
Abstract
Duchenne muscular dystrophy (DMD) is the most common muscular dystrophy and an X-linked recessive, progressive muscle wasting disease caused by the absence of a functional dystrophin protein. Dystrophin has a structural role as a cytoskeletal stabilization protein and protects cells against contraction-induced damage. Dystrophin also serves a signaling role through mechanotransduction of forces and localization of neuronal nitric oxide synthase (nNOS), which produces nitric oxide (NO) to facilitate vasorelaxation. In DMD, the signaling defects produce inadequate tissue perfusion caused by functional ischemia due to a diminished ability to respond to shear stress induced endothelium-dependent dilation. Additionally, the structural defects seen in DMD render myocytes with an increased susceptibility to mechanical stress. The combination of both defects is necessary to generate myocyte damage, which induces successive rounds of myofiber degeneration and regeneration, loss of calcium homeostasis, chronic inflammatory response, fibrosis, and myonecrosis. In individuals with DMD, these processes inevitably cause loss of ambulation shortly after the first decade and an abbreviated life with death in the third or fourth decade due to cardio-respiratory anomalies. There is no known cure for DMD, and although the culpable gene has been identified for more than twenty years, research on treatments has produced few clinically relevant results. Several recent studies on novel DMD therapeutics are vascular targeted and focused on attenuating the inherent functional ischemia. One approach improves vasorelaxation capacity through pharmaceutical inhibition of either phosphodiesterase 5 (PDE5) or angiotensin-converting enzyme (ACE). Another approach increases the density of the underlying vascular network by inducing angiogenesis, and this has been accomplished through either direct delivery of vascular endothelial growth factor (VEGF) or by downregulating the VEGF decoy-receptor type 1 (VEGFR-1 or Flt-1). The pro-angiogenic approaches also seem to be pro-myogenic and could resolve the age-related decline in satellite cell (SC) quantity seen in mdx models through expansion of the SC juxtavascular niche. Here we review these four vascular targeted treatment strategies for DMD and discuss mechanisms, proof of concept, and the potential for clinical relevance associated with each therapy.
Similar content being viewed by others


Review
Duchenne muscular dystrophy (DMD) is an X-linked recessive, progressive muscle wasting disease caused by mutations in the DMD gene that lead to absence of a functional dystrophin protein[1, 2]. Both fatal and devastating, DMD is the most common muscular dystrophy seen in children and has an annual incidence affecting one in every 3600–6000 newborn males[3]. Normally, dystrophin serves as the bridge in the dystrophin-associated glycoprotein complex (DAPC), connecting the cytoskeleton, via attachments to subsarcolemmal F-actin, to the extracellular matrix through an association with plasma membrane bound β-dystroglycan[4]. In the DAPC, dystrophin has a structural role as a cytoskeletal stabilization protein and protects cells against contraction-induced damage. Dystrophin also serves signaling roles, including mechanotransduction of forces and localization of signaling proteins, such as neuronal nitric oxide synthase (nNOS), which synthesizes nitric oxide (NO) to facilitate vasorelaxation[5–7]. Without dystrophin, the DAPC cannot completely assemble, and the supportive link between the cytoskeleton and the extracellular matrix becomes destabilized[8]. Despite normal development, the membrane in dystrophin-deficient cells is easily damaged. Membrane microlesions facilitate an influx of calcium ions, which activate proteases to begin auto-digestion of the musculature sarcoplasm[9–11]. Macrophages later arrive at the tissue to remove cellular debris, and satellite cells (SCs) are activated and proliferate to induce myofiber regeneration. This causes successive rounds of myofiber degeneration and regeneration that is exacerbated by continual membrane damage and ensuing myonecrosis. In addition, cytokines released in the process of myonecrosis recruit inflammatory cells, which release inflammatory cytokines to activate fibroblasts that lay down extracellular matrix proteins and lead to fibrosis[12]. Skeletal muscle regenerative capacity later diminishes with advancing age and decreasing numbers of SCs, and muscle tissue is steadily replaced by adipose and connective tissues[13].
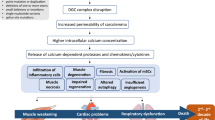
The previously described cellular events manifest themselves clinically in a devastating and progressive manner. Despite continuous contractions by the myocardium, the skeletal muscles deteriorate first in individuals with DMD, and most permanently lose ambulatory abilities shortly after the first decade[14]. Myocardial problems present later, and clinically relevant cardiomyopathy is seen in 90% of patients over 18 years old, namely due to the onset of cardiac fibrosis in addition to rhythm and conduction abnormalities[14]. Respiratory problems are also inevitable due to muscle wasting in the diaphragm and the onset of scoliosis[14]. Even with improvements in treatment, notably multidisciplinary care, the combined cardio-respiratory anomalies mean that most individuals with DMD die in their third or fourth decade of life[15, 16]. Despite knowledge of the responsible gene for over twenty years, a DMD cure remains to be found, and research on treatments has produced few clinically relevant results. Current treatment options, such as corticosteroid administration, physical therapy, nocturnal ventilation, and surgical interventions aim for symptomatic management and have been shown to improve lifespan and quality of life[16]. The clinical utility and feasibility of gene therapy and cell therapy remain to be elucidated, and other treatment areas must be sought. Our current, more holistic understanding of DMD pathogenesis, especially with more recent knowledge of the vascular role of dystrophin, implies that vascular-targeted therapies are strong candidates for future investigation. Specifically, attenuating functional ischemia could reduce myocyte damage, increase tissue perfusion, reduce cardiac workload, and prevent cardiac and skeletal muscle remodeling (Figure 1B). This review will focus on vascular-targeted treatment avenues aimed at either improving vasorelaxation capacity or increasing the underlying vascular density in order to reduce the functional ischemia and improve the DMD phenotype.

The two-hit hypothesis for myocyte damage and the proposed outcome of functional ischemia attenuation in Duchenne muscular dystrophy (DMD). (A) The combined effects from functional ischemia due to reduced nitric oxide (NO)-mediated protection and greater cellular susceptibility to metabolic stress are necessary to produce the myofiber damage observed in DMD [17]. (B) Attenuating functional ischemia by administering a vascular targeted treatment can reduce the net-combined effect of both two-hit factors and consequently curtail myofiber damage.
Defect of nitric oxide-mediated vasodilation contributes to Duchenne muscular dystrophy phenotype
The DMD pathogenesis is partially explained by the lack of the signaling role of dystrophin, which normally localizes nNOS to the sarcolemma through binding to the C-terminal region of dystrophin[6]. The nNOS is responsible for NO production to facilitate smooth muscle vasodilation in response to increased metabolic demands. During muscle contraction, NO-mediated vasodilation is important to help offset the α-adrenergic vasoconstriction in response to sympathetic activation, which optimizes muscle perfusion[18]. This functional response is intact in healthy children, but in children with DMD the sympathetic vasoconstriction in skeletal muscle is unopposed due to lack of NO-mediated vasodilation[18].
The nNOS is absent from the sarcolemma and is greatly downregulated in the cytoplasm of dystrophin-deficient muscle, which results in muscle vasoconstriction and abnormal blood flow during skeletal muscle contraction[6, 18, 19]. Specifically, loss of dystrophin in the smooth muscle results in a decreased capacity of the vasculature to respond to shear stress induced endothelium-dependent dilation, probably related to the signaling defects seen in both force transduction and inadequate NO production[19]. Furthermore, shear stress at the endothelial cell surface is a known catalyst for angiogenesis[20–23], so new blood vessel formation could be downregulated and mismatched to metabolic need in the absence of dystrophin due to defects in mechanotransduction. Lack of the signaling and structural roles of dystrophin in DMD pathogenesis have led to a two-hit hypothesis, whereby the combination of functional ischemia due to reduced capacity to benefit from NO-mediated protection and an increased susceptibility to metabolic stress are both required to cause myocyte damage (Figure 1A)[17]. So although not completely culpable for the observed pathogenesis, impaired vascular functioning seems to be both inherent to DMD and an accelerant to tissue damage in the skeletal and cardiac muscles.
