
Abstract
Although the neurobiological mechanisms underlying panic disorder (PD) are not yet clearly understood, increasing amount of evidence from animal and human studies suggests that the amygdala, which plays a pivotal role in neural network of fear and anxiety, has an important role in the pathogenesis of PD. This article aims to (1) review the findings of structural, chemical, and functional neuroimaging studies on PD, (2) relate the amygdala to panic attacks and PD development, (3) discuss the possible causes of amygdalar abnormalities in PD, (4) and suggest directions for future research.
Similar content being viewed by others


Review
Introduction
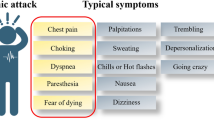
Panic disorder (PD) is characterized by repeated panic attacks, often accompanied with anticipatory anxiety, fear of losing control or sanity, or behavioral changes related to the attacks [1]. An epidemiological study conducted with a nationally representative sample estimated the lifetime prevalence of PD to be 4.5% [2]. A panic attack typically develops suddenly and reaches its peak within 10 minutes. Symptoms that accompany panic attacks include palpitations, chest pain, sweating, trembling, smothering, abdominal distress, dizziness, and fear of dying. It is estimated to be highly prevalent, with the percentage of people who experience a panic attack at least once in their lifetime reaching up to 28.3% [2–4]. Panic responses, when proximal predatory threat is approaching or is actually present, are adaptive in the sense that they prepare animals to fight vigorously or flee (“fight or flight response”) [5]. During panic attacks, however, an intense fear response to aroused sympathetic activity is manifested in the absence of actual danger [1]. Animal model studies, genetic studies, and human neuroendocrinology and neuroimaging studies have provided important insights [5–9], although the neurobiological underpinnings of the panic attack and PD are not yet completely understood [10].
Neuroendocrinological studies have implicated dysfunction of the hypothalamic-pituitary-adrenal axis, although this perturbation occurs only later in the course of the disorder, after the development of anticipatory anxiety and associated distresses [11, 12]. Apart from higher levels of COMT Val158Met polymorphism in PD patients, the role of genes in PD susceptibility has not yet been defined [7].
Neuroanatomical correlates that may be responsible for PD pathogenesis have been suggested [6, 13]. The implicated brain areas are the amygdala, thalamus, hypothalamus, and brain stem regions including the periaqueductal gray, parabrachial nucleus, and locus ceruleus [6, 13]. In particular, the amygdala has been suggested to have a critical role in PD [14], while there have been a few studies that indicate otherwise [15].
The aim of this review is to describe and discuss the neuroimaging findings and current hypotheses on the role of amygdala in the pathophysiology of PD.
The amygdala
The amygdala is known to have 13 nuclei, which can be categorized into lateral, basal and central subregions [16]. In humans, nuclei of the amygdala are usually grouped as laterobasal subgroup including both lateral and basal nuclei, centromedial subgroup, and cortical subgroup. These subgroups deal with fear [17], a process which can be schematically illustrated as follows: the lateral subgroup receives information from the cortical and subcortical areas, the basal subgroup inter-connects the lateral and central subgroups, and sends the output to the cortical areas, and the central subgroup conveys the information to the brain regions including hypothalamus and periaqueductal gray [17]. The laterobasal and central subgroups are also connected with bed nucleus of the stria terminalis, which also projects to hypothalamus, cerebellum, and brain stem areas [18].
The Figure 1 shows the simplified inputs and outputs of the amygdala, which have been reported to be associated with PD pathogenesis [13, 18]. Theoretically, disruption in any of these brain areas and connections along these areas, or any imbalance in the network can cause maladaptive and exaggerated fear responses such as panic attacks, increased basal anxiety or arousal [19], and excessive worrying [18, 20]. Consistent with this postulation, structural, chemical, and functional alterations in these amygdalar areas have been reported in neuroimaging studies in patients with PD (Tables 1, 2, 3, 4, 5; Additional files 1, 2, 3). It has been suggested that the amygdala has a critical role in the development of panic attacks and the pathogenesis of PD [14, 17, 18].

Schematic diagram of inputs to and outputs from the amygdala, relevant to panic disorder pathogenesis.
Structural abnormalities of the amygdala in patients with PD
There have been only a handful of structural neuroimaging studies that examined neuroanatomical alterations in patients with PD (Tables 1, 2, Additional file 1), relative to the number of studies in patients with other anxiety disorders such as post-traumatic stress disorder and obsessive compulsive disorder [42–45].
In earlier studies using the computerized tomography (Table 1), it may have not been possible to evaluate amygdalar structural alterations due to insufficient spatial resolution and tissue contrast.
All magnetic resonance imaging studies in which amygdalae were manually traced have consistently reported amygdalar volume reduction in patients with PD [46–48] (Additional file 1). In the report of Uchida and colleagues, there was a trend-level significance for bilateral amygdalar volume reduction [48], while two other studies reported a statistically significant bilateral amygdalar volume reduction [46, 47]. In addition to the relatively small sample size, the fact that amygdalae were traced on 2 mm-thick reformatted magnetic resonance (MR) images in the study of Uchida and colleagues [48] may have resulted in a comparatively greater error range, which could have undermined the power of detecting volume differences of small structures. In the report of Massana and colleagues, 1.2 mm-thick isocubically reformatted MR images were used for tracing [47]. Although the voxel size of images used for tracing is not described in the report of Hayano and colleagues [46], it is likely that 1.5-mm thick native images were used according to the standard protocol of manual segmentation in 3D slicer (http://www.slicer.org) which was the image analysis software adopted in the study.
Among five studies that used whole brain-wise approach of voxel-based morphometry (VBM) (Additional file 1), the report of Asami and colleagues noted amygdalar volume reduction [49]. It has been suggested that VBM approach may have limited sensitivity for identifying a priori-specified structural differences unless analytical approaches such as small volume correction are applied. This indicates that a large sample size may be required for the reliable results in VBM studies [50–52]. Thus, it is not surprising that the study of Asami et al., which had the positive finding in the amygdala, had the largest sample size among these five VBM studies.
The effect size for group differences was greater in the right amygdala than the left amygdala across all four studies that have reported the amygdalar volume reduction [46–49]. In the study of Asami et al., a statistically significant amygdalar volume reduction was noted only in the right hemisphere. For the studies of Hayano et al. and Uchida et al., Ray and Shadish’s equation 2 [53] was used to compute Cohen’s d values based on t score and sample sizes for each group presented in the paper (Cohen’s d = 0.77 for the right amygdala; 0.60 for the left amygdala in Hayano et al.; Cohen’s d = 0.78 for the right amygdala [8% volume differences]; 0.76 for the left amygdala [5% volume differences] in Uchida et al.). Regarding the report of Massana et al., Cohen’s d for amygdalar volume differences was 2.07 for the right amygdala and 1.66 for the left amygdala.
Reasons for the potentially greater deficits in the right amygdala than in the left amygdala in patients with PD may be understood in a large body of literature on hemispheric organization for processing emotions such as fear. The right hemisphere has long been considered to have dominance over the left hemisphere regarding emotional behaviors [54, 55]. Theories that suggest the lateralized role of right and left hemispheres for different aspects of emotions have arisen [56]. Sackeim and colleagues proposed that the right hemisphere may have dominance especially in processing negative emotions [57]. Regarding fear processing and the amygdala, more recent neuroimaging studies indicated that the right amygdala is primarily involved in processing acquired fear, while the left amygdala is particularly involved in processing innate fear [58]. This lateralization has been interpreted in the context that innate fear may require more conscious and linguistic processing of the stimuli. On the other hand, acquired fear may not require as much conscious elaborations of the stimuli since the response would rather be automatic [58]. In PD, enhanced conditionality of fear (i.e., a tendency to acquire fear more easily) with resistance to extinction has been considered as one of the core elements of the pathology [59, 60].
Because of the cross-sectional nature of the studies, it is not appropriate to infer the causality of the relationship between PD and reduced amygdalar volume. However, although speculative, some of the evidence indicates that amygdalar deficits, particularly the deficit in the right amygdala, may predispose to PD. Massana and colleagues found that amygdalar volume reduction was noted in both subgroups of PD patients with long duration of illness (>6 months) and those with relatively short duration of illness (<6 months). There were no associations between the magnitude of amygdalar volume reduction and clinical measures including panic symptom severity and illness duration [49]. In Uchida et al., correlational results between amgydalar volume and clinical measures were not reported [48]. In the study of Hayano et al., the right amygdalar volume of patients with PD had a negative correlation with the neuroticism score of NEO personality inventory-revised, the measure of enduring tendency toward experiencing negative emotional states, while the left amygdalar volume showed a negative correlation with the state anxiety score from the State-Trait Anxiety Inventory, the measure of anxiety severity [46]. This conjecture is entirely speculative and subsequently urges further studies.
