosolic C-terminus were recently described to mediate its lateral (i.e. cis-) oligomerization in epithelial MDCK cells [18–20]. More precisely, cystein residues in these domains are directly involved in oligomerization through disulfide bridge formation. This process being redox-sensitive, oligomerization of occludin likely contributes to the redox-dependency of the TJ assembly [20, 21]: whereas normoxia conditions support occludin oligomerization and contribute to TJ assembly, oxidative stress associated with hypoxia-reoxygenation [22] or inflammation [23, 24] results in TJ disruption. This novel concept that occludin plays a key role in the redox regulation of TJs has been very recently reviewed [25].
In addition, the second extracellular domain of occludin is required for its stable assembly in TJs [26]. Indeed, synthetic peptides corresponding to this domain were shown to perturb TJ permeability barrier in epithelial cells [27–29]. The important contribution of occludin to TJ function is illustrated by the observations that ectopic expression of chicken occludin induced the formation of TJ-like structures in Sf9 insect cells [30], while increasing electrical resistance in MDCK cells [31]. Conversely, occludin degradation induced by viruses or bacteria (like HIV-1 Tat protein or Neisseria meningitidis), is associated with increased permeability in primary or immortalized human brain microvascular endothelial cells, respectively [32, 33]. However, well-developed TJ strands were reported in cells lacking occludin (human or guinea pig testis) [34] and between adjacent occludin-deficient epithelial cells [34, 35]; together with the report that occludin deficient-mice are viable, exhibiting normal TJs morphology as well as intestinal epithelium barrier function, these observations indicate that occludin is dispensable for TJ formation [36, 37].
Claudins constitute a large family of 20-27kDa membrane proteins (with four transmembrane domains) expressed in TJs in various cell types [4, 38–40] (endothelial and epithelial cells). Brain endothelial cells predominantly express claudin-3 and claudin-5 [41, 42], claudin-12 likely being also expressed [43, 44]. A large corpus of data clearly establishes the key contribution of claudin-3 and claudin-5 to TJ formation and integrity at the BBB. Indeed, exogenous expression of claudin-5 strengthens barrier properties in cultured rat brain endothelial cells [44], whereas depletion of claudin-5 induces the disruption of the BBB in genetically-altered mice [43] and in cultured human brain endothelial cells [9]. Claudins support TJ integrity via their capacity of cis- and trans-homodimerization as well as heterodimerization, notably through their second extracellular loop, as recently reported for claudin-5 [45–47]. Claudin-5 can interact with claudin-3 [48, 49] and the selective loss of the latter during autoimmune encephalomyelitis or human glioblastoma is associated with BBB breakdown [41].
Beside occludin and claudins, JAMs, although not essential to TJ formation in epithelial and endothelial cells, may be involved in the facilitation of assembly of TJ components and in the establishment of cell polarity by recruiting the polarity complex (Par-3/Par-6/aPKC: see below) to TJs [50, 51].
Membrane-associated cytoplasmic proteins in BBB TJs
A number of cytoplasmic proteins have been described to associate with TJ transmembrane proteins and to contribute somehow to TJ integrity in epithelial and brain endothelial cells. Among them, the PDZ domain-containing, membrane-associated guanylate kinase (MAGUK) family members have been largely documented: zonula occludens-1 (ZO-1, 225kDa) [52], ZO-2 (160kDa) [53], and ZO-3 (130kDa) [54]. ZO-1 forms heterodimers with ZO-2 and ZO-3 [54–56]. ZO proteins interact with the C-terminal domain of claudins via their first PDZ domain (PDZ1) [57], to JAMs by the third PDZ domain (PDZ3) [58] and to occludin via their GUK domains [55, 56, 59]. It is well established that ZO proteins are essential to the assembly of claudins [60], occludin [35] and JAM-A [61] at TJs, then anchoring this multimolecular complex to the actin cytoskeleton [62]. Par-3 (also known as ASIP) [63] binds to JAM proteins [64–66] and recruits to TJs atypical protein kinase C [67] and Par-6 [68], the three proteins then forming a Planar Cell Polarity (PCP) complex in polarized epithelial cells [69]. Only very recently was their expression confirmed also in brain endothelial cells [70].
Among additional TJ-associated proteins, heterotrimeric G-proteins (Gαi) were first described, in association with ZO-1, to contribute to TJ biogenesis and maintenance in epithelial and brain endothelial cells [71–73]. Gαi2 proteins were reported to be involved in T-lymphocyte extravasation, including in brain [74, 75]. More recently, we reported that Gαi2 interacts with claudin-5 and that its depletion increases brain endothelial cell permeability in vitro and delays TJ reassembly after hyperosmotic shock (induced by a high concentration mannitol treatment) [9]. On the basis of these observations, we proposed that claudin-5 and Gαi2, whether they interact directly or indirectly, might control TJ integrity as components of a multiprotein complex, including caveolin, ZO-1 linked to the actin cytoskeleton and possibly also, occludin and MUPP-1.
Physiological regulation of TJ assembly by the NVU
The NVU: regulation of TJ assembly by perivascular cells
Developmental role of astrocyte and pericyte secreted proteins
Development and maintenance of the BBB requires functional interactions between endothelial cells and perivascular cells of the NVU: whereas astrocytes have been well documented to regulate BBB formation and integrity [76, 77], only recently was the role of pericytes unraveled (for reviews: [78–80]).
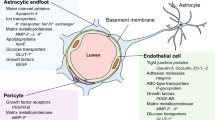
Indeed, early studies using co-culture of cerebral endothelial cells and astrocytes (or culture in the presence of astrocyte-conditioned medium) [81–87] highlighted the role of astrocyte-derived soluble factors in maintaining the specialized phenotype of brain endothelial cells (Figure 1). In addition, more recent reports established that pericytes also actively contribute to BBB formation during development by the release of several growth factors and morphogens [88–91].

Schematic representation of TJ modulation by the NVU. (a) The basal lamina protein agrin increases claudin-5 (Cld5) and occludin expression [92]; (b) Aquaporin-4 density, regulated by agrin, stabilizes TJ complexes through ZO-1 expression [93]; (c) β1-integrin engagement stabilizes Cld5 localization at the TJ [94]; (d) astrocyte/pericyte-secreted TGF-β induces Cld5 transcription through activation of Smad transcription factor [91]; (e) Shh enhances expression of TJ proteins via its membrane receptor Ptch1/Smo and the transcription factor Gli-1 [95]; (f) Endothelial PDGF-β recruits pericytes which stabilize BBB phenotype [90]; (g) Wnt 7a/7b proteins, via their membrane receptors Frizzled-4 associated to LRP5/6, induce Cld3 transcription through stabilization of β-catenin [96]; (h i j) Angiopoietin-1 (Ang1), via its membrane receptor Tie2, enhances VE-Cadherin clustering and Cld5 transcription through inhibition of FoxO1 activity by PI3K and β-catenin sequestration [83, 97] ; (k) VE-Cadherin engagement recruits CCM1 and the polarity complex (PCP) leading to TJ stabilization [98].
Astrocyte- and pericyte-derived Wnt and hedgehog morphogens were reported to control BBB formation during development and TJ integrity. Indeed, the Wnt/β-catenin pathway has been recently discovered as a major BBB-regulating pathway. Wnt ligation to its membrane receptors, Frizzled4 (Fz4) and LRP5/6 expressed by brain endothelial cells, inhibits the β-catenin repressor complex, allowing β-catenin cytoplasmic accumulation, nuclear translocation and transcription of various genes, including claudin-3 in cultured murine brain endothelial cells [96, 99] (Figure 1). Moreover, in vivo inactivation of Wnt factors (Wnt7a and Wnt7b) [100], Fz4 receptor [101] or injection of a soluble inhibitor of the Wnt/ Frizzled receptor interaction [102] lead to major vascular defects in the CNS (interestingly, not in non-neuronal tissues) and to BBB breakdown, clearly demonstrating a specific role for the Wnt/β-catenin pathway in BBB differentiation during development and for BBB maintenance in adulthood. These exciting observations (for review, see: [103]) open new research avenues for controlling BBB permeability in pathological situations as well as improving drug delivery to the CNS.
Sonic hedgehog (Shh), another well-known morphogen protein, acting through its membrane receptors Patched-1 (Ptch1)/Smoothened (Smo), was also recently shown to control BBB differentiation and to maintain the immune privilege of the CNS by inhibiting the endothelial production of chemokines and expression of adhesion proteins supporting extravasation of leukocytes to the brain [95].
In conclusion, these recent findings further document, at the molecular and cellular levels, the functional interactions between brain endothelial cells, pericytes and astrocytes and emphasise the key importance of the NVU in controlling BBB permeability and integrity. The major cellular cross-talks at the NVU are illustrated in Figure 1.
Role of basement membrane-associated proteins
The vascular basement membrane (or basal lamina) is a complex structure, composed of four glycoprotein families: laminins, collagen type IV, nidogens and heparan sulfate proteoglycans. Recent studies have unraveled the contribution of the endothelial laminin isoform α5 to the barrier property of the BBB by selectively inhibiting lymphocyte infiltration; the basement membrane thus contributes to maintain the well-known “immune privilege” of the CNS [104].
The heparan sulfate proteoglycan agrin is found in the basal lamina of brain microvessels [105]. A strict positive correlation has been reported between agrin deposition and expression of occludin [106], whereas, conversely, absence of agrin in glioblastoma vessels was shown to correlate with the lack of TJ proteins (occludin, claudin-5): these observations strongly suggest that agrin may regulate TJ formation in brain endothelium. Recently, agrin was described to be involved in the development of the BBB by contributing to astrocyte polarity [92]. Moreover, β1-integrin-mediated attachment of brain endothelial cells to the basement membrane has also been reported to be critical for stabilizing claudin-5 localization at TJs and maintaining BBB integrity in vitro and in vivo[93]. Genetic deletion of β1-integrin decreases the expression of the polarity protein Par-3, leading to the loss of endothelial cell polarity: these recent data suggest that β1-integrin-mediated brain endothelial cell adhesion to the basement membrane may lead to the development of cell polarity, TJ formation and BBB integrity [94].