Recent studies suggest that the two-hit hypothesis should migrate away from simply observational speculation towards a more widely accepted, evidence-based DMD pathogenic theory. One functional study using model DMD mdx mice showed quantitative evidence supporting the two-hit hypothesis, where inhibition of NO/EDHF (EDHF is endothelium-derived hyperpolarizing factor, another vasodilator) alone in wild-type mice caused similar functional ischemia (one-hit) to that seen in mdx mice[24]. But, the forced functional ischemia alone in the wild-type mice did not induce similar levels of muscle cell death seen in mdx mice[24]. Two-hits, consisting of severe ischemia and strenuous tetanic stimuli, were necessary to produce the same contraction-dependent myofiber damage in wild-type mice to that of mdx mice[24]. So, mdx myofibers exhibit enhanced vulnerability to metabolic and mechanical stress independently of the altered vasodilatory response, yet the combinatory effect of both factors (two-hits) is necessary to mediate the cell death numbers seen in mdx myofibers. This could also explain why nNOS knockout mice, a one-hit model, do not develop muscular dystrophy, yet myocardial specific nNOS expression prevents cardiomyopathy in mdx mice by increasing the capacity to benefit from NO-mediated protection[25, 26]. Interestingly, mdx mice that only express dystrophin in smooth muscle (SMTg/mdx), which has a prominent role in regulation of vascular tone and blood flow, have an intermediate phenotype between wild-type and mdx mice[27]. The SMTg/mdx mice showed some, albeit not total, recovery of the NO-dependent vasorelaxation mechanism in active skeletal muscle[27]. In the SMTg/mdx model, lack of dystrophin at the myofiber level and the consequent lack of complete force mechanotransduction in response to functional demands were perhaps partially responsible for the insufficient phenotypic recovery. These data overwhelmingly reveal the important role the vasculature plays in DMD pathogenesis and highlight a novel arena for therapeutic intervention.
Improved vasorelaxation capacity
Angiotensin-converting enzyme inhibitors
The renin-angiotensin-aldosterone system plays a vital role in regulating both systemic vascular resistance and total blood volume, which together impact arterial pressure and myocardial function. A key component is the angiotensin-converting enzyme (ACE), which transforms the peptide hormone angiotensin I into angiotensin II; circulating angiotensin II then stimulates vascular smooth muscle contraction, increasing vascular resistance and arterial pressure. Angiotensin II also induces the release of aldosterone, which increases sodium and water retention, and vasopressin, which increases water retention. Preventing angiotensin II production through pharmacological ACE inhibition has been shown to reduce high blood pressure and cardiac workload through enhanced vasorelaxation and prevention of downstream hormone release, and ACE inhibitors (ACEIs) are currently used to treat congestive heart failure and hypertension[28–30]. As such, improving cardiac function and enhancing systemic vasorelaxation capacity through ACE inhibition in DMD patients could have prophylactic benefit by mitigating the functional ischemia and consequently diminishing myonecrosis. In mdx mice, the ACEI captopril administered over an 8-week period and prior to the onset of cardiomyopathy was shown to reduce cardiac afterload, increase myocardial contractility, and improve cardiac hemodynamics compared to mdx control mice[31]. In clinical studies, administration of the ACEI perindopril has shown that early treatment in 9.5- to 13-year-old DMD patients with normal cardiac functioning (as measured by normal left ventricular ejection fraction or LVEF), is capable of delaying both the onset and progression of left ventricular dysfunction and significantly lowering mortality rates compared to patients starting treatment 3 years later[32, 33]. Results are less clear in DMD cases involving established cardiomyopathy where administration of the ACEI enalapril showed functional normalization in just 43% of cases, but this positive functional effect was maintained by most subjects for up to four years[34].
Combination therapy using ACEIs and β-adrenergic receptor antagonists or β-blockers (BBs) has also been investigated for use in DMD treatment. Catecholamines increase heart rate and myocardial contractility through the β-adrenergic receptors, and thus targeted BBs cause the heart to beat slower and with less force and are typically given to patients with arrhythmia or disordered automaticity. One study that utilized a combination of BBs and ACEIs in DMD patients with established cardiomyopathy found a positive effect on long-term survival, especially for individuals that showed no overt symptoms of heart failure despite documented left ventricular dysfunction[35]. A more recent study also investigated DMD patients with established cardiomyopathy but broke treatment groups down into ACEI (lisinopril) alone or ACEI plus BB (metoprolol)[36]. Both treatment groups displayed improvements in cardiac function compared to pre-therapy measurements, but no significant difference in cardiac function was seen between groups[36]. Future studies should address treatment using ACEI alone or ACEI plus BB in DMD cases where cardiomyopathy has not been fully established to assess the potential for prophylactic benefit as this could definitively rule out the need for a BB. Additionally, with regard to the two-hit hypothesis, studies that address functional ischemia attenuation to mitigate myonecrosis through enhanced tissue perfusion by ACEI-mediated vasorelaxation have not yet been performed.
Another therapeutic strategy that targets the renin-angiotensin-aldosterone system to improve vasorelaxation capacity utilizes the antihypertensive drug losartan, which is an angiotensin II type I receptor antagonist or angiotensin receptor blocker (ARB). Long-term administration of losartan in mdx mice showed improvements in myocardial function, but not skeletal muscle function, and reductions in mortality compared to control[37, 38]. Explanations as to why losartan could only ameliorate the function of cardiac muscle remain limited, but the primary mechanism could be the significant reduction in afterload seen in the hearts of losartan treated mdx mice[38]. Decreased afterload certainly reduces cardiac workload, and this may minimize mechanical injury and subsequent fibrosis to the sensitive cardiomyocytes in mdx hearts. Still, the use of losartan as a prophylactic treatment against DMD-related cardiomyopathy seems promising based on these pre-clinical studies and the current clinical availability of losartan (COZAAR™) for its use in hypertension. Future investigation should be directed at evaluating losartan in DMD patients but also, owing to pathway similarity, at comparing the effectiveness of losartan to the many FDA-approved ACEIs.
The definitive mechanism behind reducing cardiomyopathy via ARBs, ACEIs, and/or BBs in DMD patients is not completely established, but reduced aldosterone signaling through ACE inhibition could prevent fibrotic tissue development, as previous use of aldosterone-specific blockers has shown benefit in cases of heart failure[39–42]. Additionally, angiotensin II can directly induce vasoconstriction, pro-fibrotic Smad signaling, pro-fibrotic transforming growth factor beta (TGF-β) production, and the ubiquitin-proteasome pathway that has a role in the proteolysis happening in dystrophic tissues[43–46]. Angiotensin II is also known to enhance NADPH-oxidase activity, which leads to overproduction of superoxide anion and accounts for the oxidative stress in cardiac and skeletal muscle of the mdx mouse[47–52]. ACE inhibition can reduce these adverse effects, and a study with mdx mice demonstrated that the ACEI enalapril can prevent angiotensin II dependent stimulation of pro-oxidant and pro-inflammatory pathways[53]. Overall, ACEIs and/or BBs appear to be excellent candidates for DMD therapy based on current clinical availability and demonstrated ability to reduce many of the negative outcomes normally associated with the DMD pathogenic process. Still, a broader investigation regarding the potential prophylactic benefit of ACEIs should be conducted to determine an optimal age of initiation.
Phosphodiesterase 5 inhibitors
In the NO-cGMP signaling pathway, nitric oxide synthase (NOS) produces NO to activate soluble guanylyl cyclase (sGC) to synthesize cyclic guanosine monophosphate (cGMP), and cGMP activates protein kinase G to induce vasodilation. The cGMP-specific phosphodiesterases are responsible for cGMP degradation, so vasoconstriction begins as concentrations of cGMP diminish. This pathway is disrupted in dystrophin deficient membranes, as nNOS is absent from the sarcolemma and greatly downregulated, which contributes to the observed functional ischemia[6, 19]. Additionally, studies have shown greater cGMP-specific phosphodiesterase 5 (PDE5) activity in mdx skeletal muscle samples and decreased cGMP production compared with controls[54, 55]. Recently, cGMP-specific PDE5 inhibitors, specifically tadalafil (Cialis™ or Adcirca™) and sildenafil (Viagra™ or Revatio™) have been investigated for their potential in ameliorating the functional ischemia in DMD by increasing intracellular levels of cGMP to prolong vasodilation and increase blood flow to tissues.