It is less likely that amygdalar deficits in patients with PD are due to the use of antidepressants. The total daily dose of antidepressant was not associated with the amygdalar volume reduction [49]. The study of Massana and colleagues was conducted in patients who are antidepressant-naive [47]. Also, a meta-analysis in patients with major depressive disorder has suggested that antidepressant medication increases the amygdalar volume, rather than causing amygdalar deficits [61].
Since the amygdala is composed of many different subnuclei with distinctive connections and functions [62], researchers are interested in whether there would be subregional specificity of the amygdalar deficit. Hayano and colleagues investigated which subregion of the amygdala might show more deficits in patients with PD [46]. They employed the optimized voxel-based morphometry with small volume correction using bilateral amygdalar masks. Volume deficits were noted in the amygdalar subregion that might correspond to the corticomedial subregional group. Central and medial subnuclei that are parts of the corticomedial subregion have been implicated for autonomic responses to fear stimuli [63]. However, the laterobasal subregion of the amygdala has also been reported to be involved in the pathogenesis of PD [64]. Further studies that adopt strategies that may allow finer registration among inter-subject amygdalae [65–67] may be needed to confirm the subregional findings.
Evaluating the structural connectivity among the amygdala and other brain regions on diffusion tensor images would provide important additional information [68]. Relative to healthy comparison subjects, patients with PD had a greater fractional anisotropy value in the left anterior and right posterior cingulate regions [23] (Table 1). These regions may have a role in maintaining visceromotor homeostasis through its interconnections with the amygdala [69]. Recently, with the use of high quality diffusion tensor images and T1-weighted images, evaluating the connections among amygdalar subregions and other brain regions has become possible [70, 71]. Studies with this approach would also enhance the understanding on the roles of amygdalar subregions.
Functional abnormalities of the amygdala in patients with PD
Functional neuroimaging techniques that have been used for studying PD include functional magnetic resonance imaging (fMRI), positron emission tomography (PET), single photon emission computed tomography (SPECT), electroencephalography (EEG), and near-infrared spectroscopy (NIRS). Among these, NIRS and EEG may not be suitable for assessing amygdalar activity due to a relatively superficial penetration depth [72] (Tables 2 and 4).
There have been PET and SPECT studies that used neutral state paradigms. Earlier studies with a region-of-interest approach did not find significantly different amygdalar activity [73–75] (Additional file 2). As De Cristofaro and colleagues commented [74], image resolutions of SPECT and PET may not be sufficient for delineating the amygdala from other adjacent structures. A neutral-state PET study which used a whole brain-wise approach [76] also did not find amygdalar metabolic differences, while in a more recent study [77], a greater metabolism in bilateral amygdalar regions was noted in patients with PD compared with healthy comparison subjects. The authors conjectured that this discrepancy may have stemmed from differences in image resolution, sample size, and subjects’ age [77].
Among three fMRI studies that captured brain regional activation during spontaneous panic attacks, two studies conducted in patients with PD or specific phobia have found increased right amygdalar activation [78, 79]. A study conducted in an individual with restless leg syndrome but without any history of psychiatric disorders has noted the relationship of the left amygdalar activity and the heart rate [80] (Additional file 3). A PET study could not find the difference of the amygdalar blood flow in a healthy volunteer who experienced a panic attack during fear conditioning in comparison with 5 healthy volunteers who did not experience panic attacks during the same fear conditioning sessions [81].
Symptom provocation paradigms were used both in healthy volunteers and in patients with PD (Additional file 2, 3). When challenged with cholecystokinin tetrapeptide (CCK-4) or procaine, healthy volunteers showed increased amygdalar activation [82] or increased regional cerebral blood flow [83–85]. In patients with PD, CCK-4 or doxapram intravenous injection did not elicit significantly greater amygdalar activation [86, 87]. Due to relatively modest sample sizes, there is a risk for the type II error, precluding the possibility of drawing any conclusions from these negative findings. In a report of Boshuisen et al., PD patients experiencing anticipatory anxiety showed significantly decreased regional cerebral blood flow in the right amygdala [88]. The authors commented on the possibility that cortical inhibitory effects in response to the intense anticipatory anxiety may have depressed the amygdalar activity. It has also been suggested that this might reflect the functional impairment of the amygdala to cope with the anticipatory anxiety [88].
Cognitive activation probes to evaluate amygdalar function have been used in functional neuroimaging studies of PD [9]. In most studies which used fMRI, patients with PD showed altered amygdalar activation level (Additional file 3) except a study with motor activation paradigms [89] and a few with a relatively small sample size [90, 91]. In addition, Maddock et al., suggested that repeated presentation of the threat-related stimuli in their study (20 words being repeated eight times) may have habituated the amygdalar response [90]. This may have resulted in the negative finding in this region.
Among studies that report altered amygdalar activation in PD patient group relative to comparison group, both higher and lower amygdalar activation level was found [92]. While most of the studies reported increased amygdalar activation, two studies [93, 94] noted deactivated the amygdala in patients with PD relative to healthy comparison subjects. Statistical activation maps of comparison between the threat condition vs. the safe condition in the study of Tuescher and colleagues showed less activation in the amygdala of PD patients in the threat condition relative to post traumatic stress disorder patients [93]. However, this was mainly due to amygdalar hyperactivation in PD patients in the safe condition relative to threat condition, while post-traumatic stress disorder patients showed increased amygdalar activation in the threat condition. The fact that patients with PD were compared to those with post-traumatic stress disorder in this study, and the difference in the experimental paradigm from other studies precludes direct comparison of this study result with those from other studies [95]. In this study, patients were instructed that visual presentation of the square with a certain color would mean that an electrodermal stimulation can occur at any time. Actual stimulation did not happen during scanning. This may have caused anticipatory anxiety as in the study of Boshuisen and colleagues [88], in which patients with PD also exhibited decreased regional cerebral blood flow in the amygdala. Deactivation of the amygdala in response to presentation of fearful faces [94] may in part be due to the facts that most of the patients were taking antidepressants [92], since antidepressants have been reported to decrease the amygdalar activity [96]. In addition, it should be considered that the relatively lenient statistical threshold (p < 0.05) for a priori-hypothesized regions of the anterior cingulate cortex and the amygdala [94]. It is possible that only certain subgroups of patients with PD exhibit amygdalar hyperactivation [92]. Only women were responsive to fearful faces [97]. Patients with the COMT 158 val allele showed hyperactivation of the amygdala in response to fearful faces [98]. In a relatively small sample size, the heterogeneity with regard to genetic polymorphism and sex may make it hard to detect the increased amygdalar activation, if any [99]. Baseline anxiety and associated physiologic changes may also confound functional neuroimaging findings [19].
Findings regarding laterality of amygdalar activation (Additional file 3) are less consistent than those of amygdalar volume (Additional file 1). Left and right amygdalae are known to be involved in different aspects of fear processing [58]. Direct comparison of results is not appropriate since the experimental paradigms are slightly different with each other [95]. In studies with a relatively small sample size, differences in the number of subjects with right or left-handedness may confound the laterality findings [95].
There have been reports that investigated amygdalar function in relation to the treatment (Additional file 2). When untreated patients and treated patients were compared with healthy comparison subjects, patients treated with antidepressants showed no difference in amygdalar serotonin binding potential, while untreated patients showed significantly lower serotonin binding potential [100]. Treatment with paroxetine for 12 weeks in psychotropic medication-naive PD patients altered amygdalar glucose metabolism [101]. In the report of Prasko and colleagues, treatment with antidepressants did not change the amygdalar glucose metabolism [102]. This may have stemmed from exposure to antidepressants prior to study participation [102]. Cognitive behavioral therapy did not change the amygdalar glucose metabolism [102, 103]. The number of studies in regards to treatment-related changes in amygdalar function is small and more studies are required for conclusions.