VE-cadherin and β-catenin as modulators of TJs
In addition to TJs, junctional complexes between endothelial cells include adherens junctions (AJs), constituted by transmembrane proteins VE-cadherin linked to the actin cytoskeleton through catenins (eg: p120-catenin, α- and β-catenin) [107–109]. Interestingly, AJ and TJ complexes functionally interact in brain endothelial cells: indeed, VE-cadherin engagement induces claudin-5 transcription through inhibition of FoxO1 activity (a transcription repressor of claudin-5 gene) and β-catenin sequestration (a stabilizer of FoxO1 activity) in AJ complexes [97], in line with the above-mentioned capacity of β-catenin, downstream of Wnt receptor activation, to control claudin gene expression [96]. These findings clearly place VE-cadherin upstream of claudin-5 in the establishment, maturation and maintenance of endothelial cell-cell junctions.
Contribution of shear stress to TJ modulation and BBB integrity
It is established that one important mechanical stimulus contributing to BBB formation and maintenance is shear stress [110], a tangential force generated by flow across the apical surface of vascular endothelium [111, 112]. In line with the accepted concept that cerebral microcirculation is highly heterogeneous, mean shear stress levels in brain microvessels has been estimated in a range as wide as 0.01 to 10 dynes/cm2 in capillaries and 10–100 dynes/cm2 in arterioles [113–115]. Several dynamic in vitro models were developed in order to mimic a physiological situation (using laminar, steady flow) or a pathological condition (such as atherosclerosis), using an irregular flow. Interestingly, culturing human umbilical vein endothelial cells (HUVECs) in a laminar flow chamber in the presence of meningococci (N. meningitidis) was instrumental for unraveling some key molecular mechanisms of CNS invasion by these meningitis-causing human pathogens [111]. Regarding BBB differentiation, culture of brain endothelial cells under flow has been reported to induce the expression of the TJ proteins occludin and ZO-1, to promote actin cytoskeleton reorganization and to reduce endothelial permeability [113, 115–117]. In addition, very recent findings suggest that physiological shear stress (6 dynes/cm2) may increase the expression of a variety of BBB-associated genes in human brain microvascular endothelial cells, such as genes encoding for TJ proteins (ZO1, claudin-3, claudin-5), several influx transporters (Glut-1) and multidrug resistance efflux transporters (ABCB1/P-gp, ABCC5/MRP5) [116]. Nevertheless, further investigation is still required to get a better understanding of the contribution of shear stress to the maintenance of BBB integrity.
Dysregulation of the BBB via phosphorylation and relocalization of TJ proteins
Studies on CNS diseases associated with BBB dysfunctions (e.g. stroke, multiple sclerosis, cerebral infection, brain tumors, Parkinson’s and Alzheimer’s diseases) have pointed to various molecular mechanisms involved in disruption of TJ integrity, notably including Serine/Threonine (Ser/Thr)- and Tyrosine (Tyr)-phosphorylation, down-regulation, degradation or translocation of TJ proteins; a non exhaustive list of related reports are presented in Table 1. More than other TJ proteins (such as claudins or JAMs), occludin has been the focus of numerous studies investigating post-translational modifications and their consequences on TJ integrity (see for review: [118, 119]).
Ser/Thr-phosphorylation of TJ proteins and regulation of barrier permeability
Ser/Thr-phosphorylation forms of occludin are found concentrated at TJs whereas dephosphorylated occludin is rather detected on basolateral membranes and associated with disrupted TJs in epithelial cells [142, 143] as well as in brain endothelial cells in experimental autoimmune encephalomyelitis, a murine model of multiple sclerosis characterized by brain inflammation [144]. Regarding claudin-5, phosphorylation of its C-terminal domain on Thr207 residue in response to PKA or Rho kinase activation [120, 123, 145] generally affected TJ integrity in brain endothelial cells and increased permeability.
Differential regulation of TJs by Protein Kinases C (PKCs)
PKC-dependent pathways have been involved in endothelial barrier disruption, as reported following treatment by pertussis toxin, an inhibitor of Gαi heterotrimeric G proteins [146], or in response to the pro-inflammatory cytokine interleukin-6 (IL-6) which plays a critical role during hypoxia [147]. However, early reports had clearly established that PKC activity was crucial for BBB integrity in epithelial cells, inasmuch as PKC inhibitors completely blocked the formation of TJs [148, 149]; in addition, PKC-mediated phosphorylation of occludin (on residue Ser338) was involved in occludin targeting to TJs and TJ stabilization in epithelial MDCK cells [148].
At least part of the interpretation of these apparently conflicting data may be found in the heterogeneity of the PKC family. The Ser/Thr-kinases PKCs are indeed classified into conventional (cPKC: α, βI, βII and γ), novel (nPKC: δ, ε, θ, η, μ) and atypical (aPKC: λ,ζ) PKC isozymes [150] according to their modes of regulation. Accumulating evidence has pointed to a differential capacity of PKC isozymes to regulate BBB permeability. Indeed, activation of nPKC-θ and aPKC-ζ signaling by hypoxia-mediated TJ proteins results in relocalization (such as claudin-5, occludin, ZO-1) and increased BBB permeability in rat brain microvascular endothelial cells (in vitro and in vivo) [121, 151]. In human brain microvascular endothelial cells, cPKCα, cPKC βII and aPKCλ/ζ isoforms were activated by HIV-1 gp120 envelope protein, leading to BBB disruption, intracellular calcium increase and monocyte migration across cell monolayer [122]. Interestingly, when cPKC-α was found to contribute to TJ disassembly, nPKC-ε activation mediated TJ formation in epithelial MDCK cells [152]. In line with this observation, over-expression of cPKC-α in rat epididymal microvascular endothelial cells was reported to enhance thrombin-induced permeability, whereas nPKC-δ expression promoted barrier function [153]. By contrast, IL-25, expressed by mouse brain capillary endothelial cells, was shown to prevent inflammation-induced BBB disruption and down-regulation of TJ proteins (occludin, claudin-5, JAMs) through activation of the nPKC-ε pathway [154]. Altogether, these observations strongly suggest that nPKC-selective activation generally contributes to maintaining barrier integrity, whereas cPKC activation has the opposite effect, both in polarized epithelium and endothelium (Table 1).
Regarding aPKC isoforms (λ and ζ), they have been shown to contribute to the establishment of epithelial cell polarity, via participation in the PCP complex together with Par-3 and Par-6 [63, 68, 155]. As mentioned above, the PCP complex is recruited to endothelial TJs by Par-3 binding to JAM proteins [64–66]. Over-expression of a dominant negative mutant of aPKC causes mislocalization of Par-3 and affects the biogenesis of the TJs in epithelial cells [67], suggesting that Par-3 is a substrate of aPKC and that its localization in epithelial cells is dependent upon its phosphorylation. In the same line, the VE-cadherin/CCM1 (a protein encoded by the CCM1 gene which is mutated in a large proportion of patients affected by cerebral cavernous malformation) complex controls aPKC-ζ activation and Par-3 localization during early steps of brain endothelial cell polarization [98]. The participation of this PCP complex to TJ integrity was further illustrated by the recent observation that meningococcal adhesion to human cerebral endothelial cells recruited Par-3, Par-6 and aPKC-ζ under bacterial colonies and induced disruption of cell-cell junctions [156]. Surprisingly, a distinct Par-3/Par-6 complex, directly associated with VE-cadherin and lacking aPKC, has also been identified in endothelial cells [157]. Finally, although additional polarity complexes are known in epithelial cells (the apical Crumbs complex and the basolateral Scribble complex) where they also contribute to TJ formation and regulation, no similar observations have been reported, to our knowledge, in brain endothelial cells.
BBB disruption mediated by Rho/ Rho kinase and MLCK activation
The RhoA GTPase signaling pathway, activated by several membrane receptors, has been extensively documented in various cell types to induce actin cytoskeleton rearrangements involved in cell migration and proliferation. In brain endothelial cells, RhoA activation increased permeability, in response to inflammatory stimuli, through one of its major effectors Rho kinase (ROCK) [158, 159]. Among these inflammatory stimuli, chemokines like MCP-1/CCL2, acting via their seven transmembrane-domain receptors, are known to activate the RhoA/ROCK pathway in mouse brain endothelial cells, to induce occludin, claudin-5 and ZO-1 Ser/Thr-phosphorylation, followed by their delocalization from TJs, ultimately leading to increased barrier permeability [124, 160]. Similarly, enhanced monocyte migration across human brain endothelial cells was observed in an HIV-1 encephalitis model [123, 161]. Also, adhesion molecules like ICAM-1 and VCAM-1 were shown, in response to lymphocyte/monocyte adhesion, to transduce signals in rat brain endothelial cell lines including activation of the RhoA/ ROCK pathway [162, 163]: activation of this pathway ultimately leads to enhanced lymphocyte migration, suggesting that this process may be involved in the massive infiltration of immune cells into the CNS observed in multiple sclerosis. It must be mentioned, however, that lymphocyte migration across the BBB may also happen via a transcellular pathway, leaving intact endothelial TJs [164].