Asai et al. elegantly showed that tadalafil administration prior to progressive myofiber damage was able to significantly lower the net quantity of myofiber damage in mdx mice compared to placebo[24]. Essentially, attenuation of functional ischemia using tadalafil was shown to reduce the extent of contraction-induced damage[24]. Additionally, early treatment utilizing PDE5 inhibitors could have clinical prophylactic benefit, for mdx mice treated with tadalafil from conception showed improved histology[24]. Khairallah et al. showed that cardiac mRNA expression levels of atrial natriuretic factor (ANF), an early indicator for initiation of cardiomyopathic remodeling, was significantly reduced in mdx mice treated with sildenafil[56, 57]. This implies that sildenafil is capable of inhibiting the advancement of cardiomyopathic remodeling at early stages of DMD[57]. Sildenafil has also been shown to have positive functional effects in the hearts of mdx mice, notably by avoidance of cardiomyocyte damage produced in vivo via cardiac workload augmentation and maintenance of an elevated heart rate response for a significantly longer period of time compared with placebo[57]. Interestingly, administration of sildenafil is even capable of reversing cardiac dysfunction in mdx mice with established cardiomyopathy[58]. Nevertheless, the target cell and mechanism behind the reversal are still unclear.
Additionally, PDE5 inhibitors have also shown improvement in muscle tissue from other vertebrate models of DMD, namely two dystrophin deficient zebrafish models known as sapje and sapje-like mutants[59, 60]. Dystrophin-null zebrafish treated with aminophylline, a nonselective phosphodiesterase inhibitor, were able to survive significantly longer compared to controls and had restored skeletal muscle structure similar to wild-type zebrafish[61]. Furthermore, analysis of sapje mutants treated 1 to 4 days postfertilization with different phosphodiesterase inhibitors revealed that treatment with aminophylline or sildenafil citrate resulted in the lowest percentage of fish showing abnormal muscle structure[61]. These data suggest that aminophylline and sildenafil citrate are capable of preventing the onset of aberrant muscle architecture in dystrophin-null zebrafish. These findings from DMD model zebrafish are analogous to the results seen from mdx mice treated with PDE5 inhibitors, and the consistency of these results across several DMD models leaves hope that these compounds could benefit individuals living with DMD. Clinical trials assessing PDE5 inhibitors for DMD patients are currently underway, and future use of tadalafil or sildenafil for these individuals seems promising based on preclinical studies and current clinical availability of these drugs for their use in treating erectile dysfunction and pulmonary hypertension.
Increased vascular density
Vascular endothelial growth factor administration
The central paradigm behind the previously discussed PDE5 and ACEI therapies for DMD was that increasing the vasorelaxation capacity of the vasculature would be able to increase perfusion, diminish the effects from functional ischemia, and decrease myocyte damage. However, another technique to increase tissue perfusion would be to increase the density of the underlying vascular architecture that nourishes the skeletal and cardiac muscles. One method of increasing vascular density is to augment angiogenesis, which regulates the production of new vasculatures from the existing framework. The vascular endothelial growth factor (VEGF) family of signal glycoproteins acts as potent promoters of angiogenesis during embryogenesis and postnatal growth. Specifically, the binding of the VEGF-A ligand with the VEGF receptors has been shown to promote vascular permeability and also trigger endothelial cell migration, proliferation, and survival, and the newly formed endothelial cells provide the basic structure of new vasculatures[62]. The dominant VEGF signal molecule for angiogenesis, VEGF-A, mediates its signal through VEGF receptor-1 (VEGFR-1, hereafter Flt-1) and VEGF receptor-2 (VEGFR-2, hereafter Flk-1)[63]. A soluble form of Flt-1 (sFlt-1) also exists, but lacks an intracellular signaling domain and thus only serves in a regulatory capacity by sequestering VEGF-A[63]. Flt-1 and Flk-1 contain an extracellular VEGF-A-binding domain and an intracellular tyrosine kinase domain, and both show expression during the developmental stage and tissue regeneration in hemangioblasts and endothelial cell lineages[63–65]. Flt-1 has a 10 times greater binding affinity for VEGF-A (Kd approximately 2 to 10 pM) compared to Flk-1, but the weaker tyrosine kinase domain indicates that angiogenic signal transduction following VEGF-A binding to Flt-1 is comparably weaker than the Flk-1 signal (Figure 2A)[63]. As such, homozygous Flt-1 gene knockout mice die in the embryonic stage from endothelial cell overproduction and blood vessel disorganization (Figure 2B)[64–66]. Inversely, homozygous Flk-1 gene knockout mice die from defects in the development of organized blood vessels due to lack of yolk-sac blood island formation during embryogenesis (Figure 2C)[67]. Both the Flt-1 and Flk-1 receptors are needed for normal development, but selective augmentation in VEGF-A concentration should allow for greater binding to the Flk-1 receptor and induce a pro-angiogenic effect that increases capillary density.

Flt-1 is a decoy receptor for vascular endothelial growth factor (VEGF) pro-angiogenic signaling. (A) In the wild-type scenario, VEGF induces a pro-angiogenic signal by binding the Flt-1 or Flk-1 receptors [63]. Flt-1 has a higher binding affinity for VEGF but transmits a weaker angiogenic signal compared to Flk-1, which implies that Flt-1 acts a negative regulator of angiogenesis [63]. The soluble form of Flt-1 (sFlt-1) lacks the transmembrane and intracellular signaling domains of Flt-1 and only serves a regulatory role by sequestering VEGF [63]. (B) Flt-1 homozygous knockout (Flt-1−/−) mice die in the early embryonic stage from endothelial cell overproduction and blood vessel disorganization, indicating that Flt-1 is a decoy regulator for endothelial growth/differentiation [64–66]. (C) Flk-1 homozygous knockout (Flk-1−/−) mice die in the early embryonic stage from defects in the development of organized blood vessels, indicating that Flk-1 is a positive regulator for endothelial growth/differentiation [67]. (D) Developmental reduction of the Flt-1 receptor through haploinsufficiency of the Flt-1 gene (Flt-1+/−) has been shown to increase capillary density in skeletal muscle, and this same phenomenon has been demonstrated in mdx mice (mdx:Flt-1+/−) [79]. The mdx:Flt-1+/− mice also showed improved histological and functional parameters normally associated with the Duchenne muscular dystrophy (DMD) pathology [68].
Several studies have demonstrated that administration of VEGF using exogenous expression mediated through engineered myoblasts, direct systemic injections, or adeno-associated viral (AAV) vectors are capable of initiating an angiogenic signal in the myocardium and skeletal muscle in both ischemic and non-ischemic conditions[69–72]. However, these same studies do highlight the importance of precisely regulating VEGF delivery quantities for future clinical use, as overadministration has been shown to have deleterious effects in animal models, such as hemangioma formation[70]. Unfortunately, and to the best of our knowledge, functional studies that assess blood flow in DMD model organisms following VEGF-induced angiogenesis have not yet been conducted. However, one study found that four weeks following intramuscular administration of rAAV-VEGF vectors in the bicep and tibialis anterior (TA) muscles of 4-week-old mdx mice, the rAAV-VEGF-treated mdx mice showed significantly greater forelimb strength compared to pretreatment levels and AAV-LacZ-treated control mdx mice[73]. The same study confirmed the feasibility of VEGF-mediated angiogenic induction in mdx mice and showed greater capillary density, particularly in the area of regenerating fibers, as well as reduced necrotic fiber area in biceps muscle compared to AAV-LacZ-treated control mdx mice[73]. So, although this study did not directly assess reduction of functional ischemia via enhanced vascular density through VEGF-induced angiogenesis, it was able to demonstrate similar outcomes from the PDE5 inhibitor studies, especially the decrease seen in necrotic fiber area and improvement in muscle function.