Potential mechanisms underlying altered amygdalar function and structure in patients with PD
Structural and functional neuroimaging findings with regard to the amygdala in patients with PD are not perfectly consistent, but a pattern suggested by one of the neurocircuitry models may be noted [13]. Increased reactivity of the amygdala with structural deficits, similar to the findings in other anxiety disorders such as post-traumatic stress disorder [92]. Whether structural deficits of the amygdala cause its hyper-reactivity or hyperactivation of the amygdala for a prolonged period elicits overuse atrophy is not known [104].
Brain physiological processes underlying altered amygdalar function and structure may be partly expounded by the findings from molecular imaging and chemical neuroimaging studies (Additional file 2, Table 5). Decreased gamma amino-butyric acid (GABA)-benzodiazepine receptor binding has been reported in the medial temporal lobes that may include amygdalar regions [105, 106] (Additional file 2). In the amygdala, decreased 5-HT1A receptor binding has also been reported [100]. Both of these receptors are involved in inhibitory neurotransmission. Defective inhibition of the amygdalar activity may result in paroxysmal elevations in anxiety [105]. Chemical neuroimaging studies have reported lower levels of GABA in patients with PD than healthy comparison subjects [37, 40], which were not reversed with acute benzodiazepine challenge [38]. Decreases in the GABA level may cause dysfunction in GABAergic inhibition of brain activity [107]. Morever, experimentally lowered GABA level caused panic-like behaviors in rats [108].
Findings from chemical neuroimaging studies suggest metabolic disturbances in the brain of patients with PD [19] (Table 5). Hypermetabolic state was suspected based on the findings that show depletion of phosphocreatine and creatine [92, 109] in patients with PD, consistent with functional neuroimaging findings. Shioiri and colleagues reported higher level of inorganic phosphate in patients with PD [41]. Inorganic phosphate is known to be accumulated when creatine phosphate is broken down during anaerobic metabolism [110]. A rapid rise of the brain lactate level in response to physiological challenge (vigilance task with visual stimuli) also suggests an intrinsic metabolic disturbance [19, 36].
Genetic polymorphism and early-life experiences may also contribute to amygdalar abnormalities. Specially, the COMT Val158Met polymorphism that affects the amygdalar structure, function, and receptor expression [98, 111–113] has also been reported to be associated with PD development [7].
Data from the National Comorbidity Survey showed a positive association between early-life traumatic experiences and later development of PD [114]. PD patients who experienced early-life traumatic events had more severe symptom profile [115]. Relationships between early-life traumatic experiences and amygdalar dysfunction have also been reported [116].
The role of amygdalar pathology in developing PD
Electrical or chemical stimulation of the amygdalar central nucleus causes constellation of symptoms that are very similar to those of panic attacks [63]. Electrolytic lesions made in the central nucleus would also disrupt fibers connecting laterobasal nucleus and bed nucleus of the stria terminalis, which has outputs to the hypothalamus and brain stem [18], where the centers for autonomic and neuroendocrine response regulations are located. When the efferent fibers from the central nucleus are stimulated, similar effects are produced [117]. By abruptly blocking the tonic GABAergic inhibition in the laterobasal subregion of the amygdala, symptoms mimicking panic attacks are induced [64]. These animal studies suggest the role of the amygdala in the pathogenesis of PD.
Earlier models to explain the neurobiology of PD have underscored the hypersensitivity to carbon dioxide, namely “false suffocation alarm theory [118, 119],” although it has been revised since [120]. The following neurocircuitry model [13] is well suited for explaining the role of the ‘hyperactive and smaller’ amygdala [104] in the pathogenesis of PD for the fairly consistent findings.
Findings of Ziemann et al., suggest the role of the amygdala in sensing central acidosis [121] which has been considered as one of the core pathophysiological processes in PD [122]. In chemical neuroimaging studies in which neurometabolite changes were measured during induced panic attacks [31–33, 102], lactate/n-acetyl aspartate level increased during panic attacks (Table 5). Hyperventilation or sodium lactate infusion leads to a metabolic alkalosis, which presumably augments neuronal activity [123] and alters the redox state to glycolytic metabolism producing lactate [29, 31]. The report of Shioiri and colleagues which found increased level of inorganic phosphate, a potential indicator of anaerobic metabolism, indirectly supports the possibility of the redox shift to glycolysis [41]. It is also possible that lactate builds up presumably due to reduced cerebral blood flow [124]. Elevated brain lactate level, which would be associated with decreased brain pH or exaggerated alkalotic buffering [29], may trigger a panic attack by stimulating the amygdala through the chemosensing ion channel [121].
The role of the amygdala in progression to PD
Panic attacks do not necessarily progress to PD [3]. Bouton and colleagues [60] proposed that among all individuals who experience panic attacks, only those who have increased conditionality would develop PD. As panic attacks recur, the association would become even stronger [125, 126]. Consistent with this hypothesis, enhanced conditionality [127] and resistance to extinction in patients with PD have been reported. Altered anxiety neurocircuitry including the amygdala in PD may in part be responsible for this enhanced conditionality and resistance to extinction.
The amygdala projects to the striatum and cortical areas, and induce behavioral changes [62]. Phobic avoidance or agoraphobia, often associated with PD, may reflect the amygdala’s influence on these areas [128]. Unlike the general belief that ’cognition rules over emotion,’ there is evidence that emotion modulates cognition from perception and attention [17] to higher domains of judgment and reasoning [129]. Irrational worry about the connotation of the attacks, one of the PD core symptoms, may also be partly attributed to the amygdalar structural and functional abnormalities.
Summary and recommendations
The amygdala has been reported to have a crucial role in the pathophysiology of PD. In animal studies, behaviors similar to those of panic attacks were observed when the amygdala was stimulated. Increased amygdalar activity with volumetric deficits was noted in patients with PD, although not always consistent. This altered function and structure of the amygdala may be partly due to dysregulated brain metabolism [31–33, 121]. As potential causes of amygdalar abnormality in PD, COMT Val158Met polymorphism and early life traumatic experiences can also be suggested [112, 130, 131].
Enhanced conditionality and resistance to extinction are known as risk factors in progression from panic attack to PD. As an important element of the anxiety neurocircuitry, altered amygdalar function and structure may facilitate the progression to PD. Typical PD symptoms such as phobic avoidance and irrational worry of panic attacks may also be attributed to amygdalar structural and functional abnormalities.
Partly inconsistent findings from human neuroimaging studies are possibly due to sample heterogeneity, small sample size, and limitations of neuroimaging techniques. More studies with a larger sample size are warranted. In terms of technical difficulties, subregional analysis of the amygdala, which detects alterations in subregions of the amygdala separately, could be a novel strategy for future research, since each nucleus might have a distinct role in the pathophysiology of PD. This approach has already shown its potential in structural neuroimaging studies [65, 66] and functional neuroimaging studies, examining neural circuitry in healthy individuals [132, 133]. A postmortem study, investigating association between pathology in amygdalar subdivisions of patients with Parkinson’s disease [134] and premortem anxiety symptoms, provides another insight for a new strategy.
Conclusions
The amygdala, the hub of fear processing networks, is closely associated with the pathogenesis of PD as well as panic attack. Further studies in well-defined larger samples, with more sophisticated research designs and advanced technologies would promise a better understanding on the role of the amygdala in the pathophysiology of PD.
Authors’ information
JEK is an assistant professor at the Department of Brain & Cognitive Sciences, Ewha Womans University Graduate School, Seoul, South Korea. SRD and IKL are professors at the Department of Radiology, University of Washington School of Medicine, Seattle, USA. IKL is a professor at the Division of Life and Pharmaceutical Sciences, Ewha Womans University College of Pharmacy and a director of Ewha Brain Institute, Ewha Womans University, Seoul, South Korea.
Abbreviations
- PD:
-
Panic disorder
- MR:
-
Magnetic resonance
- VBM:
-
Voxel-based morphometry
- fMRI:
-
Functional magnetic resonance imaging
- PET:
-
Positron emission tomography
- SPECT:
-
Single photon emission computed tomography
- EEG:
-
Electroencephalography
- NIRS:
-
near-infrared spectroscopy
- CCK-4:
-
cholecystokinin tetrapeptide
- GABA:
-
gamma amino-butyric acid
References
American Psychiatric Association: Diagnostic criteria from DSM-IV-TR. 2000, Washington, D.C: American Psychiatric Association
Kessler RC, Chiu WT, Jin R, Ruscio AM, Shear K, Walters EE: The epidemiology of panic attacks, panic disorder, and agoraphobia in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2006, 63: 415-424. 10.1001/archpsyc.63.4.415.