Rearrangements of the actin cytoskeleton have long been recognized to be regulated, not only by the RhoA/ROCK pathway, but also, often in a coordinated manner, by the myosin light chain kinase (MLCK): MLCK directly phosphorylates the myosin light chain, leading to actomyosin contraction and endothelial barrier disruption [165–167]. In the same line, inhibition of MLCK in bovine brain endothelial cells was more recently reported to prevent hypoxia-induced BBB disruption [127], whereas alcohol increased human brain endothelial cell permeability via activation of MLCK and phosphorylation of occludin and claudin-5 [125, 126]. Recently, pro-inflammatory cytokines (IL1β and TNFα), secreted by lymphocytes chronically infected by the HTLV-1 retrovirus, were reported to induce barrier disruption in the human brain endothelial cell line hCMEC/D3, associated with loss of occludin and ZO-1 through activation of the MLCK pathway [140].
In conclusion, as summarized in Table 1, inflammation- or infection-induced actin cytoskeleton rearrangements in brain endothelial cells, mediated by the RhoA/ROCK and/or MLCK pathways, are associated with the phosphorylation, followed by delocalization or degradation of TJ proteins, and BBB disruption.
BBB dysregulation by Tyr-phosphorylation of TJ proteins
Early studies with cultured bovine brain endothelial cells and MDCK cells had pointed to Tyr-phosphorylation as a mechanism for increasing TJ permeability [131]. Accumulating evidence demonstrated that Tyr-phosphorylation of TJ proteins, as well as AJ proteins, was directly involved in BBB disruption, as observed in various pathological situations, although the identity of the Tyr-kinases involved often remained unknown. Unlike occludin Ser/Thr phosphorylation associated with barrier formation, as mentioned above, occludin Tyr-phosphorylation was reported to be associated with increased permeability of cultured rat brain endothelial cells exposed to glutamate, as a way to mimic cerebral ischemia [129] (Table 1). Like other pro-inflammatory cytokines, transforming growth factor (TGF)-β1 is known to increase BBB permeability: as recently reported in bovine retinal and human brain endothelial cells, this effect was mediated by Tyr-phosphorylation of both claudin-5 and VE-cadherin [130]. Vascular endothelial growth factor (VEGF), a major angiogenic factor, which is drastically enhanced in response to hypoxia, promotes Tyr-phosphorylation of TJ proteins (ZO-1, occludin) in mouse brain and retinal endothelial cells [168, 169] either directly via its membrane receptor tyrosine kinase VEGFR2 or via the activation of the cytosolic tyrosine kinase c-src [128, 170]. VEGF-mediated Tyr-phosphorylation of TJ proteins in brain endothelial cells was often followed by their down-regulation and/or re-localization, leading to TJ destabilization and permeability increase [136–138, 171].
Alterations of expression and localization of TJ proteins
Caveolae are specialized plasma membrane microdomains, abundantly found in endothelial cells where they mediate various biological events such as transcytosis, vascular permeability and angiogenesis [172, 173]. They are enriched in the small membrane protein caveolin-1 which has been shown to recruit TJ proteins [9, 174]. Caveolae-mediated endocytosis induced by actin depolymerization was reported to evoke occludin internalization in MDCK cells [175]. Interestingly, exposure of cultured rat brain endothelial cells to the HIV-1 Tat protein was reported to increase TJ permeability, through alterations in expression and distribution of TJs proteins: occludin, claudin-5, ZO1, ZO2 [134, 176]. In the same line, the increase in TJ permeability observed in mouse brain endothelial cell response to the inflammatory cytokine CCL2 was recently shown to be associated with claudin-5 and occludin internalization in a caveolae-dependent manner [132]. Altogether these results strongly support the conclusion that alterations in expression and localization of TJ proteins, associated or not with their phosphorylation in response to various pathological stimuli, directly contribute to TJ disruption and BBB permeability increase (Table 1); in addition, they suggest a role of caveolin-1/caveolae in such TJ remodeling.
Conclusion
The brain endothelial TJ complex, which constitutes a key feature of the BBB, is now understood as a scaffolding and signaling platform in close interaction with the actin cytoskeleton and the AJ complex. It also appears as a dynamic complex, submitted to post-translational modifications in response to physiological and pathological stimuli. Indeed, perivascular cells of the NVU, notably astrocytes and pericytes, secrete multiple growth factors and morphogens that contribute to TJ formation and integrity. Conversely, various pathological situations associated with the presence of inflammatory cytokines, reactive oxygen species or pathogens, lead to TJ disruption following phosphorylation and/or internalization of TJ proteins.
Although our understanding of TJ architecture and function has significantly increased over the last ten years, a number of issues will have to be addressed in the next future, in particular taking advantage of new and/or global analysis technologies. For example, super-resolution light microscopy (time-lapse stimulated emission depletion (STED) imaging) recently appeared as a very powerful approach to unravel synapse assembly and plasticity [177]; in the same line, super-resolution microscopy of TJs (with a resolution down to 50–80 nm) of cerebral microvessels in brain slices should provide a more accurate understanding of TJ organization and dynamics. Also, thanks to the availability of validated BBB in vitro models, identification by mass spectrometry (MS/MS analysis) of the secreted proteins (so-called ‘secretome’) from brain endothelial co-cultures with astrocytes or pericytes may unravel new paracrine signaling pathways in the NVU which contribute to the stabilization of TJs at the BBB; in addition, similar analyses in the presence of inflammatory agents or pathogens [178] may highlight unsuspected mechanisms of TJ disruption. This approach will complement quantitative targeted absolute proteomics (also known as selected reaction monitoring (SRM)), an emerging approach to quantify membrane proteins [179]. This technology will also greatly benefit the field, allowing absolute quantification of TJ proteins in physiological and various pharmacological situations, as recently proposed [180]. The treatment of neurological diseases is currently hampered by difficulties encountered in delivering therapeutic compounds to the brain, across the BBB. Because previous drug delivery strategies based on transcellular transport machinery have shown limited efficacy so far, it is tempting to propose that transient modulation of TJs at the BBB, using in vitro models of the BBB and in vivo models of human pathologies, may constitute an alternative approach for drug delivery to the brain. Clearly, this field will benefit greatly from an in-depth understanding of TJ architecture and functional regulatory mechanisms.
Abbreviations
- AJ:
-
Adherens junction
- BBB:
-
Blood brain barrier
- CNS:
-
Central nervous system
- ECL:
-
Extracellular loop
- Fz:
-
Frizzled
- HUVEC:
-
Human umbilical vein endothelial cell
- IL:
-
Interleukin
- JAM:
-
Junctional adhesion molecule
- MAGUK:
-
Membrane associated guanylate kinase
- MLCK:
-
Myosin light chain kinase
- NVU:
-
Neurovascular unit
- PCP:
-
Planar cell polarity
- PKC:
-
Protein kinase C
- Ptch1:
-
Patched-1
- ROCK:
-
Rho kinase
- Shh:
-
Sonic hedgehog
- Smo:
-
Smoothened
- TAMP:
-
Tight junction-associated marvel proteins
- TGF:
-
Transforming growth factor
- TJ:
-
Tight junction
- VEGF:
-
Vascular endothelial growth factor
- ZO:
-
Zonula occludens.
References
Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ: Structure and function of the blood–brain barrier. Neurobiol Dis. 2010, 37: 13-25. 10.1016/j.nbd.2009.07.030.
Begley DJ, Brightman MW: Structural and functional aspects of the blood–brain barrier. Prog Drug Res. 2003, 61: 39-78.
Wolburg H, Noell S, Mack A, Wolburg-Buchholz K, Fallier-Becker P: Brain endothelial cells and the glio-vascular complex. Cell Tissue Res. 2009, 335: 75-96. 10.1007/s00441-008-0658-9.
Tsukita S, Furuse M, Itoh M: Multifunctional strands in tight junctions. Nat Rev Mol Cell Biol. 2001, 2: 285-293. 10.1038/35067088.
Cereijido M, Valdes J, Shoshani L, Contreras RG: Role of tight junctions in establishing and maintaining cell polarity. Annu Rev Physiol. 1998, 60: 161-177. 10.1146/annurev.physiol.60.1.161.
Gumbiner BM: Breaking through the tight junction barrier. J Cell Biol. 1993, 123: 1631-1633. 10.1083/jcb.123.6.1631.
Schneeberger EE, Lynch RD: Structure, function, and regulation of cellular tight junctions. Am J Physiol. 1992, 262: L647-661.
Vorbrodt AW, Dobrogowska DH: Molecular anatomy of intercellular junctions in brain endothelial and epithelial barriers: electron microscopist’s view. Brain Res Brain Res Rev. 2003, 42: 221-242.
Luissint AC, Federici C, Guillonneau F, Chretien F, Camoin L, Glacial F, Ganeshamoorthy K, Couraud PO: Guanine nucleotide-binding protein Galphai2: a new partner of claudin-5 that regulates tight junction integrity in human brain endothelial cells. J Cereb Blood Flow Metab. 2012, 32: 860-873. 10.1038/jcbfm.2011.202.
Aijaz S, Balda MS, Matter K: Tight junctions: molecular architecture and function. Int Rev Cytol. 2006, 248: 261-298.
Redzic Z: Molecular biology of the blood–brain and the blood-cerebrospinal fluid barriers: similarities and differences. Fluids Barriers CNS. 2011, 8: 3-10.1186/2045-8118-8-3.
Ando-Akatsuka Y, Saitou M, Hirase T, Kishi M, Sakakibara A, Itoh M, Yonemura S, Furuse M, Tsukita S: Interspecies diversity of the occludin sequence: cDNA cloning of human, mouse, dog, and rat-kangaroo homologues. J Cell Biol. 1996, 133: 43-47. 10.1083/jcb.133.1.43.
Furuse M, Hirase T, Itoh M, Nagafuchi A, Yonemura S, Tsukita S: Occludin: a novel integral membrane protein localizing at tight junctions. J Cell Biol. 1993, 123: 1777-1788. 10.1083/jcb.123.6.1777.