But apart from the documented pro-angiogenic effect, VEGF delivery also has a powerful pro-myogenic effect. In normal skeletal muscle tissues, VEGF administration induces muscle fiber regeneration and promotes muscle recovery after ischemic and chemical damage[74]. In in vitro studies using C2C12 myoblast cell line and primary mouse myoblasts derived from cultured SCs, VEGF was shown to promote growth and protect cells from apoptosis[74]. Similar effects have been documented in dystrophin deficient muscle tissues, where rAAV-VEGF-treated mdx mice showed an increase in the area occupied by regenerating fibers and an increased number of activated SCs and developmental myosin-heavy chain-positive fibers in skeletal muscles[73]. In vivo transplantation of muscle-derived stem cells (MDSCs) engineered to overexpress VEGF into dystrophic skeletal muscle results in an increase in angiogenesis and endogenous muscle regeneration along with reduction in fibrosis both two and four weeks following transplantation[75]. Thus, the dual functionalities of VEGF, especially the pro-angiogenic and pro-myogenic effects, are capable of improving both the histological and functional parameters normally associated with mdx muscle pathophysiology.
These data seem logical because developmentally reducing angiogenesis in mdx mice through ablation of matrix metalloproteinase-2 impairs the growth of regenerated myofibers and decreases VEGF expression, further complementing current theories about the close developmental relationship between angiogenesis and myogenesis[76]. But what is the pro-myogenic mechanism of VEGF delivery? SCs are clearly the dominant muscle-specific stem cells utilized for muscle growth, repair, and regeneration, and the number of SCs parallels muscle capillary quantity, largely because SCs reside in a juxtavascular niche[77, 78]. In fact, most SCs maintain tight locality to capillaries regardless of character, including quiescent SCs, proliferating SCs (myogenic precursor cells), and differentiating SCs (myocytes), and differentiating myogenin-positive myocytes assessed from DMD muscle biopsies show spatiotemporal association with new capillary growth[78]. A recent in vitro study specifically showed that endothelial cells augment myogenic precursor cell growth while differentiating SCs display pro-angiogenic characteristics, thus demonstrating a complementary angio-myogenesis signaling system[78]. Specifically, VEGF stimulated in vitro myogenic precursor cell growth, which supports the notion that VEGF is a co-regulatory substance for both angiogenesis and myogenesis[78, 79]. Recent work demonstrates that myofibers, SCs or myogenic precursor cells are negative for both Flt-1 and Flk-1, indicating that the effects of VEGF on these cells may be mediated through other VEGF receptors, such as neuropilin-1 (NRP1) and neuropilin-2 (NRP2)[68]. NRP1 and NRP2 can bind to VEGF with a high affinity and act as co-receptors for Flk-1 and Flt-3, respectively[80]. However, other studies have shown that in some developmental cases, the cellular proliferation functions of VEGF via binding to NRP1 are independent of Flk-1[81]. Thus, the exact mechanism of Flt-1/Flk-1 independent VEGF signaling remains to be elucidated.
An additional element of pro-angiogenic induction is that the expansion of the juxtavascular niche of SCs also serves to increase the basal number of SCs, and this is something not seen in the previously described vasorelaxation strategies[68]. Normally, the successive rounds of regeneration and degeneration seen in mdx tissue serves to exhaust the SC pool and reduce the regenerative capacity of the muscle tissue, which decreases SC quantity over time[82, 83]. This predetermined decline can be attenuated by improving the regenerative capacity of the muscle through an increase in the number of SCs present, which can be accomplished through expansion of the SC juxtavascular niche, including microcapillaries[78, 84, 85].
As for the future of VEGF therapies in DMD patients, more data is certainly needed before clinical benefit can be realized. Most important is deciphering effective dosing levels and delivery vehicles, which are both extraordinarily difficult owing to the fact that the body of pharmacology knowledge has shown us that inhibition is easier than introduction. Similarly, there is no agreed upon clinical standard for what constitutes a therapeutic or pathologic increase in the density of the vasculatures. Also, growth factors like VEGF need to be closely monitored due to carcinogenic properties. Similar to the PDE5 inhibitor studies, the effects of VEGF-induced angiogenesis to enhance capillary quantity and mitigate contraction-induced muscle damage via functional ischemia reduction should be further investigated to validate this hypothesis. Moreover, data regarding the functional aspects of the myocardium in dystrophin deficient tissues, the developmental effects of earlier VEGF administration, and the basal quantity of SCs following VEGF-induced angiogenesis in organisms lacking dystrophin all could help ready this therapy for clinical trials in DMD patients.
VEGF receptor regulation
A different pro-angiogenic approach that also increases vascular density to ameliorate the functional ischemia in DMD would be to modulate the VEGF receptors. As previously described, Flt-1 acts as a decoy receptor and modulates angiogenesis through its ability to sequester VEGF-A, which reduces signaling through Flk-1. So, although both Flt-1 and Flk-1 are inherently pro-angiogenic, due to the high affinity and low tyrosine kinase activity of Flt-1 over Flk-1 with respect to VEGF-A, Flt-1 acts as a VEGF-A sink preventing Flk-1 access to VEGF-A and thereby functioning as a negative regulator of angiogenesis. This phenomenon has been previously discussed in more detail, and it implies that reduced levels of Flt-1 and/or sFlt-1 could increase the serum concentration of free VEGF-A available to bind Flk-1 and induce a pro-angiogenic response[86].
Interestingly, Flt-1 reduction through haploinsufficiency of Flt-1 (Flt-1+/−) produced increased capillary density in skeletal and cardiac muscle compared with control (Flt-1+/+) in a murine model (Figure 2D)[68]. More significant to this discussion, developmentally reducing Flt-1 through the same genetic method was also shown to further enhance capillary density in the skeletal muscle of mdx mice (mdx:Flt-1+/−) compared to controls (mdx:Flt-1+/+)[68]. The increased capillary density seen in mdx:Flt-1+/− mice serve as a proof of concept that regulating VEGF-A receptors can stimulate a similar pro-angiogenic effect to that seen with direct VEGF-A administration in dystrophin-deficient organisms. More importantly, the changes seen in the mdx:Flt-1+/− mice persist into adulthood, yet it remains unclear if this effect must be initiated during developmental stages or if it can be recapitulated in postnatal models.
Analysis of the mdx:Flt-1+/− mice with increased capillary density demonstrated improved skeletal muscle histology with a reduction in both myocyte damage and fibers with centrally located nuclei, which strongly suggests that mdx:Flt-1+/− fibers display less fiber turnover compared to the mdx:Flt-1+/+ controls[68]. Additionally, mdx:Flt-1+/− fibers show less calcification, fibrosis, and membrane permeability, all of which are downstream effects of dystrophin deficiency[68]. These histological improvements also translated to improved functional parameters such that increased capillary density resulted in increased muscle tissue perfusion and improved skeletal muscle contractile function[68]. Furthermore, Flt-1 has also been assessed in another DMD mouse model: mdx:utrophin (utrn)−/− mice. These mice, deficient for both dystrophin and the dystrophin-related protein utrophin, display a more severe, progressive form of muscular dystrophy as compared with the mdx mice[87, 88]. Long term studies using the mdx:utrn−/−:Flt-1+/− mice with increased capillary density showed significant increases in body mass and survival compared to the mdx:utrn−/−:Flt-1+/+ control mice[68].