Katerndahl DA, Realini JP: Lifetime prevalence of panic states. Am J Psychiatry. 1993, 150: 246-249.
Norton PJ, Zvolensky MJ, Bonn-Miller MO, Cox BJ, Norton GR: Use of the Panic Attack Questionnaire-IV to assess non-clinical panic attacks and limited symptom panic attacks in student and community samples. J Anxiety Disord. 2008, 22: 1159-1171. 10.1016/j.janxdis.2007.12.002.
Graeff FG, Del-Ben CM: Neurobiology of panic disorder: from animal models to brain neuroimaging. Neurosci Biobehav Rev. 2008, 32: 1326-1335. 10.1016/j.neubiorev.2008.05.017.
Coplan JD, Lydiard RB: Brain circuits in panic disorder. Biol Psychiatry. 1998, 44: 1264-1276. 10.1016/S0006-3223(98)00300-X.
Maron E, Hettema JM, Shlik J: Advances in molecular genetics of panic disorder. Mol Psychiatry. 2010, 15: 681-701. 10.1038/mp.2009.145.
Martin EI, Ressler KJ, Binder E, Nemeroff CB: The neurobiology of anxiety disorders: brain imaging, genetics, and psychoneuroendocrinology. Psychiatr Clin North Am. 2009, 32: 549-575. 10.1016/j.psc.2009.05.004.
Rauch SL, Shin LM, Wright CI: Neuroimaging studies of amygdala function in anxiety disorders. Ann N Y Acad Sci. 2003, 985: 389-410.
Ninan PT, Dunlop BW: Neurobiology and etiology of panic disorder. J Clin Psychiatry. 2005, 66 (Suppl 4): 3-7.
Battaglia M, Ogliari A: Anxiety and panic: from human studies to animal research and back. Neurosci Biobehav Rev. 2005, 29: 169-179. 10.1016/j.neubiorev.2004.06.013.
Sinha SS, Coplan JD, Pine DS, Martinez JA, Klein DF, Gorman JM: Panic induced by carbon dioxide inhalation and lack of hypothalamic-pituitary-adrenal axis activation. Psychiatry Res. 1999, 86: 93-98. 10.1016/S0165-1781(99)00029-3.
Gorman JM, Kent JM, Sullivan GM, Coplan JD: Neuroanatomical hypothesis of panic disorder, revised. Am J Psychiatry. 2000, 157: 493-505. 10.1176/appi.ajp.157.4.493.
Shekhar A, Sajdyk TJ, Gehlert DR, Rainnie DG: The amygdala, panic disorder, and cardiovascular responses. Ann N Y Acad Sci. 2003, 985: 308-325.
Wiest G, Lehner-Baumgartner E, Baumgartner C: Panic attacks in an individual with bilateral selective lesions of the amygdala. Arch Neurol. 2006, 63: 1798-1801. 10.1001/archneur.63.12.1798.
Whalen PJ, Phelps EA: The human amygdala. 2009, New York: Guilford Press
Phelps EA, LeDoux JE: Contributions of the amygdala to emotion processing: from animal models to human behavior. Neuron. 2005, 48: 175-187. 10.1016/j.neuron.2005.09.025.
Davis M, Whalen PJ: The amygdala: vigilance and emotion. Mol Psychiatry. 2001, 6: 13-34. 10.1038/sj.mp.4000812.
Dager SR: The vexing role of baseline: implications for neuroimaging studies of panic disorder. Int J Psychophysiol. 2010, 78: 20-26. 10.1016/j.ijpsycho.2010.01.007.
Mohlman J, Price RB, Eldreth DA, Chazin D, Glover DM, Kates WR: The relation of worry to prefrontal cortex volume in older adults with and without generalized anxiety disorder. Psychiatry Res. 2009, 173: 121-127. 10.1016/j.pscychresns.2008.09.010.
Wurthmann C, Gregor J, Baumann B, Effenberger O, Dohring W, Bogerts B: [Qualitative evaluation of brain structure in CT in panic disorders]. Nervenarzt. 1998, 69: 763-768. 10.1007/s001150050340.
Lepola U, Nousiainen U, Puranen M, Riekkinen P, Rimon R: EEG and CT findings in patients with panic disorder. Biol Psychiatry. 1990, 28: 721-727. 10.1016/0006-3223(90)90458-E.
Han DH, Renshaw PF, Dager SR, Chung A, Hwang J, Daniels MA, Lee YS, Lyoo IK: Altered cingulate white matter connectivity in panic disorder patients. J Psychiatr Res. 2008, 42: 399-407. 10.1016/j.jpsychires.2007.03.002.
Tanii H, Nishimura Y, Inoue K, Koshimizu H, Matsumoto R, Takami T, Hara N, Nishida A, Okada M, Kaiya H, Okazaki Y: Asymmetry of prefrontal cortex activities and catechol-O-methyltransferase Val158Met genotype in patients with panic disorder during a verbal fluency task: near-infrared spectroscopy study. Neurosci Lett. 2009, 452: 63-67. 10.1016/j.neulet.2009.01.044.
Dresler T, Ehlis AC, Plichta MM, Richter MM, Jabs B, Lesch KP, Fallgatter AJ: Panic disorder and a possible treatment approach by means of high-frequency rTMS: a case report. World J Biol Psychiatry. 2009, 10: 991-997. 10.1080/15622970902898147.
Akiyoshi J, Hieda K, Aoki Y, Nagayama H: Frontal brain hypoactivity as a biological substrate of anxiety in patients with panic disorders. Neuropsychobiology. 2003, 47: 165-170. 10.1159/000070587.
Eser D, Leicht G, Baghai T, Pogarell O, Schule C, Karch S, Nothdurfter C, Rupprecht R, Mulert C: Impact of loudness dependency of auditory evoked potentials on the panic response to CCK-4. J Psychiatr Res. 2009, 43: 393-400. 10.1016/j.jpsychires.2008.04.010.
Stein MB, Uhde TW: Infrequent occurrence of EEG abnormalities in panic disorder. Am J Psychiatry. 1989, 146: 517-520.
Friedman SD, Mathis CM, Hayes C, Renshaw P, Dager SR: Brain pH response to hyperventilation in panic disorder: preliminary evidence for altered acid–base regulation. Am J Psychiatry. 2006, 163: 710-715. 10.1176/appi.ajp.163.4.710.
Layton ME, Friedman SD, Dager SR: Brain metabolic changes during lactate-induced panic: effects of gabapentin treatment. Depress Anxiety. 2001, 14: 251-254. 10.1002/da.1076.
Dager SR, Friedman SD, Heide A, Layton ME, Richards T, Artru A, Strauss W, Hayes C, Posse S: Two-dimensional proton echo-planar spectroscopic imaging of brain metabolic changes during lactate-induced panic. Arch Gen Psychiatry. 1999, 56: 70-77. 10.1001/archpsyc.56.1.70.
Dager SR, Richards T, Strauss W, Artru A: Single-voxel 1 H-MRS investigation of brain metabolic changes during lactate-induced panic. Psychiatry Res. 1997, 76: 89-99. 10.1016/S0925-4927(97)00066-8.
Dager SR, Marro KI, Richards TL, Metzger GD: Preliminary application of magnetic resonance spectroscopy to investigate lactate-induced panic. Am J Psychiatry. 1994, 151: 57-63.
Trzesniak C, Uchida RR, Araujo D, Guimaraes FS, Freitas-Ferrari MC, Filho AS, Santos AC, Busatto GF, Zuardi AW, Del-Ben CM: (1)H magnetic resonance spectroscopy imaging of the hippocampus in patients with panic disorder. Psychiatry Res. 2010, 182: 261-265. 10.1016/j.pscychresns.2010.03.008.
Hasler G, van der Veen JW, Geraci M, Shen J, Pine D, Drevets WC: Prefrontal cortical gamma-aminobutyric Acid levels in panic disorder determined by proton magnetic resonance spectroscopy. Biol Psychiatry. 2009, 65: 273-275. 10.1016/j.biopsych.2008.06.023.
Maddock RJ, Buonocore MH, Copeland LE, Richards AL: Elevated brain lactate responses to neural activation in panic disorder: a dynamic 1 H-MRS study. Mol Psychiatry. 2009, 14: 537-545. 10.1038/sj.mp.4002137.