Balda MS, Whitney JA, Flores C, Gonzalez S, Cereijido M, Matter K: Functional dissociation of paracellular permeability and transepithelial electrical resistance and disruption of the apical-basolateral intramembrane diffusion barrier by expression of a mutant tight junction membrane protein. J Cell Biol. 1996, 134: 1031-1049. 10.1083/jcb.134.4.1031.
Ikenouchi J, Furuse M, Furuse K, Sasaki H, Tsukita S: Tricellulin constitutes a novel barrier at tricellular contacts of epithelial cells. J Cell Biol. 2005, 171: 939-945. 10.1083/jcb.200510043.
Raleigh DR, Marchiando AM, Zhang Y, Shen L, Sasaki H, Wang Y, Long M, Turner JR: Tight junction-associated MARVEL proteins marveld3, tricellulin, and occludin have distinct but overlapping functions. Mol Biol Cell. 2010, 21: 1200-1213. 10.1091/mbc.E09-08-0734.
Steed E, Rodrigues NT, Balda MS, Matter K: Identification of MarvelD3 as a tight junction-associated transmembrane protein of the occludin family. BMC Cell Biol. 2009, 10: 95-10.1186/1471-2121-10-95.
Yaffe Y, Shepshelovitch J, Nevo-Yassaf I, Yeheskel A, Shmerling H, Kwiatek JM, Gaus K, Pasmanik-Chor M, Hirschberg K: The MARVEL transmembrane motif of occludin mediates oligomerization and targeting to the basolateral surface in epithelia. J Cell Sci. 2012, 125: 3545-3556. 10.1242/jcs.100289.
Blasig IE, Winkler L, Lassowski B, Mueller SL, Zuleger N, Krause E, Krause G, Gast K, Kolbe M, Piontek J: On the self-association potential of transmembrane tight junction proteins. Cell Mol Life Sci. 2006, 63: 505-514. 10.1007/s00018-005-5472-x.
Walter JK, Castro V, Voss M, Gast K, Rueckert C, Piontek J, Blasig IE: Redox-sensitivity of the dimerization of occludin. Cell Mol Life Sci. 2009, 66: 3655-3662. 10.1007/s00018-009-0150-z.
Walter JK, Rueckert C, Voss M, Mueller SL, Piontek J, Gast K, Blasig IE: The oligomerization of the coiled coil-domain of occludin is redox sensitive. Ann N Y Acad Sci. 2009, 1165: 19-27. 10.1111/j.1749-6632.2009.04058.x.
Lochhead JJ, McCaffrey G, Quigley CE, Finch J, DeMarco KM, Nametz N, Davis TP: Oxidative stress increases blood–brain barrier permeability and induces alterations in occludin during hypoxia-reoxygenation. J Cereb Blood Flow Metab. 2010, 30: 1625-1636. 10.1038/jcbfm.2010.29.
Lochhead JJ, McCaffrey G, Sanchez-Covarrubias L, Finch JD, Demarco KM, Quigley CE, Davis TP, Ronaldson PT: Tempol modulates changes in xenobiotic permeability and occludin oligomeric assemblies at the blood–brain barrier during inflammatory pain. Am J Physiol Heart Circ Physiol. 2012, 302: H582-593. 10.1152/ajpheart.00889.2011.
McCaffrey G, Seelbach MJ, Staatz WD, Nametz N, Quigley C, Campos CR, Brooks TA, Davis TP: Occludin oligomeric assembly at tight junctions of the blood–brain barrier is disrupted by peripheral inflammatory hyperalgesia. J Neurochem. 2008, 106: 2395-2409. 10.1111/j.1471-4159.2008.05582.x.
Blasig IE, Bellmann C, Cording J, Del Vecchio G, Zwanziger D, Huber O, Haseloff RF: Occludin protein family: oxidative stress and reducing conditions. Antioxid Redox Signal. 2011, 15: 1195-1219. 10.1089/ars.2010.3542.
Medina R, Rahner C, Mitic LL, Anderson JM, Van Itallie CM: Occludin localization at the tight junction requires the second extracellular loop. J Membr Biol. 2000, 178: 235-247. 10.1007/s002320010031.
Wong V, Gumbiner BM: A synthetic peptide corresponding to the extracellular domain of occludin perturbs the tight junction permeability barrier. J Cell Biol. 1997, 136: 399-409. 10.1083/jcb.136.2.399.
Tavelin S, Hashimoto K, Malkinson J, Lazorova L, Toth I, Artursson P: A new principle for tight junction modulation based on occludin peptides. Mol Pharmacol. 2003, 64: 1530-1540. 10.1124/mol.64.6.1530.
Everett RS, Vanhook MK, Barozzi N, Toth I, Johnson LG: Specific modulation of airway epithelial tight junctions by apical application of an occludin peptide. Mol Pharmacol. 2006, 69: 492-500.
Furuse M, Fujimoto K, Sato N, Hirase T, Tsukita S: Overexpression of occludin, a tight junction-associated integral membrane protein, induces the formation of intracellular multilamellar bodies bearing tight junction-like structures. J Cell Sci. 1996, 109 (Pt 2): 429-435.
McCarthy KM, Skare IB, Stankewich MC, Furuse M, Tsukita S, Rogers RA, Lynch RD, Schneeberger EE: Occludin is a functional component of the tight junction. J Cell Sci. 1996, 109 (Pt 9): 2287-2298.
Schubert-Unkmeir A, Konrad C, Slanina H, Czapek F, Hebling S, Frosch M: Neisseria meningitidis induces brain microvascular endothelial cell detachment from the matrix and cleavage of occludin: a role for MMP-8. PLoS Pathog. 2010, 6: e1000874-10.1371/journal.ppat.1000874.
Xu R, Feng X, Xie X, Zhang J, Wu D, Xu L: HIV-1 Tat protein increases the permeability of brain endothelial cells by both inhibiting occludin expression and cleaving occludin via matrix metalloproteinase-9. Brain Res. 2012, 1436: 13-19.
Moroi S, Saitou M, Fujimoto K, Sakakibara A, Furuse M, Yoshida O, Tsukita S: Occludin is concentrated at tight junctions of mouse/rat but not human/guinea pig Sertoli cells in testes. Am J Physiol. 1998, 274: C1708-1717.
Saitou M, Fujimoto K, Doi Y, Itoh M, Fujimoto T, Furuse M, Takano H, Noda T, Tsukita S: Occludin-deficient embryonic stem cells can differentiate into polarized epithelial cells bearing tight junctions. J Cell Biol. 1998, 141: 397-408. 10.1083/jcb.141.2.397.
Saitou M, Furuse M, Sasaki H, Schulzke JD, Fromm M, Takano H, Noda T, Tsukita S: Complex phenotype of mice lacking occludin, a component of tight junction strands. Mol Biol Cell. 2000, 11: 4131-4142.
Schulzke JD, Gitter AH, Mankertz J, Spiegel S, Seidler U, Amasheh S, Saitou M, Tsukita S, Fromm M: Epithelial transport and barrier function in occludin-deficient mice. Biochim Biophys Acta. 2005, 1669: 34-42. 10.1016/j.bbamem.2005.01.008.
Furuse M, Sasaki H, Tsukita S: Manner of interaction of heterogeneous claudin species within and between tight junction strands. J Cell Biol. 1999, 147: 891-903. 10.1083/jcb.147.4.891.
Mineta K, Yamamoto Y, Yamazaki Y, Tanaka H, Tada Y, Saito K, Tamura A, Igarashi M, Endo T, Takeuchi K, Tsukita S: Predicted expansion of the claudin multigene family. FEBS Lett. 2011, 585: 606-612. 10.1016/j.febslet.2011.01.028.
Morita K, Furuse M, Fujimoto K, Tsukita S: Claudin multigene family encoding four-transmembrane domain protein components of tight junction strands. Proc Natl Acad Sci USA. 1999, 96: 511-516. 10.1073/pnas.96.2.511.
Wolburg H, Wolburg-Buchholz K, Kraus J, Rascher-Eggstein G, Liebner S, Hamm S, Duffner F, Grote EH, Risau W, Engelhardt B: Localization of claudin-3 in tight junctions of the blood–brain barrier is selectively lost during experimental autoimmune encephalomyelitis and human glioblastoma multiforme. Acta Neuropathol. 2003, 105: 586-592.
Morita K, Sasaki H, Furuse M, Tsukita S: Endothelial claudin: claudin-5/TMVCF constitutes tight junction strands in endothelial cells. J Cell Biol. 1999, 147: 185-194. 10.1083/jcb.147.1.185.
Nitta T, Hata M, Gotoh S, Seo Y, Sasaki H, Hashimoto N, Furuse M, Tsukita S: Size-selective loosening of the blood–brain barrier in claudin-5-deficient mice. J Cell Biol. 2003, 161: 653-660. 10.1083/jcb.200302070.
Ohtsuki S, Sato S, Yamaguchi H, Kamoi M, Asashima T, Terasaki T: Exogenous expression of claudin-5 induces barrier properties in cultured rat brain capillary endothelial cells. J Cell Physiol. 2007, 210: 81-86. 10.1002/jcp.20823.
Piehl C, Piontek J, Cording J, Wolburg H, Blasig IE: Participation of the second extracellular loop of claudin-5 in paracellular tightening against ions, small and large molecules. Cell Mol Life Sci. 2010, 67: 2131-2140. 10.1007/s00018-010-0332-8.
Piontek J, Winkler L, Wolburg H, Muller SL, Zuleger N, Piehl C, Wiesner B, Krause G, Blasig IE: Formation of tight junction: determinants of homophilic interaction between classic claudins. FASEB J. 2008, 22: 146-158.