Absolute mechanisms that explain the improved phenotype and survival seen in mdx:Flt-1+/− mice and mdx:utrn−/−:Flt-1+/− mice remain to be elucidated. But owing to pathway similarity, the explanation is probably similar to descriptions of VEGF-induced angiogenesis in mdx mice, namely the close developmental relationship between myogenesis and angiogenesis. Increasing tissue perfusion may compensate for lack of NO-mediated vasodilation, which would attenuate one of the proposed ‘two-hits’ required for myocyte damage. Another theory behind the progressive nature of the DMD pathology is the SC exhaustion model[83]. This theory states that easily damaged DMD myofibers are constantly replaced by endogenous SCs, yet the constant SC cycling leads to rapid shortening of telomerase length and eventual exhaustion of the SC pool[82, 83]. Interestingly, mdx:Flt-1+/− were shown to have developmentally increased numbers of SCs, perhaps mediated through an expanded SC vascular niche. Thus, enhancement in the basal number of SCs could mitigate the accelerated age-related decline seen among SCs from dystrophin-deficient muscle tissue[13].
Overall, Flt-1 is a novel target for pro-angiogenic therapy in DMD. Greater blood perfusion alone seems to compensate for the functional ischemic phenotype in mdx mice, but definitive studies showing attenuation of functional ischemia through Flt-1 signal mitigation to reduce the effects of contraction induced damage have not been shown. Flt-1 has also been investigated for its role in cancer where it acts as a positive regulator of the pathological angiogenesis seen with tumor formation[89], which opposes the physiological role of Flt-1 as a negative angiogenic regulator. Thus, numerous small molecules have already been investigated and verified (in vivo and in vitro) that can antagonize Flt-1 binding to VEGF, reduce angiogenesis, and prevent tumor growth[90]. These same small molecules could be used to selectively block Flt-1 function and promote angiogenesis in dystrophin-deficient tissue. Still, more screening studies could be needed to decipher substances that are viable in vivo and can reduce Flt-1 function in order to fully translate the results seen from the developmental studies with mdx:Flt-1+/− and mdx:utrn−/−:Flt-1+/− mice using a pharmaceutical agent.
Conclusions
The role of the vasculature in DMD can no longer be ignored in light of the mounting evidence for its role in the pathogenic process. With this new knowledge in mind and with the dearth of current treatments, this review focused on a variety of new therapeutic options that specifically target these DMD vascular defects, namely attenuation of the functional ischemia (see Table 1 for summary of therapies). One therapy improves systemic vasorelaxation capacity using ACEIs with or without BBs, and this method has shown clinical utility in both preventing and improving the adverse cardiac events normally associated with the DMD phenotype. Treatment using PDE5 inhibitors also improves systemic vasorelaxation capacity, and preclinical evidence from DMD murine models demonstrates the ability of PDE5 inhibitors to prevent skeletal and cardiac muscle damage and even reverse the functional parameters associated with established cardiomyopathy. Both PDE5 and ACEI therapies have a clear practical advantage as they have extensive clinical safety records and many of the drugs are clinically available. There are a wide variety of ACEIs that are FDA approved to treat heart failure and hypertension, including benazepril (Lotensin™), captopril (Capoten™), enalapril (Vasotec™), fosinopril (Monopril™), lisinopril (Prinivil™, Zestril™) moexipril (Univasc™), perindopril (Aceon™), quinapril (Accupril™), ramipril (Altace™), and trandolapril (Mavik™). There are also several PDE5 inhibitors that received FDA approval for treating erectile dysfunction or hypertension, including tadalafil (Cialis™ or Adcirca™), sildenafil (Viagra™ or Revatio™), and vardenafil (Levitra™ or STAXYN™). Additionally, sildenafil has already been extensively studied in a pediatric population and was found to be safe for pulmonary hypertension treatment[91]. Both tadalafil and sildenafil are currently in a phase 1 clinical trial (NCT01580501) that will assess these drugs ability to attenuate functional ischemia in boys with DMD, and other future clinical studies could address the ability of ACEIs to mitigate the same effect.
Therapies that enhance the underlying vascular architecture through pro-angiogenic induction include VEGF administration and also VEGF receptor modulation. The pro-angiogenic therapies have shown exciting preclinical proof of concept evidence in DMD murine models, especially the expansion in the basal number of SCs mediated through a larger juxtavascular SC niche and the documented pro-myogenic effects. Still, the pro-angiogenic strategies are in early stages and both methods need definitive means of achieving their desired result and more safety information before clinical trial initiation. In all, the hope is that at least some or combinations of these vascular-targeted therapies will soon have clinical utility and provide current and future human beings living with DMD enhanced control over their own destiny.
Authors’ information
JPE is a staff researcher in the laboratory of Dr. Atsushi Asakura at the University of Minnesota Stem Cell Institute.
MV is an MD/PhD candidate through the Medical Scientist Training Program at the University of Minnesota Medical School.
AA is an Assistant Professor of Neurology and a faculty member of the Stem Cell Institute in the University of Minnesota Medical School. He also belongs to the Paul & Sheila Wellstone Muscular Dystrophy Center in the University of Minnesota Medical School.
Abbreviations
- AAV:
-
Adeno-associated virus
- ACE:
-
Angiotensin-converting enzyme
- ACEI:
-
Angiotensin-converting enzyme inhibitor
- ANF:
-
Atrial natriuretic factor
- ARB:
-
Angiotensin receptor blocker
- BB:
-
β-blocker
- BMD:
-
Becker muscular dystrophy
- cGMP:
-
Cyclic guanosine monophosphate
- CNS:
-
Central nervous system
- DAPC:
-
Dystrophin-associated glycoprotein complex
- DMD:
-
Duchenne muscular dystrophy
- EDHF:
-
Endothelium-derived hyperpolarizing factor
- LVEF:
-
Left ventricular ejection fraction
- MDSCs:
-
Muscle-derived stem cells
- nNOS:
-
Neuronal nitric oxide synthase
- NO:
-
Nitric oxide
- NOS:
-
Nitric oxide synthase
- NRP1:
-
Neuropilin-1
- NRP2:
-
Neuropilin-2
- PDE5:
-
Phosphodiesterase 5
- sGC:
-
Soluble guanylyl cyclase
- SC:
-
Satellite cell
- rAAV:
-
Recombinant adeno-associated virus
- TA:
-
tibialis anterior
- TGF- β:
-
Transforming growth factor-β
- VEGF:
-
Vascular endothelial growth factor
- VEGFR:
-
Vascular endothelial growth factor receptor.
References
Monaco AP, Neve RL, Colletti-Feener C, Bertelson CJ, Kurnit DM, Kunkel LM: Isolation of candidate cDNAs for portions of the Duchenne muscular dystrophy gene. Nature. 1986, 323: 646-650. 10.1038/323646a0.
Hoffman EP, Kunkel LM: Dystrophin abnormalities in Duchenne/Becker muscular dystrophy. Neuron. 1989, 2: 1019-1029. 10.1016/0896-6273(89)90226-2.
Bushby K, Finkel R, Birnkrant DJ, Case LE, Clemens PR, Cripe L, Kaul A, Kinnett K, McDonald C, Pandya S, Poysky J, Shapiro F, Tomezsko J, Constantin C, DMD Care Considerations Working Group: Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010, 9: 77-93. 10.1016/S1474-4422(09)70271-6.
Ervasti JM, Campbell KP: A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J Cell Biol. 1993, 122: 809-823. 10.1083/jcb.122.4.809.
Davies KE, Nowak KJ: Molecular mechanisms of muscular dystrophies: old and new players. Nat Rev Mol Cell Biol. 2006, 7: 762-773. 10.1038/nrm2024.
Brenman JE, Chao DS, Xia H, Aldape K, Bredt DS: Nitric oxide synthase complexed with dystrophin and absent from skeletal muscle sarcolemma in Duchenne muscular dystrophy. Cell. 1995, 82: 743-752. 10.1016/0092-8674(95)90471-9.
Goldspink G: Changes in muscle mass and phenotype and the expression of autocrine and systemic growth factors by muscle in response to stretch and overload. J Anat. 1999, 194 (Pt 3): 323-334.
Rybakova IN, Patel JR, Ervasti JM: The dystrophin complex forms a mechanically strong link between the sarcolemma and costameric actin. J Cell Biol. 2000, 150: 1209-1214. 10.1083/jcb.150.5.1209.