Ham BJ, Sung Y, Kim N, Kim SJ, Kim JE, Kim DJ, Lee JY, Kim JH, Yoon SJ, Lyoo IK: Decreased GABA levels in anterior cingulate and basal ganglia in medicated subjects with panic disorder: a proton magnetic resonance spectroscopy (1 H-MRS) study. Prog Neuropsychopharmacol Biol Psychiatry. 2007, 31: 403-411. 10.1016/j.pnpbp.2006.10.011.
Goddard AW, Mason GF, Appel M, Rothman DL, Gueorguieva R, Behar KL, Krystal JH: Impaired GABA neuronal response to acute benzodiazepine administration in panic disorder. Am J Psychiatry. 2004, 161: 2186-2193. 10.1176/appi.ajp.161.12.2186.
Massana G, Gasto C, Junque C, Mercader JM, Gomez B, Massana J, Torres X, Salamero M: Reduced levels of creatine in the right medial temporal lobe region of panic disorder patients detected with (1)H magnetic resonance spectroscopy. Neuroimage. 2002, 16: 836-842. 10.1006/nimg.2002.1083.
Goddard AW, Mason GF, Almai A, Rothman DL, Behar KL, Petroff OA, Charney DS, Krystal JH: Reductions in occipital cortex GABA levels in panic disorder detected with 1 h-magnetic resonance spectroscopy. Arch Gen Psychiatry. 2001, 58: 556-561. 10.1001/archpsyc.58.6.556.
Shioiri T, Kato T, Murashita J, Hamakawa H, Inubushi T, Takahashi S: High-energy phosphate metabolism in the frontal lobes of patients with panic disorder detected by phase-encoded 31P-MRS. Biol Psychiatry. 1996, 40: 785-793. 10.1016/0006-3223(95)00487-4.
Karl A, Schaefer M, Malta LS, Dorfel D, Rohleder N, Werner A: A meta-analysis of structural brain abnormalities in PTSD. Neurosci Biobehav Rev. 2006, 30: 1004-1031. 10.1016/j.neubiorev.2006.03.004.
Rotge JY, Guehl D, Dilharreguy B, Tignol J, Bioulac B, Allard M, Burbaud P, Aouizerate B: Meta-analysis of brain volume changes in obsessive-compulsive disorder. Biol Psychiatry. 2009, 65: 75-83. 10.1016/j.biopsych.2008.06.019.
Rotge JY, Langbour N, Guehl D, Bioulac B, Jaafari N, Allard M, Aouizerate B, Burbaud P: Gray matter alterations in obsessive-compulsive disorder: an anatomic likelihood estimation meta-analysis. Neuropsychopharmacology. 2010, 35: 686-691. 10.1038/npp.2009.175.
Woon FL, Hedges DW: Amygdala volume in adults with posttraumatic stress disorder: a meta-analysis. J Neuropsychiatry Clin Neurosci. 2009, 21: 5-12. 10.1176/appi.neuropsych.21.1.5.
Hayano F, Nakamura M, Asami T, Uehara K, Yoshida T, Roppongi T, Otsuka T, Inoue T, Hirayasu Y: Smaller amygdala is associated with anxiety in patients with panic disorder. Psychiatry Clin Neurosci. 2009, 63: 266-276. 10.1111/j.1440-1819.2009.01960.x.
Massana G, Serra-Grabulosa JM, Salgado-Pineda P, Gasto C, Junque C, Massana J, Mercader JM, Gomez B, Tobena A, Salamero M: Amygdalar atrophy in panic disorder patients detected by volumetric magnetic resonance imaging. Neuroimage. 2003, 19: 80-90. 10.1016/S1053-8119(03)00036-3.
Uchida RR, Del-Ben CM, Santos AC, Araujo D, Crippa JA, Guimaraes FS, Graeff FG: Decreased left temporal lobe volume of panic patients measured by magnetic resonance imaging. Braz J Med Biol Res. 2003, 36: 925-929. 10.1590/S0100-879X2003000700014.
Asami T, Yamasue H, Hayano F, Nakamura M, Uehara K, Otsuka T, Roppongi T, Nihashi N, Inoue T, Hirayasu Y: Sexually dimorphic gray matter volume reduction in patients with panic disorder. Psychiatry Res. 2009, 173: 128-134. 10.1016/j.pscychresns.2008.10.004.
Desseilles M, Dang-Vu T, Schabus M, Sterpenich V, Maquet P, Schwartz S: Neuroimaging insights into the pathophysiology of sleep disorders. Sleep. 2008, 31: 777-794.
Giuliani NR, Calhoun VD, Pearlson GD, Francis A, Buchanan RW: Voxel-based morphometry versus region of interest: a comparison of two methods for analyzing gray matter differences in schizophrenia. Schizophr Res. 2005, 74: 135-147. 10.1016/j.schres.2004.08.019.
Kubicki M, Shenton ME, Salisbury DF, Hirayasu Y, Kasai K, Kikinis R, Jolesz FA, McCarley RW: Voxel-based morphometric analysis of gray matter in first episode schizophrenia. Neuroimage. 2002, 17: 1711-1719. 10.1006/nimg.2002.1296.
Ray JW, Shadish WR: How interchangeable are different estimators of effect size?. J Consult Clin Psychol. 1996, 64: 1316-1325.
Boller F: Handbook of neuropsychology. 1988, Amsterdam: Elsevier
Ley RG, Bryden MP: Hemispheric differences in processing emotions and faces. Brain Lang. 1979, 7: 127-138. 10.1016/0093-934X(79)90010-5.
Kolb B, Taylor L: Affective behavior in patients with localized cortical excisions: role of lesion site and side. Science. 1981, 214: 89-91. 10.1126/science.7280683.
Sackeim HA, Greenberg MS, Weiman AL, Gur RC, Hungerbuhler JP, Geschwind N: Hemispheric asymmetry in the expression of positive and negative emotions. Neurologic evidence. Arch Neurol. 1982, 39: 210-218. 10.1001/archneur.1982.00510160016003.
Lane RD, Nadel L, Ahern G: Cognitive neuroscience of emotion. 2000, New York: Oxford University Press
Acheson DT, Forsyth JP, Moses E: Interoceptive fear conditioning and panic disorder: the role of conditioned stimulus-unconditioned stimulus predictability. Behav Ther. 2012, 43: 174-189. 10.1016/j.beth.2011.06.001.
Bouton ME, Mineka S, Barlow DH: A modern learning theory perspective on the etiology of panic disorder. Psychol Rev. 2001, 108: 4-32.
Hamilton JP, Siemer M, Gotlib IH: Amygdala volume in major depressive disorder: a meta-analysis of magnetic resonance imaging studies. Mol Psychiatry. 2008, 13: 993-1000. 10.1038/mp.2008.57.
Aggleton JP: The amygdala: a functional analysis. 2000, Oxford: Oxford University Press, 2
Davis M: The role of the amygdala in fear and anxiety. Annu Rev Neurosci. 1992, 15: 353-375. 10.1146/annurev.ne.15.030192.002033.
Shekhar A, Sajdyk TS, Keim SR, Yoder KK, Sanders SK: Role of the basolateral amygdala in panic disorder. Ann N Y Acad Sci. 1999, 877: 747-750. 10.1111/j.1749-6632.1999.tb09315.x.
Chung MK, Worsley KJ, Nacewicz BM, Dalton KM, Davidson RJ: General multivariate linear modeling of surface shapes using SurfStat. Neuroimage. 2010, 53: 491-505. 10.1016/j.neuroimage.2010.06.032.
Kim JE, Lyoo IK, Estes AM, Renshaw PF, Shaw DW, Friedman SD, Kim DJ, Yoon SJ, Hwang J, Dager SR: Laterobasal amygdalar enlargement in 6- to 7-year-old children with autism spectrum disorder. Arch Gen Psychiatry. 2010, 67: 1187-1197. 10.1001/archgenpsychiatry.2010.148.
Klein A, Andersson J, Ardekani BA, Ashburner J, Avants B, Chiang MC, Christensen GE, Collins DL, Gee J, Hellier P: Evaluation of 14 nonlinear deformation algorithms applied to human brain MRI registration. Neuroimage. 2009, 46: 786-802. 10.1016/j.neuroimage.2008.12.037.
Ayling E, Aghajani M, Fouche JP, van der Wee N: Diffusion tensor imaging in anxiety disorders. Curr Psychiatry Rep. 2012, 14: 197-202. 10.1007/s11920-012-0273-z.