Zhang J, Piontek J, Wolburg H, Piehl C, Liss M, Otten C, Christ A, Willnow TE, Blasig IE, Abdelilah-Seyfried S: Establishment of a neuroepithelial barrier by Claudin5a is essential for zebrafish brain ventricular lumen expansion. Proc Natl Acad Sci USA. 2010, 107: 1425-1430. 10.1073/pnas.0911996107.
Coyne CB, Gambling TM, Boucher RC, Carson JL, Johnson LG: Role of claudin interactions in airway tight junctional permeability. Am J Physiol Lung Cell Mol Physiol. 2003, 285: L1166-1178.
Piontek J, Fritzsche S, Cording J, Richter S, Hartwig J, Walter M, Yu D, Turner JR, Gehring C, Rahn HP, et al: Elucidating the principles of the molecular organization of heteropolymeric tight junction strands. Cell Mol Life Sci. 2011, 68: 3903-3918. 10.1007/s00018-011-0680-z.
Bazzoni G: Pathobiology of junctional adhesion molecules. Antioxid Redox Signal. 2011, 15: 1221-1234. 10.1089/ars.2010.3867.
Ebnet K, Suzuki A, Ohno S, Vestweber D: Junctional adhesion molecules (JAMs): more molecules with dual functions?. J Cell Sci. 2004, 117: 19-29. 10.1242/jcs.00930.
Stevenson BR, Siliciano JD, Mooseker MS, Goodenough DA: Identification of ZO-1: a high molecular weight polypeptide associated with the tight junction (zonula occludens) in a variety of epithelia. J Cell Biol. 1986, 103: 755-766. 10.1083/jcb.103.3.755.
Gumbiner B, Lowenkopf T, Apatira D: Identification of a 160-kDa polypeptide that binds to the tight junction protein ZO-1. Proc Natl Acad Sci USA. 1991, 88: 3460-3464. 10.1073/pnas.88.8.3460.
Haskins J, Gu L, Wittchen ES, Hibbard J, Stevenson BR: ZO-3, a novel member of the MAGUK protein family found at the tight junction, interacts with ZO-1 and occludin. J Cell Biol. 1998, 141: 199-208. 10.1083/jcb.141.1.199.
Fanning AS, Jameson BJ, Jesaitis LA, Anderson JM: The tight junction protein ZO-1 establishes a link between the transmembrane protein occludin and the actin cytoskeleton. J Biol Chem. 1998, 273: 29745-29753. 10.1074/jbc.273.45.29745.
Itoh M, Morita K, Tsukita S: Characterization of ZO-2 as a MAGUK family member associated with tight as well as adherens junctions with a binding affinity to occludin and alpha catenin. J Biol Chem. 1999, 274: 5981-5986. 10.1074/jbc.274.9.5981.
Itoh M, Furuse M, Morita K, Kubota K, Saitou M, Tsukita S: Direct binding of three tight junction-associated MAGUKs, ZO-1, ZO-2, and ZO-3, with the COOH termini of claudins. J Cell Biol. 1999, 147: 1351-1363. 10.1083/jcb.147.6.1351.
Bazzoni G, Martinez-Estrada OM, Orsenigo F, Cordenonsi M, Citi S, Dejana E: Interaction of junctional adhesion molecule with the tight junction components ZO-1, cingulin, and occludin. J Biol Chem. 2000, 275: 20520-20526. 10.1074/jbc.M905251199.
Furuse M, Itoh M, Hirase T, Nagafuchi A, Yonemura S, Tsukita S: Direct association of occludin with ZO-1 and its possible involvement in the localization of occludin at tight junctions. J Cell Biol. 1994, 127: 1617-1626. 10.1083/jcb.127.6.1617.
Umeda K, Ikenouchi J, Katahira-Tayama S, Furuse K, Sasaki H, Nakayama M, Matsui T, Tsukita S, Furuse M: ZO-1 and ZO-2 independently determine where claudins are polymerized in tight-junction strand formation. Cell. 2006, 126: 741-754. 10.1016/j.cell.2006.06.043.
Nusrat A, Brown GT, Tom J, Drake A, Bui TT, Quan C, Mrsny RJ: Multiple protein interactions involving proposed extracellular loop domains of the tight junction protein occludin. Mol Biol Cell. 2005, 16: 1725-1734. 10.1091/mbc.E04-06-0465.
Fanning AS, Ma TY, Anderson JM: Isolation and functional characterization of the actin binding region in the tight junction protein ZO-1. FASEB J. 2002, 16: 1835-1837.
Izumi Y, Hirose T, Tamai Y, Hirai S, Nagashima Y, Fujimoto T, Tabuse Y, Kemphues KJ, Ohno S: An atypical PKC directly associates and colocalizes at the epithelial tight junction with ASIP, a mammalian homologue of caenorhabditis elegans polarity protein PAR-3. J Cell Biol. 1998, 143: 95-106. 10.1083/jcb.143.1.95.
Ebnet K, Aurrand-Lions M, Kuhn A, Kiefer F, Butz S, Zander K, Meyer zu Brickwedde MK, Suzuki A, Imhof BA, Vestweber D: The junctional adhesion molecule (JAM) family members JAM-2 and JAM-3 associate with the cell polarity protein PAR-3: a possible role for JAMs in endothelial cell polarity. J Cell Sci. 2003, 116: 3879-3891. 10.1242/jcs.00704.
Ebnet K, Suzuki A, Horikoshi Y, Hirose T, Meyer Zu Brickwedde MK, Ohno S, Vestweber D: The cell polarity protein ASIP/PAR-3 directly associates with junctional adhesion molecule (JAM). EMBO J. 2001, 20: 3738-3748. 10.1093/emboj/20.14.3738.
Itoh M, Sasaki H, Furuse M, Ozaki H, Kita T, Tsukita S: Junctional adhesion molecule (JAM) binds to PAR-3: a possible mechanism for the recruitment of PAR-3 to tight junctions. J Cell Biol. 2001, 154: 491-497. 10.1083/jcb.200103047.
Suzuki A, Yamanaka T, Hirose T, Manabe N, Mizuno K, Shimizu M, Akimoto K, Izumi Y, Ohnishi T, Ohno S: Atypical protein kinase C is involved in the evolutionarily conserved par protein complex and plays a critical role in establishing epithelia-specific junctional structures. J Cell Biol. 2001, 152: 1183-1196. 10.1083/jcb.152.6.1183.
Joberty G, Petersen C, Gao L, Macara IG: The cell-polarity protein Par6 links Par3 and atypical protein kinase C to Cdc42. Nat Cell Biol. 2000, 2: 531-539. 10.1038/35019573.
Lin D, Edwards AS, Fawcett JP, Mbamalu G, Scott JD, Pawson T: A mammalian PAR-3-PAR-6 complex implicated in Cdc42/Rac1 and aPKC signalling and cell polarity. Nat Cell Biol. 2000, 2: 540-547. 10.1038/35019582.
Daneman R, Zhou L, Agalliu D, Cahoy JD, Kaushal A, Barres BA: The mouse blood–brain barrier transcriptome: a new resource for understanding the development and function of brain endothelial cells. PLoS One. 2010, 5: e13741-10.1371/journal.pone.0013741.
Denker BM, Saha C, Khawaja S, Nigam SK: Involvement of a heterotrimeric G protein alpha subunit in tight junction biogenesis. J Biol Chem. 1996, 271: 25750-25753. 10.1074/jbc.271.42.25750.
Saha C, Nigam SK, Denker BM: Involvement of Galphai2 in the maintenance and biogenesis of epithelial cell tight junctions. J Biol Chem. 1998, 273: 21629-21633. 10.1074/jbc.273.34.21629.
Fabian G, Szabo CA, Bozo B, Greenwood J, Adamson P, Deli MA, Joo F, Krizbai IA, Szucs M: Expression of G-protein subtypes in cultured cerebral endothelial cells. Neurochem Int. 1998, 33: 179-185. 10.1016/S0197-0186(98)00008-4.
Adamson P, Wilbourn B, Etienne-Manneville S, Calder V, Beraud E, Milligan G, Couraud PO, Greenwood J: Lymphocyte trafficking through the blood–brain barrier is dependent on endothelial cell heterotrimeric G-protein signaling. FASEB J. 2002, 16: 1185-1194. 10.1096/fj.02-0035com.
Pero RS, Borchers MT, Spicher K, Ochkur SI, Sikora L, Rao SP, Abdala-Valencia H, O’Neill KR, Shen H, McGarry MP, et al: Galphai2-mediated signaling events in the endothelium are involved in controlling leukocyte extravasation. Proc Natl Acad Sci USA. 2007, 104: 4371-4376. 10.1073/pnas.0700185104.
Abbott NJ, Ronnback L, Hansson E: Astrocyte-endothelial interactions at the blood–brain barrier. Nat Rev Neurosci. 2006, 7: 41-53. 10.1038/nrn1824.
Janzer RC, Raff MC: Astrocytes induce blood–brain barrier properties in endothelial cells. Nature. 1987, 325: 253-257. 10.1038/325253a0.
Bonkowski D, Katyshev V, Balabanov RD, Borisov A, Dore-Duffy P: The CNS microvascular pericyte: pericyte-astrocyte crosstalk in the regulation of tissue survival. Fluids Barriers CNS. 2011, 8: 8-10.1186/2045-8118-8-8.
Sa-Pereira I, Brites D, Brito MA: Neurovascular unit: a focus on pericytes. Mol Neurobiol. 2012, 45: 327-347. 10.1007/s12035-012-8244-2.
Winkler EA, Bell RD, Zlokovic BV: Pericyte-specific expression of PDGF beta receptor in mouse models with normal and deficient PDGF beta receptor signaling. Mol Neurodegener. 2010, 5: 32-10.1186/1750-1326-5-32.