Duncan CJ: Role of intracellular calcium in promoting muscle damage: a strategy for controlling the dystrophic condition. Experientia. 1978, 34: 1531-1535. 10.1007/BF02034655.
Spencer MJ, Croall DE, Tidball JG: Calpains are activated in necrotic fibers from mdx dystrophic mice. J Biol Chem. 1995, 270: 10909-10914. 10.1074/jbc.270.18.10909.
Hopf FW, Turner PR, Steinhardt RA: Calcium misregulation and the pathogenesis of muscular dystrophy. Subcell Biochem. 2007, 45: 429-464. 10.1007/978-1-4020-6191-2_16.
Spencer MJ, Montecino-Rodriguez E, Dorshkind K, Tidball JG: Helper (CD4(+)) and cytotoxic (CD8(+)) T cells promote the pathology of dystrophin-deficient muscle. Clin Immunol. 2001, 98: 235-243. 10.1006/clim.2000.4966.
Jejurikar SS, Kuzon WM: Satellite cell depletion in degenerative skeletal muscle. Apoptosis. 2003, 8: 573-578.
Townsend D, Yasuda S, Metzger J: Cardiomyopathy of Duchenne muscular dystrophy: pathogenesis and prospect of membrane sealants as a new therapeutic approach. Expert Rev Cardiovasc Ther. 2007, 5: 99-109. 10.1586/14779072.5.1.99.
Eagle M, Baudouin SV, Chandler C, Giddings DR, Bullock R, Bushby K: Survival in Duchenne muscular dystrophy: improvements in life expectancy since 1967 and the impact of home nocturnal ventilation. Neuromuscul Disord. 2002, 12: 926-929. 10.1016/S0960-8966(02)00140-2.
Eagle M, Bourke J, Bullock R, Gibson M, Mehta J, Giddings D, Straub V, Bushby K: Managing Duchenne muscular dystrophy–the additive effect of spinal surgery and home nocturnal ventilation in improving survival. Neuromuscul Disord. 2007, 17: 470-475. 10.1016/j.nmd.2007.03.002.
Rando TA: Role of nitric oxide in the pathogenesis of muscular dystrophies: a "two hit" hypothesis of the cause of muscle necrosis. Microsc Res Tech. 2001, 55: 223-235. 10.1002/jemt.1172.
Sander M, Chavoshan B, Harris SA, Iannaccone ST, Stull JT, Thomas GD, Victor RG: Functional muscle ischemia in neuronal nitric oxide synthase-deficient skeletal muscle of children with Duchenne muscular dystrophy. Proc Natl Acad Sci USA. 2000, 97: 13818-13823. 10.1073/pnas.250379497.
Loufrani L, Matrougui K, Gorny D, Duriez M, Blanc I, Levy BI, Henrion D: Flow (shear stress)-induced endothelium-dependent dilation is altered in mice lacking the gene encoding for dystrophin. Circulation. 2001, 103: 864-870. 10.1161/01.CIR.103.6.864.
Davies PF: Flow-mediated endothelial mechanotransduction. Physiol Rev. 1995, 75: 519-560.
Friebel M, Klotz KF, Ley K, Gaehtgens P, Pries AR: Flow-dependent regulation of arteriolar diameter in rat skeletal muscle in situ: role of endothelium-derived relaxing factor and prostanoids. J Physiol. 1995, 483 (Pt 3): 715-726.
Ichioka S, Shibata M, Kosaki K, Sato Y, Harii K, Kamiya A: Effects of shear stress on wound-healing angiogenesis in the rabbit ear chamber. J Surg Res. 1997, 72: 29-35. 10.1006/jsre.1997.5170.
Ando J, Kamiya A: Blood flow and vascular endothelial cell function. Front Med Biol Eng. 1993, 5: 245-264.
Asai A, Sahani N, Kaneki M, Ouchi Y, Martyn JA, Yasuhara SE: Primary role of functional ischemia, quantitative evidence for the two-hit mechanism, and phosphodiesterase-5 inhibitor therapy in mouse muscular dystrophy. PLoS One. 2007, 2: e806-10.1371/journal.pone.0000806.
Huang PL, Dawson TM, Bredt DS, Snyder SH, Fishman MC: Targeted disruption of the neuronal nitric oxide synthase gene. Cell. 1993, 75: 1273-1286. 10.1016/0092-8674(93)90615-W.
Wehling-Henricks M, Jordan MC, Roos KP, Deng B, Tidball JG: Cardiomyopathy in dystrophin-deficient hearts is prevented by expression of a neuronal nitric oxide synthase transgene in the myocardium. Hum Mol Genet. 2005, 14: 1921-1933. 10.1093/hmg/ddi197.
Ito K, Kimura S, Ozasa S, Matsukura M, Ikezawa M, Yoshioka K, Ueno H, Suzuki M, Araki K, Yamamura K, Miwa T, Dickson G, Thomas GD, Miike T: Smooth muscle-specific dystrophin expression improves aberrant vasoregulation in mdx mice. Hum Mol Genet. 2006, 15: 2266-2275. 10.1093/hmg/ddl151.
Garg R, Yusuf S: Overview of randomized trials of angiotensin-converting enzyme inhibitors on mortality and morbidity in patients with heart failure. Collaborative Group on ACE Inhibitor Trials. JAMA. 1995, 273: 1450-1456.
Mant J, Al-Mohammad A, Swain S, Laramee P, Guideline Development G: Management of chronic heart failure in adults: synopsis of the National Institute For Health and clinical excellence guideline. Ann Intern Med. 2011, 155: 252-259. 10.7326/0003-4819-155-4-201108160-00009.
Krause T, Lovibond K, Caulfield M, McCormack T, Williams B, Guideline Development G: Management of hypertension: summary of NICE guidance. BMJ. 2011, 343: d4891-10.1136/bmj.d4891.
Bauer R, Straub V, Blain A, Bushby K, MacGowan GA: Contrasting effects of steroids and angiotensin-converting-enzyme inhibitors in a mouse model of dystrophin-deficient cardiomyopathy. Eur J Heart Fail. 2009, 11: 463-471. 10.1093/eurjhf/hfp028.
Duboc D, Meune C, Lerebours G, Devaux JY, Vaksmann G, Becane HM: Effect of perindopril on the onset and progression of left ventricular dysfunction in Duchenne muscular dystrophy. J Am Coll Cardiol. 2005, 45: 855-857. 10.1016/j.jacc.2004.09.078.
Duboc D, Meune C, Pierre B, Wahbi K, Eymard B, Toutain A, Berard C, Vaksmann G, Weber S, Becane HM: Perindopril preventive treatment on mortality in Duchenne muscular dystrophy: 10 years' follow-up. Am Heart J. 2007, 154: 596-602. 10.1016/j.ahj.2007.05.014.
Ramaciotti C, Heistein LC, Coursey M, Lemler MS, Eapen RS, Iannaccone ST, Scott WA: Left ventricular function and response to enalapril in patients with duchenne muscular dystrophy during the second decade of life. Am J Cardiol. 2006, 98: 825-827. 10.1016/j.amjcard.2006.04.020.
Ogata H, Ishikawa Y, Minami R: Beneficial effects of beta-blockers and angiotensin-converting enzyme inhibitors in Duchenne muscular dystrophy. J Cardiol. 2009, 53: 72-78. 10.1016/j.jjcc.2008.08.013.
Viollet L, Thrush PT, Flanigan KM, Mendell JR, Allen HD: Effects of Angiotensin-Converting Enzyme Inhibitors and/or Beta Blockers on the Cardiomyopathy in Duchenne Muscular Dystrophy. Am J Cardiol. 2012, 110: 98-102. 10.1016/j.amjcard.2012.02.064.
Bish LT, Yarchoan M, Sleeper MM, Gazzara JA, Morine KJ, Acosta P, Barton ER, Sweeney HL: Chronic losartan administration reduces mortality and preserves cardiac but not skeletal muscle function in dystrophic mice. PLoS One. 2011, 6: e20856-10.1371/journal.pone.0020856.