LeDoux JE: Emotion circuits in the brain. Annu Rev Neurosci. 2000, 23: 155-184. 10.1146/annurev.neuro.23.1.155.
Bach DR, Behrens TE, Garrido L, Weiskopf N, Dolan RJ: Deep and superficial amygdala nuclei projections revealed in vivo by probabilistic tractography. J Neurosci. 2011, 31: 618-623. 10.1523/JNEUROSCI.2744-10.2011.
Solano-Castiella E, Anwander A, Lohmann G, Weiss M, Docherty C, Geyer S, Reimer E, Friederici AD, Turner R: Diffusion tensor imaging segments the human amygdala in vivo. Neuroimage. 2010, 49: 2958-2965. 10.1016/j.neuroimage.2009.11.027.
Strangman G, Boas DA, Sutton JP: Non-invasive neuroimaging using near-infrared light. Biol Psychiatry. 2002, 52: 679-693. 10.1016/S0006-3223(02)01550-0.
Eren I, Tukel R, Polat A, Karaman R, Unal S: Evaluation of regional cerebral blood flow changes in panic disorder with Tc99m-HMPAO SPECT. Psychiatry Res. 2003, 123: 135-143. 10.1016/S0925-4927(03)00062-3.
De Cristofaro MT, Sessarego A, Pupi A, Biondi F, Faravelli C: Brain perfusion abnormalities in drug-naive, lactate-sensitive panic patients: a SPECT study. Biol Psychiatry. 1993, 33: 505-512. 10.1016/0006-3223(93)90004-W.
Reiman EM, Raichle ME, Robins E, Butler FK, Herscovitch P, Fox P, Perlmutter J: The application of positron emission tomography to the study of panic disorder. Am J Psychiatry. 1986, 143: 469-477.
Bisaga A, Katz JL, Antonini A, Wright CE, Margouleff C, Gorman JM, Eidelberg D: Cerebral glucose metabolism in women with panic disorder. Am J Psychiatry. 1998, 155: 1178-1183.
Sakai Y, Kumano H, Nishikawa M, Sakano Y, Kaiya H, Imabayashi E, Ohnishi T, Matsuda H, Yasuda A, Sato A: Cerebral glucose metabolism associated with a fear network in panic disorder. Neuroreport. 2005, 16: 927-931. 10.1097/00001756-200506210-00010.
Dresler T, Hahn T, Plichta MM, Ernst LH, Tupak SV, Ehlis AC, Warrings B, Deckert J, Fallgatter AJ: Neural correlates of spontaneous panic attacks. J Neural Transm. 2011, 118: 263-269. 10.1007/s00702-010-0540-2.
Pfleiderer B, Zinkirciran S, Arolt V, Heindel W, Deckert J, Domschke K: fMRI amygdala activation during a spontaneous panic attack in a patient with panic disorder. World J Biol Psychiatry. 2007, 8: 269-272. 10.1080/15622970701216673.
Spiegelhalder K, Hornyak M, Kyle SD, Paul D, Blechert J, Seifritz E, Hennig J, Tebartz van Elst L, Riemann D, Feige B: Cerebral correlates of heart rate variations during a spontaneous panic attack in the fMRI scanner. Neurocase. 2009, 15: 527-534. 10.1080/13554790903066909.
Fischer H, Andersson JL, Furmark T, Fredrikson M: Brain correlates of an unexpected panic attack: a human positron emission tomographic study. Neurosci Lett. 1998, 251: 137-140. 10.1016/S0304-3940(98)00503-5.
Eser D, Leicht G, Lutz J, Wenninger S, Kirsch V, Schule C, Karch S, Baghai T, Pogarell O, Born C: Functional neuroanatomy of CCK-4-induced panic attacks in healthy volunteers. Hum Brain Mapp. 2009, 30: 511-522. 10.1002/hbm.20522.
Javanmard M, Shlik J, Kennedy SH, Vaccarino FJ, Houle S, Bradwejn J: Neuroanatomic correlates of CCK-4-induced panic attacks in healthy humans: a comparison of two time points. Biol Psychiatry. 1999, 45: 872-882. 10.1016/S0006-3223(98)00348-5.
Servan-Schreiber D, Perlstein WM, Cohen JD, Mintun M: Selective pharmacological activation of limbic structures in human volunteers: a positron emission tomography study. J Neuropsychiatry Clin Neurosci. 1998, 10: 148-159.
Benkelfat C, Bradwejn J, Meyer E, Ellenbogen M, Milot S, Gjedde A, Evans A: Functional neuroanatomy of CCK4-induced anxiety in normal healthy volunteers. Am J Psychiatry. 1995, 152: 1180-1184.
Kent JM, Coplan JD, Mawlawi O, Martinez JM, Browne ST, Slifstein M, Martinez D, Abi-Dargham A, Laruelle M, Gorman JM: Prediction of panic response to a respiratory stimulant by reduced orbitofrontal cerebral blood flow in panic disorder. Am J Psychiatry. 2005, 162: 1379-1381. 10.1176/appi.ajp.162.7.1379.
Schunck T, Erb G, Mathis A, Gilles C, Namer IJ, Hode Y, Demaziere A, Luthringer R, Macher JP: Functional magnetic resonance imaging characterization of CCK-4-induced panic attack and subsequent anticipatory anxiety. Neuroimage. 2006, 31: 1197-1208. 10.1016/j.neuroimage.2006.01.035.
Boshuisen ML, Ter Horst GJ, Paans AM, Reinders AA, den Boer JA: rCBF differences between panic disorder patients and control subjects during anticipatory anxiety and rest. Biol Psychiatry. 2002, 52: 126-135. 10.1016/S0006-3223(02)01355-0.
Marchand WR, Lee JN, Healy L, Thatcher JW, Rashkin E, Starr J, Hsu E: An fMRI motor activation paradigm demonstrates abnormalities of putamen activation in females with panic disorder. J Affect Disord. 2009, 116: 121-125. 10.1016/j.jad.2008.10.026.
Maddock RJ, Buonocore MH, Kile SJ, Garrett AS: Brain regions showing increased activation by threat-related words in panic disorder. Neuroreport. 2003, 14: 325-328.
Bystritsky A, Pontillo D, Powers M, Sabb FW, Craske MG, Bookheimer SY: Functional MRI changes during panic anticipation and imagery exposure. Neuroreport. 2001, 12: 3953-3957. 10.1097/00001756-200112210-00020.
Shin LM, Liberzon I: The neurocircuitry of fear, stress, and anxiety disorders. Neuropsychopharmacology. 2010, 35: 169-191. 10.1038/npp.2009.83.
Tuescher O, Protopopescu X, Pan H, Cloitre M, Butler T, Goldstein M, Root JC, Engelien A, Furman D, Silverman M: Differential activity of subgenual cingulate and brainstem in panic disorder and PTSD. J Anxiety Disord. 2011, 25: 251-257. 10.1016/j.janxdis.2010.09.010.
Pillay SS, Gruber SA, Rogowska J, Simpson N, Yurgelun-Todd DA: fMRI of fearful facial affect recognition in panic disorder: the cingulate gyrus-amygdala connection. J Affect Disord. 2006, 94: 173-181. 10.1016/j.jad.2006.04.007.
Dias MRC, Silveira ACD, de Melo Neto VL, Bevilaqua MCN, Gardino PF, Nardi AE: Current methodological designs of fMRI studies of panic disorder: Can data be compared?. Psychology & Neuroscience. 2011, 4: 391-407. 10.3922/j.psns.2011.3.013.
Harmer CJ, Mackay CE, Reid CB, Cowen PJ, Goodwin GM: Antidepressant drug treatment modifies the neural processing of nonconscious threat cues. Biol Psychiatry. 2006, 59: 816-820. 10.1016/j.biopsych.2005.10.015.
Ohrmann P, Pedersen A, Braun M, Bauer J, Kugel H, Kersting A, Domschke K, Deckert J, Suslow T: Effect of gender on processing threat-related stimuli in patients with panic disorder: sex does matter. Depress Anxiety. 2010, 27: 1034-1043. 10.1002/da.20721.
Domschke K, Ohrmann P, Braun M, Suslow T, Bauer J, Hohoff C, Kersting A, Engelien A, Arolt V, Heindel W: Influence of the catechol-O-methyltransferase val158met genotype on amygdala and prefrontal cortex emotional processing in panic disorder. Psychiatry Res. 2008, 163: 13-20. 10.1016/j.pscychresns.2007.04.016.