Arthur FE, Shivers RR, Bowman PD: Astrocyte-mediated induction of tight junctions in brain capillary endothelium: an efficient in vitro model. Brain Res. 1987, 433: 155-159.
Colgan OC, Collins NT, Ferguson G, Murphy RP, Birney YA, Cahill PA, Cummins PM: Influence of basolateral condition on the regulation of brain microvascular endothelial tight junction properties and barrier function. Brain Res. 2008, 1193: 84-92.
Lee SW, Kim WJ, Choi YK, Song HS, Son MJ, Gelman IH, Kim YJ, Kim KW: SSeCKS regulates angiogenesis and tight junction formation in blood–brain barrier. Nat Med. 2003, 9: 900-906. 10.1038/nm889.
Neuhaus J, Risau W, Wolburg H: Induction of blood–brain barrier characteristics in bovine brain endothelial cells by rat astroglial cells in transfilter coculture. Ann N Y Acad Sci. 1991, 633: 578-580. 10.1111/j.1749-6632.1991.tb15667.x.
Rubin LL, Hall DE, Porter S, Barbu K, Cannon C, Horner HC, Janatpour M, Liaw CW, Manning K, Morales J, et al: A cell culture model of the blood–brain barrier. J Cell Biol. 1991, 115: 1725-1735. 10.1083/jcb.115.6.1725.
Tao-Cheng JH, Nagy Z, Brightman MW: Tight junctions of brain endothelium in vitro are enhanced by astroglia. J Neurosci. 1987, 7: 3293-3299.
Wolburg H, Neuhaus J, Kniesel U, Krauss B, Schmid EM, Ocalan M, Farrell C, Risau W: Modulation of tight junction structure in blood–brain barrier endothelial cells. Effects of tissue culture, second messengers and cocultured astrocytes. J Cell Sci. 1994, 107 (Pt 5): 1347-1357.
Hori S, Ohtsuki S, Hosoya K, Nakashima E, Terasaki T: A pericyte-derived angiopoietin-1 multimeric complex induces occludin gene expression in brain capillary endothelial cells through Tie-2 activation in vitro. J Neurochem. 2004, 89: 503-513. 10.1111/j.1471-4159.2004.02343.x.
Daneman R, Zhou L, Kebede AA, Barres BA: Pericytes are required for blood–brain barrier integrity during embryogenesis. Nature. 2010, 468: 562-566. 10.1038/nature09513.
Armulik A, Genove G, Mae M, Nisancioglu MH, Wallgard E, Niaudet C, He L, Norlin J, Lindblom P, Strittmatter K, et al: Pericytes regulate the blood–brain barrier. Nature. 2010, 468: 557-561. 10.1038/nature09522.
Dohgu S, Takata F, Yamauchi A, Nakagawa S, Egawa T, Naito M, Tsuruo T, Sawada Y, Niwa M, Kataoka Y: Brain pericytes contribute to the induction and up-regulation of blood–brain barrier functions through transforming growth factor-beta production. Brain Res. 2005, 1038: 208-215. 10.1016/j.brainres.2005.01.027.
Liebner S, Czupalla CJ, Wolburg H: Current concepts of blood–brain barrier development. Int J Dev Biol. 2011, 55: 467-476. 10.1387/ijdb.103224sl.
Osada T, Gu YH, Kanazawa M, Tsubota Y, Hawkins BT, Spatz M, Milner R, del Zoppo GJ: Interendothelial claudin-5 expression depends on cerebral endothelial cell-matrix adhesion by beta(1)-integrins. J Cereb Blood Flow Metab. 2011, 31: 1972-1985. 10.1038/jcbfm.2011.99.
Zovein AC, Luque A, Turlo KA, Hofmann JJ, Yee KM, Becker MS, Fassler R, Mellman I, Lane TF, Iruela-Arispe ML: Beta1 integrin establishes endothelial cell polarity and arteriolar lumen formation via a Par3-dependent mechanism. Dev Cell. 2010, 18: 39-51. 10.1016/j.devcel.2009.12.006.
Alvarez JI, Dodelet-Devillers A, Kebir H, Ifergan I, Fabre PJ, Terouz S, Sabbagh M, Wosik K, Bourbonniere L, Bernard M, et al: The Hedgehog pathway promotes blood–brain barrier integrity and CNS immune quiescence. Science. 2011, 334: 1727-1731. 10.1126/science.1206936.
Liebner S, Corada M, Bangsow T, Babbage J, Taddei A, Czupalla CJ, Reis M, Felici A, Wolburg H, Fruttiger M, et al: Wnt/beta-catenin signaling controls development of the blood–brain barrier. J Cell Biol. 2008, 183: 409-417. 10.1083/jcb.200806024.
Taddei A, Giampietro C, Conti A, Orsenigo F, Breviario F, Pirazzoli V, Potente M, Daly C, Dimmeler S, Dejana E: Endothelial adherens junctions control tight junctions by VE-cadherin-mediated upregulation of claudin-5. Nat Cell Biol. 2008, 10: 923-934. 10.1038/ncb1752.
Lampugnani MG, Orsenigo F, Rudini N, Maddaluno L, Boulday G, Chapon F, Dejana E: CCM1 regulates vascular-lumen organization by inducing endothelial polarity. J Cell Sci. 2010, 123: 1073-1080. 10.1242/jcs.059329.
Chien AJ, Conrad WH, Moon RT: A Wnt survival guide: from flies to human disease. J Invest Dermatol. 2009, 129: 1614-1627. 10.1038/jid.2008.445.
Stenman JM, Rajagopal J, Carroll TJ, Ishibashi M, McMahon J, McMahon AP: Canonical Wnt signaling regulates organ-specific assembly and differentiation of CNS vasculature. Science. 2008, 322: 1247-1250. 10.1126/science.1164594.
Ye X, Wang Y, Cahill H, Yu M, Badea TC, Smallwood PM, Peachey NS, Nathans J: Norrin, frizzled-4, and Lrp5 signaling in endothelial cells controls a genetic program for retinal vascularization. Cell. 2009, 139: 285-298. 10.1016/j.cell.2009.07.047.
Daneman R, Agalliu D, Zhou L, Kuhnert F, Kuo CJ, Barres BA: Wnt/beta-catenin signaling is required for CNS, but not non-CNS, angiogenesis. Proc Natl Acad Sci USA. 2009, 106: 641-646. 10.1073/pnas.0805165106.
Paolinelli R, Corada M, Orsenigo F, Dejana E: The molecular basis of the blood brain barrier differentiation and maintenance. Is it still a mystery?. Pharmacol Res. 2011, 63: 165-171. 10.1016/j.phrs.2010.11.012.
Wu C, Ivars F, Anderson P, Hallmann R, Vestweber D, Nilsson P, Robenek H, Tryggvason K, Song J, Korpos E, et al: Endothelial basement membrane laminin alpha5 selectively inhibits T lymphocyte extravasation into the brain. Nat Med. 2009, 15: 519-527. 10.1038/nm.1957.
Barber AJ, Lieth E: Agrin accumulates in the brain microvascular basal lamina during development of the blood–brain barrier. Dev Dyn. 1997, 208: 62-74. 10.1002/(SICI)1097-0177(199701)208:1<62::AID-AJA6>3.0.CO;2-#.
Rascher G, Fischmann A, Kroger S, Duffner F, Grote EH, Wolburg H: Extracellular matrix and the blood–brain barrier in glioblastoma multiforme: spatial segregation of tenascin and agrin. Acta Neuropathol. 2002, 104: 85-91. 10.1007/s00401-002-0524-x.
Gavard J: Breaking the VE-cadherin bonds. FEBS Lett. 2009, 583: 1-6. 10.1016/j.febslet.2008.11.032.
Dejana E, Giampietro C: Vascular endothelial-cadherin and vascular stability. Curr Opin Hematol. 2012, 19: 218-223. 10.1097/MOH.0b013e3283523e1c.
Harris ES, Nelson WJ: VE-cadherin: at the front, center, and sides of endothelial cell organization and function. Curr Opin Cell Biol. 2010, 22: 651-658. 10.1016/j.ceb.2010.07.006.
Stanness KA, Westrum LE, Fornaciari E, Mascagni P, Nelson JA, Stenglein SG, Myers T, Janigro D: Morphological and functional characterization of an in vitro blood–brain barrier model. Brain Res. 1997, 771: 329-342. 10.1016/S0006-8993(97)00829-9.
Mairey E, Genovesio A, Donnadieu E, Bernard C, Jaubert F, Pinard E, Seylaz J, Olivo-Marin JC, Nassif X, Dumenil G: Cerebral microcirculation shear stress levels determine Neisseria meningitidis attachment sites along the blood–brain barrier. J Exp Med. 2006, 203: 1939-1950. 10.1084/jem.20060482.
Santaguida S, Janigro D, Hossain M, Oby E, Rapp E, Cucullo L: Side by side comparison between dynamic versus static models of blood–brain barrier in vitro: a permeability study. Brain Res. 2006, 1109: 1-13. 10.1016/j.brainres.2006.06.027.
Colgan OC, Ferguson G, Collins NT, Murphy RP, Meade G, Cahill PA, Cummins PM: Regulation of bovine brain microvascular endothelial tight junction assembly and barrier function by laminar shear stress. Am J Physiol Heart Circ Physiol. 2007, 292: H3190-3197. 10.1152/ajpheart.01177.2006.
Krizanac-Bengez L, Mayberg MR, Cunningham E, Hossain M, Ponnampalam S, Parkinson FE, Janigro D: Loss of shear stress induces leukocyte-mediated cytokine release and blood–brain barrier failure in dynamic in vitro blood–brain barrier model. J Cell Physiol. 2006, 206: 68-77. 10.1002/jcp.20429.