Spurney CF, Sali A, Guerron AD, Iantorno M, Yu Q, Gordish-Dressman H, Rayavarapu S, van der Meulen J, Hoffman EP, Nagaraju K: Losartan decreases cardiac muscle fibrosis and improves cardiac function in dystrophin-deficient mdx mice. J Cardiovasc Pharmacol Ther. 2011, 16: 87-95. 10.1177/1074248410381757.
Delcayre C, Swynghedauw B: Molecular mechanisms of myocardial remodeling. The role of aldosterone. J Mol Cell Cardiol. 2002, 34: 1577-1584. 10.1006/jmcc.2002.2088.
Lijnen P, Petrov V: Induction of cardiac fibrosis by aldosterone. J Mol Cell Cardiol. 2000, 32: 865-879. 10.1006/jmcc.2000.1129.
Pitt B, Zannad F, Remme WJ, Cody R, Castaigne A, Perez A, Palensky J, Wittes J: The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med. 1999, 341: 709-717. 10.1056/NEJM199909023411001.
Pitt B, Remme W, Zannad F, Neaton J, Martinez F, Roniker B, Bittman R, Hurley S, Kleiman J, Gatlin M, Eplerenone post-acute myocardial infarction heart failure efficacy and survival study I: Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med. 2003, 348: 1309-1321. 10.1056/NEJMoa030207.
Wynn TA: Cellular and molecular mechanisms of fibrosis. J Pathol. 2008, 214: 199-210. 10.1002/path.2277.
Martin MM, Buckenberger JA, Jiang J, Malana GE, Knoell DL, Feldman DS, Elton TS: TGF-beta1 stimulates human AT1 receptor expression in lung fibroblasts by cross talk between the Smad, p38 MAPK, JNK, and PI3K signaling pathways. Am J Physiol Lung Cell Mol Physiol. 2007, 293: L790-L799. 10.1152/ajplung.00099.2007.
Gazzerro E, Assereto S, Bonetto A, Sotgia F, Scarfi S, Pistorio A, Bonuccelli G, Cilli M, Bruno C, Zara F, Lisanti MP, Minetti C: Therapeutic potential of proteasome inhibition in Duchenne and Becker muscular dystrophies. Am J Pathol. 2010, 176: 1863-1877. 10.2353/ajpath.2010.090468.
Russell ST, Wyke SM, Tisdale MJ: Mechanism of induction of muscle protein degradation by angiotensin II. Cell Signal. 2006, 18: 1087-1096. 10.1016/j.cellsig.2005.09.009.
Rando TA, Disatnik MH, Yu Y, Franco A: Muscle cells from mdx mice have an increased susceptibility to oxidative stress. Neuromuscul Disord. 1998, 8: 14-21. 10.1016/S0960-8966(97)00124-7.
Wei Y, Sowers JR, Clark SE, Li W, Ferrario CM, Stump CS: Angiotensin II-induced skeletal muscle insulin resistance mediated by NF-kappaB activation via NADPH oxidase. Am J Physiol Endocrinol Metab. 2008, 294: E345-E351.
Burdi R, Rolland JF, Fraysse B, Litvinova K, Cozzoli A, Giannuzzi V, Liantonio A, Camerino GM, Sblendorio V, Capogrosso RF, Palmieri B, Andreetta F, Confalonieri P, De Benedictis L, Montagnani M, De Luca A: Multiple pathological events in exercised dystrophic mdx mice are targeted by pentoxifylline: outcome of a large array of in vivo and ex vivo tests. J Appl Physiol. 2009, 106: 1311-1324. 10.1152/japplphysiol.90985.2008.
Shkryl VM, Martins AS, Ullrich ND, Nowycky MC, Niggli E, Shirokova N: Reciprocal amplification of ROS and Ca(2+) signals in stressed mdx dystrophic skeletal muscle fibers. Pflugers Arch. 2009, 458: 915-928. 10.1007/s00424-009-0670-2.
Spurney CF, Knoblach S, Pistilli EE, Nagaraju K, Martin GR, Hoffman EP: Dystrophin-deficient cardiomyopathy in mouse: expression of Nox4 and Lox are associated with fibrosis and altered functional parameters in the heart. Neuromuscul Disord. 2008, 18: 371-381. 10.1016/j.nmd.2008.03.008.
Williams IA, Allen DG: The role of reactive oxygen species in the hearts of dystrophin-deficient mdx mice. Am J Physiol Heart Circ Physiol. 2007, 293: H1969-H1977. 10.1152/ajpheart.00489.2007.
Cozzoli A, Nico B, Sblendorio VT, Capogrosso RF, Dinardo MM, Longo V, Gagliardi S, Montagnani M, De Luca A: Enalapril treatment discloses an early role of angiotensin II in inflammation- and oxidative stress-related muscle damage in dystrophic mdx mice. Pharmacol Res. 2011, 64: 482-492. 10.1016/j.phrs.2011.06.002.
Bloom TJ: Cyclic nucleotide phosphodiesterase isozymes expressed in mouse skeletal muscle. Can J Physiol Pharmacol. 2002, 80: 1132-1135. 10.1139/y02-149.
Lau KS, Grange RW, Chang WJ, Kamm KE, Sarelius I, Stull JT: Skeletal muscle contractions stimulate cGMP formation and attenuate vascular smooth muscle myosin phosphorylation via nitric oxide. FEBS Lett. 1998, 431: 71-74. 10.1016/S0014-5793(98)00728-5.
Khairallah M, Khairallah R, Young ME, Dyck JR, Petrof BJ, Des Rosiers C: Metabolic and signaling alterations in dystrophin-deficient hearts precede overt cardiomyopathy. J Mol Cell Cardiol. 2007, 43: 119-129. 10.1016/j.yjmcc.2007.05.015.
Khairallah M, Khairallah RJ, Young ME, Allen BG, Gillis MA, Danialou G, Deschepper CF, Petrof BJ, Des Rosiers C: Sildenafil and cardiomyocyte-specific cGMP signaling prevent cardiomyopathic changes associated with dystrophin deficiency. Proc Natl Acad Sci USA. 2008, 105: 7028-7033. 10.1073/pnas.0710595105.
Adamo CM, Dai DF, Percival JM, Minami E, Willis MS, Patrucco E, Froehner SC, Beavo JA: Sildenafil reverses cardiac dysfunction in the mdx mouse model of Duchenne muscular dystrophy. Proc Natl Acad Sci USA. 2010, 107: 19079-19083. 10.1073/pnas.1013077107.
Bassett DI, Currie PD: The zebrafish as a model for muscular dystrophy and congenital myopathy. Hum Mol Genet. 2003, 12: R265-R270. 10.1093/hmg/ddg279.
Guyon JR, Goswami J, Jun SJ, Thorne M, Howell M, Pusack T, Kawahara G, Steffen LS, Galdzicki M, Kunkel LM: Genetic isolation and characterization of a splicing mutant of zebrafish dystrophin. Hum Mol Genet. 2009, 18: 202-211. 10.1093/hmg/ddp366.
Kawahara G, Karpf JA, Myers JA, Alexander MS, Guyon JR, Kunkel LM: Drug screening in a zebrafish model of Duchenne muscular dystrophy. Proc Natl Acad Sci USA. 2011, 108: 5331-5336. 10.1073/pnas.1102116108.
Ferrara N: Role of vascular endothelial growth factor in physiologic and pathologic angiogenesis: therapeutic implications. Semin Oncol. 2002, 29: 10-14.
Matsumoto T, Claesson-Welsh L: VEGF receptor signal transduction. Sci STKE. 2001, 2001: re21-
Fong GH, Rossant J, Gertsenstein M, Breitman ML: Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature. 1995, 376: 66-70. 10.1038/376066a0.
Fong GH, Zhang L, Bryce DM, Peng J: Increased hemangioblast commitment, not vascular disorganization, is the primary defect in flt-1 knock-out mice. Development. 1999, 126: 3015-3025.