Pillay SS, Rogowska J, Gruber SA, Simpson N, Yurgelun-Todd DA: Recognition of happy facial affect in panic disorder: an fMRI study. J Anxiety Disord. 2007, 21: 381-393. 10.1016/j.janxdis.2006.04.001.
Nash JR, Sargent PA, Rabiner EA, Hood SD, Argyropoulos SV, Potokar JP, Grasby PM, Nutt DJ: Serotonin 5-HT1A receptor binding in people with panic disorder: positron emission tomography study. Br J Psychiatry. 2008, 193: 229-234. 10.1192/bjp.bp.107.041186.
Sim HB, Kang EH, Yu BH: Changes in cerebral cortex and limbic brain functions after short-term paroxetine treatment in panic disorder: an [F]FDG-PET pilot study. Psychiatry Investig. 2010, 7: 215-219. 10.4306/pi.2010.7.3.215.
Prasko J, Horacek J, Zalesky R, Kopecek M, Novak T, Paskova B, Skrdlantova L, Belohlavek O, Hoschl C: The change of regional brain metabolism (18FDG PET) in panic disorder during the treatment with cognitive behavioral therapy or antidepressants. Neuro Endocrinol Lett. 2004, 25: 340-348.
Sakai Y, Kumano H, Nishikawa M, Sakano Y, Kaiya H, Imabayashi E, Ohnishi T, Matsuda H, Yasuda A, Sato A: Changes in cerebral glucose utilization in patients with panic disorder treated with cognitive-behavioral therapy. Neuroimage. 2006, 33: 218-226. 10.1016/j.neuroimage.2006.06.017.
McEwen BS: Mood disorders and allostatic load. Biol Psychiatry. 2003, 54: 200-207. 10.1016/S0006-3223(03)00177-X.
Malizia AL, Cunningham VJ, Bell CJ, Liddle PF, Jones T, Nutt DJ: Decreased brain GABA(A)-benzodiazepine receptor binding in panic disorder: preliminary results from a quantitative PET study. Arch Gen Psychiatry. 1998, 55: 715-720. 10.1001/archpsyc.55.8.715.
Kaschka W, Feistel H, Ebert D: Reduced benzodiazepine receptor binding in panic disorders measured by iomazenil SPECT. J Psychiatr Res. 1995, 29: 427-434. 10.1016/0022-3956(95)00019-2.
Novotny EJ, Fulbright RK, Pearl PL, Gibson KM, Rothman DL: Magnetic resonance spectroscopy of neurotransmitters in human brain. Ann Neurol. 2003, 54 (Suppl 6): S25-S31.
Shekhar A, Keim SR, Simon JR, McBride WJ: Dorsomedial hypothalamic GABA dysfunction produces physiological arousal following sodium lactate infusions. Pharmacol Biochem Behav. 1996, 55: 249-256. 10.1016/S0091-3057(96)00077-9.
Maddock RJ: The lactic acid response to alkalosis in panic disorder: an integrative review. J Neuropsychiatry Clin Neurosci. 2001, 13: 22-34. 10.1176/appi.neuropsych.13.1.22.
Westerblad H, Allen DG, Lannergren J: Muscle fatigue: lactic acid or inorganic phosphate the major cause?. News Physiol Sci. 2002, 17: 17-21.
Kempton MJ, Haldane M, Jogia J, Christodoulou T, Powell J, Collier D, Williams SC, Frangou S: The effects of gender and COMT Val158Met polymorphism on fearful facial affect recognition: a fMRI study. Int J Neuropsychopharmacol. 2009, 12: 371-381. 10.1017/S1461145708009395.
Taylor WD, Zuchner S, Payne ME, Messer DF, Doty TJ, MacFall JR, Beyer JL, Krishnan KR: The COMT Val158Met polymorphism and temporal lobe morphometry in healthy adults. Psychiatry Res. 2007, 155: 173-177. 10.1016/j.pscychresns.2007.01.005.
Zinkstok J, Schmitz N, van Amelsvoort T, de Win M, van den Brink W, Baas F, Linszen D: The COMT val158met polymorphism and brain morphometry in healthy young adults. Neurosci Lett. 2006, 405: 34-39. 10.1016/j.neulet.2006.06.034.
Leskin GA, Sheikh JI: Lifetime trauma history and panic disorder: findings from the National Comorbidity Survey. J Anxiety Disord. 2002, 16: 599-603. 10.1016/S0887-6185(02)00125-1.
Friedman S, Smith L, Fogel D, Paradis C, Viswanathan R, Ackerman R, Trappler B: The incidence and influence of early traumatic life events in patients with panic disorder: a comparison with other psychiatric outpatients. J Anxiety Disord. 2002, 16: 259-272. 10.1016/S0887-6185(02)00097-X.
Anda RF, Felitti VJ, Bremner JD, Walker JD, Whitfield C, Perry BD, Dube SR, Giles WH: The enduring effects of abuse and related adverse experiences in childhood. A convergence of evidence from neurobiology and epidemiology. Eur Arch Psychiatry Clin Neurosci. 2006, 256: 174-186. 10.1007/s00406-005-0624-4.
Lewis SJ, Verberne AJ, Robinson TG, Jarrott B, Louis WJ, Beart PM: Excitotoxin-induced lesions of the central but not basolateral nucleus of the amygdala modulate the baroreceptor heart rate reflex in conscious rats. Brain Res. 1989, 494: 232-240. 10.1016/0006-8993(89)90591-X.
Etkin A: Functional neuroanatomy of anxiety: a neural circuit perspective. Curr Top Behav Neurosci. 2010, 2: 251-277.
Klein DF: False suffocation alarms, spontaneous panics, and related conditions. An integrative hypothesis. Arch Gen Psychiatry. 1993, 50: 306-317. 10.1001/archpsyc.1993.01820160076009.
Nutt DJ, Ballenger JC: Anxiety disorders. 2003, Malden, Mass: Blackwell Science
Ziemann AE, Allen JE, Dahdaleh NS, Drebot , Coryell MW, Wunsch AM, Lynch CM, Faraci FM, Howard MA, Welsh MJ, Wemmie JA: The amygdala is a chemosensor that detects carbon dioxide and acidosis to elicit fear behavior. Cell. 2009, 139: 1012-1021. 10.1016/j.cell.2009.10.029.
Noyes R, Hoehn-Saric R: The anxiety disorders. 1998, Cambridge, UK: Cambridge University Press
Huttunen J, Tolvanen H, Heinonen E, Voipio J, Wikstrom H, Ilmoniemi RJ, Hari R, Kaila K: Effects of voluntary hyperventilation on cortical sensory responses. Electroencephalographic and magnetoencephalographic studies. Exp Brain Res. 1999, 125: 248-254. 10.1007/s002210050680.
Artru AA: Reduction of cerebrospinal fluid pressure by hypocapnia: changes in cerebral blood volume, cerebrospinal fluid volume and brain tissue water and electrolytes. II. Effects of anesthetics. J Cereb Blood Flow Metab. 1988, 8: 750-756. 10.1038/jcbfm.1988.123.
Forsyth JP, Eifert GH: Response intensity in content-specific fear conditioning comparing 20% versus 13% CO2-enriched air as unconditioned stimuli. J Abnorm Psychol. 1998, 107: 291-304.
Godemann F, Ahrens B, Behrens S, Berthold R, Gandor C, Lampe F, Linden M: Classic conditioning and dysfunctional cognitions in patients with panic disorder and agoraphobia treated with an implantable cardioverter/defibrillator. Psychosom Med. 2001, 63: 231-238.
Michael T, Blechert J, Vriends N, Margraf J, Wilhelm FH: Fear conditioning in panic disorder: Enhanced resistance to extinction. J Abnorm Psychol. 2007, 116: 612-617.
LeDoux JE, Gorman JM: A call to action: overcoming anxiety through active coping. Am J Psychiatry. 2001, 158: 1953-1955. 10.1176/appi.ajp.158.12.1953.
Dolan RJ: Emotion, cognition, and behavior. Science. 2002, 298: 1191-1194. 10.1126/science.1076358.