Siddharthan V, Kim YV, Liu S, Kim KS: Human astrocytes/astrocyte-conditioned medium and shear stress enhance the barrier properties of human brain microvascular endothelial cells. Brain Res. 2007, 1147: 39-50.
Cucullo L, Hossain M, Puvenna V, Marchi N, Janigro D: The role of shear stress in blood–brain barrier endothelial physiology. BMC Neurosci. 2011, 12: 40-10.1186/1471-2202-12-40.
Walsh TG, Murphy RP, Fitzpatrick P, Rochfort KD, Guinan AF, Murphy A, Cummins PM: Stabilization of brain microvascular endothelial barrier function by shear stress involves VE-cadherin signaling leading to modulation of pTyr-occludin levels. J Cell Physiol. 2011, 226: 3053-3063. 10.1002/jcp.22655.
Cummins PM: Occludin: one protein, many forms. Mol Cell Biol. 2012, 32: 242-250. 10.1128/MCB.06029-11.
Dorfel MJ, Huber O: Modulation of tight junction structure and function by kinases and phosphatases targeting occludin. J Biomed Biotechnol. 2012, 2012: 807356-
Soma T, Chiba H, Kato-Mori Y, Wada T, Yamashita T, Kojima T, Sawada N: Thr(207) of claudin-5 is involved in size-selective loosening of the endothelial barrier by cyclic AMP. Exp Cell Res. 2004, 300: 202-212. 10.1016/j.yexcr.2004.07.012.
Willis CL, Meske DS, Davis TP: Protein kinase C activation modulates reversible increase in cortical blood–brain barrier permeability and tight junction protein expression during hypoxia and posthypoxic reoxygenation. J Cereb Blood Flow Metab. 2010, 30: 1847-1859. 10.1038/jcbfm.2010.119.
Kanmogne GD, Schall K, Leibhart J, Knipe B, Gendelman HE, Persidsky Y: HIV-1 gp120 compromises blood–brain barrier integrity and enhances monocyte migration across blood–brain barrier: implication for viral neuropathogenesis. J Cereb Blood Flow Metab. 2007, 27: 123-134. 10.1038/sj.jcbfm.9600330.
Yamamoto M, Ramirez SH, Sato S, Kiyota T, Cerny RL, Kaibuchi K, Persidsky Y, Ikezu T: Phosphorylation of claudin-5 and occludin by rho kinase in brain endothelial cells. Am J Pathol. 2008, 172: 521-533. 10.2353/ajpath.2008.070076.
Stamatovic SM, Dimitrijevic OB, Keep RF, Andjelkovic AV: Protein kinase Calpha-RhoA cross-talk in CCL2-induced alterations in brain endothelial permeability. J Biol Chem. 2006, 281: 8379-8388. 10.1074/jbc.M513122200.
Haorah J, Knipe B, Leibhart J, Ghorpade A, Persidsky Y: Alcohol-induced oxidative stress in brain endothelial cells causes blood–brain barrier dysfunction. J Leukoc Biol. 2005, 78: 1223-1232. 10.1189/jlb.0605340.
Haorah J, Heilman D, Knipe B, Chrastil J, Leibhart J, Ghorpade A, Miller DW, Persidsky Y: Ethanol-induced activation of myosin light chain kinase leads to dysfunction of tight junctions and blood–brain barrier compromise. Alcohol Clin Exp Res. 2005, 29: 999-1009. 10.1097/01.ALC.0000166944.79914.0A.
Kuhlmann CR, Tamaki R, Gamerdinger M, Lessmann V, Behl C, Kempski OS, Luhmann HJ: Inhibition of the myosin light chain kinase prevents hypoxia-induced blood–brain barrier disruption. J Neurochem. 2007, 102: 501-507. 10.1111/j.1471-4159.2007.04506.x.
Takenaga Y, Takagi N, Murotomi K, Tanonaka K, Takeo S: Inhibition of Src activity decreases tyrosine phosphorylation of occludin in brain capillaries and attenuates increase in permeability of the blood–brain barrier after transient focal cerebral ischemia. J Cereb Blood Flow Metab. 2009, 29: 1099-1108. 10.1038/jcbfm.2009.30.
Andras IE, Deli MA, Veszelka S, Hayashi K, Hennig B, Toborek M: The NMDA and AMPA/KA receptors are involved in glutamate-induced alterations of occludin expression and phosphorylation in brain endothelial cells. J Cereb Blood Flow Metab. 2007, 27: 1431-1443. 10.1038/sj.jcbfm.9600445.
Shen W, Li S, Chung SH, Zhu L, Stayt J, Su T, Couraud PO, Romero IA, Weksler B, Gillies MC: Tyrosine phosphorylation of VE-cadherin and claudin-5 is associated with TGF-beta1-induced permeability of centrally derived vascular endothelium. Eur J Cell Biol. 2011, 90: 323-332. 10.1016/j.ejcb.2010.10.013.
Staddon JM, Herrenknecht K, Smales C, Rubin LL: Evidence that tyrosine phosphorylation may increase tight junction permeability. J Cell Sci. 1995, 108 (Pt 2): 609-619.
Stamatovic SM, Keep RF, Wang MM, Jankovic I, Andjelkovic AV: Caveolae-mediated internalization of occludin and claudin-5 during CCL2-induced tight junction remodeling in brain endothelial cells. J Biol Chem. 2009, 284: 19053-19066. 10.1074/jbc.M109.000521.
Tai LM, Holloway KA, Male DK, Loughlin AJ, Romero IA: Amyloid-beta-induced occludin down-regulation and increased permeability in human brain endothelial cells is mediated by MAPK activation. J Cell Mol Med. 2010, 14: 1101-1112.
Andras IE, Pu H, Tian J, Deli MA, Nath A, Hennig B, Toborek M: Signaling mechanisms of HIV-1 Tat-induced alterations of claudin-5 expression in brain endothelial cells. J Cereb Blood Flow Metab. 2005, 25: 1159-1170. 10.1038/sj.jcbfm.9600115.
Fischer S, Wiesnet M, Marti HH, Renz D, Schaper W: Simultaneous activation of several second messengers in hypoxia-induced hyperpermeability of brain derived endothelial cells. J Cell Physiol. 2004, 198: 359-369. 10.1002/jcp.10417.
Mark KS, Davis TP: Cerebral microvascular changes in permeability and tight junctions induced by hypoxia-reoxygenation. Am J Physiol Heart Circ Physiol. 2002, 282: H1485-1494.
Koto T, Takubo K, Ishida S, Shinoda H, Inoue M, Tsubota K, Okada Y, Ikeda E: Hypoxia disrupts the barrier function of neural blood vessels through changes in the expression of claudin-5 in endothelial cells. Am J Pathol. 2007, 170: 1389-1397. 10.2353/ajpath.2007.060693.
Argaw AT, Gurfein BT, Zhang Y, Zameer A, John GR: VEGF-mediated disruption of endothelial CLN-5 promotes blood–brain barrier breakdown. Proc Natl Acad Sci USA. 2009, 106: 1977-1982. 10.1073/pnas.0808698106.
Jiao H, Wang Z, Liu Y, Wang P, Xue Y: Specific role of tight junction proteins claudin-5, occludin, and ZO-1 of the blood–brain barrier in a focal cerebral ischemic insult. J Mol Neurosci. 2011, 44: 130-139. 10.1007/s12031-011-9496-4.
Afonso PV, Ozden S, Prevost MC, Schmitt C, Seilhean D, Weksler B, Couraud PO, Gessain A, Romero IA, Ceccaldi PE: Human blood–brain barrier disruption by retroviral-infected lymphocytes: role of myosin light chain kinase in endothelial tight-junction disorganization. J Immunol. 2007, 179: 2576-2583.
Schreibelt G, Kooij G, Reijerkerk A, van Doorn R, Gringhuis SI, van der Pol S, Weksler BB, Romero IA, Couraud PO, Piontek J, et al: Reactive oxygen species alter brain endothelial tight junction dynamics via RhoA, PI3 kinase, and PKB signaling. FASEB J. 2007, 21: 3666-3676. 10.1096/fj.07-8329com.
Clarke H, Soler AP, Mullin JM: Protein kinase C activation leads to dephosphorylation of occludin and tight junction permeability increase in LLC-PK1 epithelial cell sheets. J Cell Sci. 2000, 113 (Pt 18): 3187-3196.
Sakakibara A, Furuse M, Saitou M, Ando-Akatsuka Y, Tsukita S: Possible involvement of phosphorylation of occludin in tight junction formation. J Cell Biol. 1997, 137: 1393-1401. 10.1083/jcb.137.6.1393.
Morgan L, Shah B, Rivers LE, Barden L, Groom AJ, Chung R, Higazi D, Desmond H, Smith T, Staddon JM: Inflammation and dephosphorylation of the tight junction protein occludin in an experimental model of multiple sclerosis. Neuroscience. 2007, 147: 664-673. 10.1016/j.neuroscience.2007.04.051.
Ishizaki T, Chiba H, Kojima T, Fujibe M, Soma T, Miyajima H, Nagasawa K, Wada I, Sawada N: Cyclic AMP induces phosphorylation of claudin-5 immunoprecipitates and expression of claudin-5 gene in blood–brain-barrier endothelial cells via protein kinase A-dependent and -independent pathways. Exp Cell Res. 2003, 290: 275-288. 10.1016/S0014-4827(03)00354-9.
Bruckener KE, El Baya A, Galla HJ, Schmidt MA: Permeabilization in a cerebral endothelial barrier model by pertussis toxin involves the PKC effector pathway and is abolished by elevated levels of cAMP. J Cell Sci. 2003, 116: 1837-1846. 10.1242/jcs.00378.