Ferrara N, Gerber HP, LeCouter J: The biology of VEGF and its receptors. Nat Med. 2003, 9: 669-676. 10.1038/nm0603-669.
Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu XF, Breitman ML, Schuh AC: Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature. 1995, 376: 62-66. 10.1038/376062a0.
Verma M, Asakura Y, Hirai H, Watanabe S, Tastad C, Fong GH, Ema M, Call JA, Lowe DA, Asakura A: Flt-1 haploinsufficiency ameliorates muscular dystrophy phenotype by developmentally increased vasculature in mdx mice. Hum Mol Genet. 2010, 19: 4145-4159. 10.1093/hmg/ddq334.
Bauters C, Asahara T, Zheng LP, Takeshita S, Bunting S, Ferrara N, Symes JF, Isner JM: Site-specific therapeutic angiogenesis after systemic administration of vascular endothelial growth factor. J Vasc Surg. 1995, 21: 314-324. 10.1016/S0741-5214(95)70272-5. discussion 324–315
Springer ML, Chen AS, Kraft PE, Bednarski M, Blau HM: VEGF gene delivery to muscle: potential role for vasculogenesis in adults. Mol Cell. 1998, 2: 549-558. 10.1016/S1097-2765(00)80154-9.
Lee RJ, Springer ML, Blanco-Bose WE, Shaw R, Ursell PC, Blau HM: VEGF gene delivery to myocardium: deleterious effects of unregulated expression. Circulation. 2000, 102: 898-901. 10.1161/01.CIR.102.8.898.
Su H, Lu R, Kan YW: Adeno-associated viral vector-mediated vascular endothelial growth factor gene transfer induces neovascular formation in ischemic heart. Proc Natl Acad Sci USA. 2000, 97: 13801-13806. 10.1073/pnas.250488097.
Messina S, Mazzeo A, Bitto A, Aguennouz M, Migliorato A, De Pasquale MG, Minutoli L, Altavilla D, Zentilin L, Giacca M, Squadrito F, Vita G: VEGF overexpression via adeno-associated virus gene transfer promotes skeletal muscle regeneration and enhances muscle function in mdx mice. FASEB J. 2007, 21: 3737-3746. 10.1096/fj.07-8459com.
Arsic N, Zacchigna S, Zentilin L, Ramirez-Correa G, Pattarini L, Salvi A, Sinagra G, Giacca M: Vascular endothelial growth factor stimulates skeletal muscle regeneration in vivo. Mol Ther. 2004, 10: 844-854. 10.1016/j.ymthe.2004.08.007.
Deasy BM, Feduska JM, Payne TR, Li Y, Ambrosio F, Huard J: Effect of VEGF on the regenerative capacity of muscle stem cells in dystrophic skeletal muscle. Mol Ther. 2009, 17: 1788-1798. 10.1038/mt.2009.136.
Miyazaki D, Nakamura A, Fukushima K, Yoshida K, Takeda S, Ikeda S: Matrix metalloproteinase-2 ablation in dystrophin-deficient mdx muscles reduces angiogenesis resulting in impaired growth of regenerated muscle fibers. Hum Mol Genet. 2011, 20: 1787-1799. 10.1093/hmg/ddr062.
Dhawan J, Rando TA: Stem cells in postnatal myogenesis: molecular mechanisms of satellite cell quiescence, activation and replenishment. Trends Cell Biol. 2005, 15: 666-673. 10.1016/j.tcb.2005.10.007.
Christov C, Chretien F, Abou-Khalil R, Bassez G, Vallet G, Authier FJ, Bassaglia Y, Shinin V, Tajbakhsh S, Chazaud B, Gherardi RK: Muscle satellite cells and endothelial cells: close neighbors and privileged partners. Mol Biol Cell. 2007, 18: 1397-1409. 10.1091/mbc.E06-08-0693.
Williams RS, Annex BH: Plasticity of myocytes and capillaries: a possible coordinating role for VEGF. Circ Res. 2004, 95: 7-8. 10.1161/01.RES.0000136345.81719.37.
Koch S: Neuropilin signalling in angiogenesis. Biochem Soc Trans. 2012, 40: 20-25. 10.1042/BST20110689.
Zachary IC: How neuropilin-1 regulates receptor tyrosine kinase signalling: the knowns and known unknowns. Biochem Soc Trans. 2011, 39: 1583-1591. 10.1042/BST20110697.
Brack AS, Bildsoe H, Hughes SM: Evidence that satellite cell decrement contributes to preferential decline in nuclear number from large fibres during murine age-related muscle atrophy. J Cell Sci. 2005, 118: 4813-4821. 10.1242/jcs.02602.
Sacco A, Mourkioti F, Tran R, Choi J, Llewellyn M, Kraft P, Shkreli M, Delp S, Pomerantz JH, Artandi SE, Blau HM: Short telomeres and stem cell exhaustion model Duchenne muscular dystrophy in mdx/mTR mice. Cell. 2010, 143: 1059-1071. 10.1016/j.cell.2010.11.039.
Stark DA, Karvas RM, Siegel AL, Cornelison DD: Eph/ephrin interactions modulate muscle satellite cell motility and patterning. Development. 2011, 138: 5279-5289. 10.1242/dev.068411.
Mounier R, Chretien F, Chazaud B: Blood vessels and the satellite cell niche. Curr Top Dev Biol. 2011, 96: 121-138.
Shibuya M: Vascular endothelial growth factor receptor-1 (VEGFR-1/Flt-1): a dual regulator for angiogenesis. Angiogenesis. 2006, 9: 225-230. 10.1007/s10456-006-9055-8.
Deconinck AE, Rafael JA, Skinner JA, Brown SC, Potter AC, Metzinger L, Watt DJ, Dickson JG, Tinsley JM, Davies KE: Utrophin-dystrophin-deficient mice as a model for Duchenne muscular dystrophy. Cell. 1997, 90: 717-727. 10.1016/S0092-8674(00)80532-2.
Grady RM, Teng H, Nichol MC, Cunningham JC, Wilkinson RS, Sanes JR: Skeletal and cardiac myopathies in mice lacking utrophin and dystrophin: a model for Duchenne muscular dystrophy. Cell. 1997, 90: 729-738. 10.1016/S0092-8674(00)80533-4.
Hiratsuka S, Maru Y, Okada A, Seiki M, Noda T, Shibuya M, Hiratsuka S, Maru Y, Okada A, Seiki M, Noda T, Shibuya M: Involvement of Flt-1 Tyrosine Kinase (Vascular Endothelial Growth Factor Receptor-1) in Pathological Angiogenesis. Cancer Res. 2001, 61: 1207-1213.
Bae DG, Kim TD, Li G, Yoon WH, Chae CB: Anti-flt1 peptide, a vascular endothelial growth factor receptor 1-specific hexapeptide, inhibits tumor growth and metastasis. Clin Cancer Res. 2005, 11: 2651-2661. 10.1158/1078-0432.CCR-04-1564.
Huddleston AJ, Knoderer CA, Morris JL, Ebenroth ES: Sildenafil for the treatment of pulmonary hypertension in pediatric patients. Pediatr Cardiol. 2009, 30: 871-882. 10.1007/s00246-009-9523-1.
Acknowledgements
We thank Dr. Lawrence Charnas and Dr. Dennis Keefe for critical reading of this manuscript. This work was supported by grants to AA from Grant-in-Aid of the University of Minnesota and the Muscular Dystrophy Association (MDA). The work was also supported by the NIH-T32-GM008244 grant to MV.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors have no financial competing interests.
Authors’ contributions
JPE completed the literature review, developed the figures, and prepared the review. MV revised the manuscript. AA advised the literature review process and revised the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Ennen, J.P., Verma, M. & Asakura, A. Vascular-targeted therapies for Duchenne muscular dystrophy. Skeletal Muscle 3, 9 (2013). https://doi.org/10.1186/2044-5040-3-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2044-5040-3-9