Bremner JD, Randall P, Vermetten E, Staib L, Bronen RA, Mazure C, Capelli S, McCarthy G, Innis RB, Charney DS: Magnetic resonance imaging-based measurement of hippocampal volume in posttraumatic stress disorder related to childhood physical and sexual abuse–a preliminary report. Biol Psychiatry. 1997, 41: 23-32. 10.1016/S0006-3223(96)00162-X.
De Bellis MD, Keshavan MS, Clark DB, Casey BJ, Giedd JN, Boring AM, Frustaci K, Ryan ND: A.E. Bennett Research Award. Developmental traumatology. Part II: Brain development. Biol Psychiatry. 1999, 45: 1271-1284. 10.1016/S0006-3223(99)00045-1.
Anderson AK, Sobel N: Dissociating intensity from valence as sensory inputs to emotion. Neuron. 2003, 39: 581-583. 10.1016/S0896-6273(03)00504-X.
Roy AK, Shehzad Z, Margulies DS, Kelly AM, Uddin LQ, Gotimer K, Biswal BB, Castellanos FX, Milham MP: Functional connectivity of the human amygdala using resting state fMRI. Neuroimage. 2009, 45: 614-626. 10.1016/j.neuroimage.2008.11.030.
Harding AJ, Stimson E, Henderson JM, Halliday GM: Clinical correlates of selective pathology in the amygdala of patients with Parkinson’s disease. Brain. 2002, 125: 2431-2445. 10.1093/brain/awf251.
Roppongi T, Nakamura M, Asami T, Hayano F, Otsuka T, Uehara K, Fujiwara A, Saeki T, Hayasaka S, Yoshida T: Posterior orbitofrontal sulcogyral pattern associated with orbitofrontal cortex volume reduction and anxiety trait in panic disorder. Psychiatry Clin Neurosci. 2010, 64: 318-326. 10.1111/j.1440-1819.2010.02085.x.
Asami T, Hayano F, Nakamura M, Yamasue H, Uehara K, Otsuka T, Roppongi T, Nihashi N, Inoue T, Hirayasu Y: Anterior cingulate cortex volume reduction in patients with panic disorder. Psychiatry Clin Neurosci. 2008, 62: 322-330. 10.1111/j.1440-1819.2008.01800.x.
Uchida RR, Del-Ben CM, Busatto GF, Duran FL, Guimaraes FS, Crippa JA, Araujo D, Santos AC, Graeff FG: Regional gray matter abnormalities in panic disorder: a voxel-based morphometry study. Psychiatry Res. 2008, 163: 21-29. 10.1016/j.pscychresns.2007.04.015.
Protopopescu X, Pan H, Tuescher O, Cloitre M, Goldstein M, Engelien A, Yang Y, Gorman J, LeDoux J, Stern E, Silbersweig D: Increased brainstem volume in panic disorder: a voxel-based morphometric study. Neuroreport. 2006, 17: 361-363. 10.1097/01.wnr.0000203354.80438.1.
Yoo HK, Kim MJ, Kim SJ, Sung YH, Sim ME, Lee YS, Song SY, Kee BS, Lyoo IK: Putaminal gray matter volume decrease in panic disorder: an optimized voxel-based morphometry study. Eur J Neurosci. 2005, 22: 2089-2094. 10.1111/j.1460-9568.2005.04394.x.
Massana G, Serra-Grabulosa JM, Salgado-Pineda P, Gasto C, Junque C, Massana J, Mercader JM: Parahippocampal gray matter density in panic disorder: a voxel-based morphometric study. Am J Psychiatry. 2003, 160: 566-568. 10.1176/appi.ajp.160.3.566.
Vythilingam M, Anderson ER, Goddard A, Woods SW, Staib LH, Charney DS, Bremner JD: Temporal lobe volume in panic disorder–a quantitative magnetic resonance imaging study. Psychiatry Res. 2000, 99: 75-82. 10.1016/S0925-4927(00)00055-X.
Fontaine R, Breton G, Dery R, Fontaine S, Elie R: Temporal lobe abnormalities in panic disorder: an MRI study. Biol Psychiatry. 1990, 27: 304-310. 10.1016/0006-3223(90)90004-L.
Nordahl TE, Semple WE, Gross M, Mellman TA, Stein MB, Goyer P, King AC, Uhde TW, Cohen RM: Cerebral glucose metabolic differences in patients with panic disorder. Neuropsychopharmacology. 1990, 3: 261-272.
Reiman EM, Raichle ME, Butler FK, Herscovitch P, Robins E: A focal brain abnormality in panic disorder, a severe form of anxiety. Nature. 1984, 310: 683-685. 10.1038/310683a0.
Reiman EM, Raichle ME, Robins E, Mintun MA, Fusselman MJ, Fox PT, Price JL, Hackman KA: Neuroanatomical correlates of a lactate-induced anxiety attack. Arch Gen Psychiatry. 1989, 46: 493-500. 10.1001/archpsyc.1989.01810060013003.
Stewart RS, Devous MD, Rush AJ, Lane L, Bonte FJ: Cerebral blood flow changes during sodium-lactate-induced panic attacks. Am J Psychiatry. 1988, 145: 442-449.
Woods SW, Koster K, Krystal JK, Smith EO, Zubal IG, Hoffer PB, Charney DS: Yohimbine alters regional cerebral blood flow in panic disorder. Lancet. 1988, 2: 678-
Nordahl TE, Stein MB, Benkelfat C, Semple WE, Andreason P, Zametkin A, Uhde TW, Cohen RM: Regional cerebral metabolic asymmetries replicated in an independent group of patients with panic disorders. Biol Psychiatry. 1998, 44: 998-1006. 10.1016/S0006-3223(98)00026-2.
Beutel ME, Stark R, Pan H, Silbersweig D, Dietrich S: Changes of brain activation pre- post short-term psychodynamic inpatient psychotherapy: an fMRI study of panic disorder patients. Psychiatry Res. 2010, 184: 96-104. 10.1016/j.pscychresns.2010.06.005.
Pfleiderer B, Zinkirciran S, Michael N, Hohoff C, Kersting A, Arolt V, Deckert J, Domschke K: Altered auditory processing in patients with panic disorder: a pilot study. World J Biol Psychiatry. 2010, 11: 945-955. 10.3109/15622975.2010.490273.
Chechko N, Wehrle R, Erhardt A, Holsboer F, Czisch M, Samann PG: Unstable prefrontal response to emotional conflict and activation of lower limbic structures and brainstem in remitted panic disorder. PLoS One. 2009, 4: e5537-10.1371/journal.pone.0005537.
Domschke K, Braun M, Ohrmann P, Suslow T, Kugel H, Bauer J, Hohoff C, Kersting A, Engelien A, Arolt V: Association of the functional -1019C/G 5-HT1A polymorphism with prefrontal cortex and amygdala activation measured with 3 T fMRI in panic disorder. Int J Neuropsychopharmacol. 2006, 9: 349-355. 10.1017/S1461145705005869.
van den Heuvel OA, Veltman DJ, Groenewegen HJ, Witter MP, Merkelbach J, Cath DC, van Balkom AJ, van Oppen P, van Dyck R: Disorder-specific neuroanatomical correlates of attentional bias in obsessive-compulsive disorder, panic disorder, and hypochondriasis. Arch Gen Psychiatry. 2005, 62: 922-933. 10.1001/archpsyc.62.8.922.
Acknowledgments
Taeyoung Choi, MD, PhD, Hyeonseok S Jeong, BS, Jiyoung Jung, MD, Yera Choi, BA, Ekyung Shin, BS, and Jooyeon Jamie Im, BA have provided technical support. This work was supported in part by grant KRF-2008-220-E00021 (IKL); and grant 2009–0074584 (JEK) funded by the National Research Foundation of Korea; grant A112009 (JEK) from the Korean Ministry of Health and Welfare; and grant 2012K001117 (IKL) from the Korean Ministry of Education, Science, and Technology.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
IKL has received research support from AstraZeneca, Lundbeck, and GSK. All other authors declare that they have no competing interests.
Authors’ contributions
JEK, SRD and IKL have contributed to conception and design of the review. JEK drafted the manuscript. All authors have done a critical revision of the manuscript and approved the final version of the manuscript.
Electronic supplementary material
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Kim, J.E., Dager, S.R. & Lyoo, I.K. The role of the amygdala in the pathophysiology of panic disorder: evidence from neuroimaging studies. Biol Mood Anxiety Disord 2, 20 (2012). https://doi.org/10.1186/2045-5380-2-20
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2045-5380-2-20