Desai TR, Leeper NJ, Hynes KL, Gewertz BL: Interleukin-6 causes endothelial barrier dysfunction via the protein kinase C pathway. J Surg Res. 2002, 104: 118-123. 10.1006/jsre.2002.6415.
Andreeva AY, Krause E, Muller EC, Blasig IE, Utepbergenov DI: Protein kinase C regulates the phosphorylation and cellular localization of occludin. J Biol Chem. 2001, 276: 38480-38486. 10.1074/jbc.M104923200.
Stuart RO, Nigam SK: Regulated assembly of tight junctions by protein kinase C. Proc Natl Acad Sci USA. 1995, 92: 6072-6076. 10.1073/pnas.92.13.6072.
Hofmann J: The potential for isoenzyme-selective modulation of protein kinase C. FASEB J. 1997, 11: 649-669.
Fleegal MA, Hom S, Borg LK, Davis TP: Activation of PKC modulates blood–brain barrier endothelial cell permeability changes induced by hypoxia and posthypoxic reoxygenation. Am J Physiol Heart Circ Physiol. 2005, 289: H2012-2019. 10.1152/ajpheart.00495.2005.
Andreeva AY, Piontek J, Blasig IE, Utepbergenov DI: Assembly of tight junction is regulated by the antagonism of conventional and novel protein kinase C isoforms. Int J Biochem Cell Biol. 2006, 38: 222-233.
Harrington EO, Brunelle JL, Shannon CJ, Kim ES, Mennella K, Rounds S: Role of protein kinase C isoforms in rat epididymal microvascular endothelial barrier function. Am J Respir Cell Mol Biol. 2003, 28: 626-636. 10.1165/rcmb.2002-0085OC.
Sonobe Y, Takeuchi H, Kataoka K, Li H, Jin S, Mimuro M, Hashizume Y, Sano Y, Kanda T, Mizuno T, Suzumura A: Interleukin-25 expressed by brain capillary endothelial cells maintains blood–brain barrier function in a protein kinase Cepsilon-dependent manner. J Biol Chem. 2009, 284: 31834-31842. 10.1074/jbc.M109.025940.
Goldstein B, Macara IG: The PAR proteins: fundamental players in animal cell polarization. Dev Cell. 2007, 13: 609-622. 10.1016/j.devcel.2007.10.007.
Coureuil M, Mikaty G, Miller F, Lecuyer H, Bernard C, Bourdoulous S, Dumenil G, Mege RM, Weksler BB, Romero IA, et al: Meningococcal type IV pili recruit the polarity complex to cross the brain endothelium. Science. 2009, 325: 83-87. 10.1126/science.1173196.
Iden S, Rehder D, August B, Suzuki A, Wolburg-Buchholz K, Wolburg H, Ohno S, Behrens J, Vestweber D, Ebnet K: A distinct PAR complex associates physically with VE-cadherin in vertebrate endothelial cells. EMBO Rep. 2006, 7: 1239-1246. 10.1038/sj.embor.7400819.
Birukova AA, Smurova K, Birukov KG, Kaibuchi K, Garcia JG, Verin AD: Role of Rho GTPases in thrombin-induced lung vascular endothelial cells barrier dysfunction. Microvasc Res. 2004, 67: 64-77. 10.1016/j.mvr.2003.09.007.
Boivin D, Bilodeau D, Beliveau R: Regulation of cytoskeletal functions by Rho small GTP-binding proteins in normal and cancer cells. Can J Physiol Pharmacol. 1996, 74: 801-810. 10.1139/y96-083.
Stamatovic SM, Keep RF, Kunkel SL, Andjelkovic AV: Potential role of MCP-1 in endothelial cell tight junction ‘opening’: signaling via Rho and Rho kinase. J Cell Sci. 2003, 116: 4615-4628. 10.1242/jcs.00755.
Persidsky Y, Heilman D, Haorah J, Zelivyanskaya M, Persidsky R, Weber GA, Shimokawa H, Kaibuchi K, Ikezu T: Rho-mediated regulation of tight junctions during monocyte migration across the blood–brain barrier in HIV-1 encephalitis (HIVE). Blood. 2006, 107: 4770-4780. 10.1182/blood-2005-11-4721.
Adamson P, Etienne S, Couraud PO, Calder V, Greenwood J: Lymphocyte migration through brain endothelial cell monolayers involves signaling through endothelial ICAM-1 via a rho-dependent pathway. J Immunol. 1999, 162: 2964-2973.
Etienne S, Adamson P, Greenwood J, Strosberg AD, Cazaubon S, Couraud PO: ICAM-1 signaling pathways associated with Rho activation in microvascular brain endothelial cells. J Immunol. 1998, 161: 5755-5761.
Engelhardt B, Wolburg H: Mini-review: Transendothelial migration of leukocytes: through the front door or around the side of the house?. Eur J Immunol. 2004, 34: 2955-2963. 10.1002/eji.200425327.
Goeckeler ZM, Wysolmerski RB: Myosin light chain kinase-regulated endothelial cell contraction: the relationship between isometric tension, actin polymerization, and myosin phosphorylation. J Cell Biol. 1995, 130: 613-627. 10.1083/jcb.130.3.613.
Hixenbaugh EA, Goeckeler ZM, Papaiya NN, Wysolmerski RB, Silverstein SC, Huang AJ: Stimulated neutrophils induce myosin light chain phosphorylation and isometric tension in endothelial cells. Am J Physiol. 1997, 273: H981-988.
Moy AB, Shasby SS, Scott BD, Shasby DM: The effect of histamine and cyclic adenosine monophosphate on myosin light chain phosphorylation in human umbilical vein endothelial cells. J Clin Invest. 1993, 92: 1198-1206. 10.1172/JCI116690.
Antonetti DA, Barber AJ, Hollinger LA, Wolpert EB, Gardner TW: Vascular endothelial growth factor induces rapid phosphorylation of tight junction proteins occludin and zonula occluden 1. A potential mechanism for vascular permeability in diabetic retinopathy and tumors. J Biol Chem. 1999, 274: 23463-23467. 10.1074/jbc.274.33.23463.
Nico B, Mangieri D, Crivellato E, Longo V, De Giorgis M, Capobianco C, Corsi P, Benagiano V, Roncali L, Ribatti D: HIF activation and VEGF overexpression are coupled with ZO-1 up-phosphorylation in the brain of dystrophic mdx mouse. Brain Pathol. 2007, 17: 399-406. 10.1111/j.1750-3639.2007.00090.x.
Kago T, Takagi N, Date I, Takenaga Y, Takagi K, Takeo S: Cerebral ischemia enhances tyrosine phosphorylation of occludin in brain capillaries. Biochem Biophys Res Commun. 2006, 339: 1197-1203. 10.1016/j.bbrc.2005.11.133.
Wang W, Dentler WL, Borchardt RT: VEGF increases BMEC monolayer permeability by affecting occludin expression and tight junction assembly. Am J Physiol Heart Circ Physiol. 2001, 280: H434-440.
Frank PG, Woodman SE, Park DS, Lisanti MP: Caveolin, caveolae, and endothelial cell function. Arterioscler Thromb Vasc Biol. 2003, 23: 1161-1168. 10.1161/01.ATV.0000070546.16946.3A.
Sprenger RR, Fontijn RD, van Marle J, Pannekoek H, Horrevoets AJ: Spatial segregation of transport and signalling functions between human endothelial caveolae and lipid raft proteomes. Biochem J. 2006, 400: 401-410. 10.1042/BJ20060355.
Nusrat A, Parkos CA, Verkade P, Foley CS, Liang TW, Innis-Whitehouse W, Eastburn KK, Madara JL: Tight junctions are membrane microdomains. J Cell Sci. 2000, 113 (Pt 10): 1771-1781.
Shen L, Turner JR: Actin depolymerization disrupts tight junctions via caveolae-mediated endocytosis. Mol Biol Cell. 2005, 16: 3919-3936. 10.1091/mbc.E04-12-1089.
Andras IE, Pu H, Deli MA, Nath A, Hennig B, Toborek M: HIV-1 Tat protein alters tight junction protein expression and distribution in cultured brain endothelial cells. J Neurosci Res. 2003, 74: 255-265. 10.1002/jnr.10762.
Urban NT, Willig KI, Hell SW, Nagerl UV: STED nanoscopy of actin dynamics in synapses deep inside living brain slices. Biophys J. 2011, 101: 1277-1284. 10.1016/j.bpj.2011.07.027.
Keene SD, Greco TM, Parastatidis I, Lee SH, Hughes EG, Balice-Gordon RJ, Speicher DW, Ischiropoulos H: Mass spectrometric and computational analysis of cytokine-induced alterations in the astrocyte secretome. Proteomics. 2009, 9: 768-782. 10.1002/pmic.200800385.
Picotti P, Aebersold R: Selected reaction monitoring-based proteomics: workflows, potential, pitfalls and future directions. Nat Methods. 2012, 9: 555-566. 10.1038/nmeth.2015.
Uchida Y, Ohtsuki S, Katsukura Y, Ikeda C, Suzuki T, Kamiie J, Terasaki T: Quantitative targeted absolute proteomics of human blood–brain barrier transporters and receptors. J Neurochem. 2011, 117: 333-345. 10.1111/j.1471-4159.2011.07208.x.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare no conflict of interest.
Authors’ contributions
ACL and CA: were responsible for the collection of data and references, and for the drafting of the document. FG and KG were involved in the collection of data and drafting of the document. POC was responsible for the drafting and editing of the document, and for the discussion of the data. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Luissint, AC., Artus, C., Glacial, F. et al. Tight junctions at the blood brain barrier: physiological architecture and disease-associated dysregulation. Fluids Barriers CNS 9, 23 (2012). https://doi.org/10.1186/2045-8118-9-23
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2045-8118-9-23