
Abstract
Background
Peripheral arterial disease (PAD) is a relatively common manifestation of systemic atherosclerosis that leads to progressive narrowing of the lumen of leg arteries. Circulating monocytes are in contact with the arterial wall and can serve as reporters of vascular pathology in the setting of PAD. We performed gene expression analysis of peripheral blood mononuclear cells (PBMC) in patients with PAD and controls without PAD to identify differentially regulated genes.
Methods
PAD was defined as an ankle brachial index (ABI) ≤0.9 (n = 19) while age and gender matched controls had an ABI > 1.0 (n = 18). Microarray analysis was performed using Affymetrix HG-U133 plus 2.0 gene chips and analyzed using GeneSpring GX 11.0. Gene expression data was normalized using Robust Multichip Analysis (RMA) normalization method, differential expression was defined as a fold change ≥1.5, followed by unpaired Mann-Whitney test (P < 0.05) and correction for multiple testing by Benjamini and Hochberg False Discovery Rate. Meta-analysis of differentially expressed genes was performed using an integrated bioinformatics pipeline with tools for enrichment analysis using Gene Ontology (GO) terms, pathway analysis using Kyoto Encyclopedia of Genes and Genomes (KEGG), molecular event enrichment using Reactome annotations and network analysis using Ingenuity Pathway Analysis suite. Extensive biocuration was also performed to understand the functional context of genes.
Results
We identified 87 genes differentially expressed in the setting of PAD; 40 genes were upregulated and 47 genes were downregulated. We employed an integrated bioinformatics pipeline coupled with literature curation to characterize the functional coherence of differentially regulated genes.
Conclusion
Notably, upregulated genes mediate immune response, inflammation, apoptosis, stress response, phosphorylation, hemostasis, platelet activation and platelet aggregation. Downregulated genes included several genes from the zinc finger family that are involved in transcriptional regulation. These results provide insights into molecular mechanisms relevant to the pathophysiology of PAD.
Similar content being viewed by others


Introduction
Peripheral arterial disease (PAD) affects more than eight million adults in the United States and is associated with significant mortality and morbidity [1–6]. PAD is a surrogate for diffuse atherosclerosis but is often underdiagnosed [4, 6]. Identification of differentially regulated genes in the setting of PAD may lead to potential biomarkers for the earlier detection and prognostication of this disease and provide insights into its pathophysiology.
Gene expression analysis of peripheral blood mononuclear cells (PBMC) in asymptomatic individuals has previously revealed individual genetic variation and differentially regulated expression patterns [7, 8]. Circulating peripheral blood cells have been used to examine differentially regulated genes in several cardiovascular disorders. For example, gene expression profiling studies of blood cells have identified differentially regulated genes and pathways in hypertension [9], coronary artery disease [10, 11] and ischemic stroke [1, 10, 12–16]. However, genes differentially expressed in PBMC in the setting of PAD have yet to be identified. Circulating PBMC are in contact with the arterial wall and may be useful in investigating molecular mechanisms relevant to PAD. We therefore performed gene expression analysis to identify differentially expressed genes in PBMC in the setting of PAD.
Materials and methods
Participant recruitment and sample characteristics
The Mayo Clinic Institutional Review Board approved the study and all participants provided written informed consent. The participants were recruited from the Mayo non-invasive vascular laboratory and PAD was defined as an ankle brachial index (ABI) ≤0.9 (n = 19) while age and gender matched controls had an ABI > 1.0 (n = 18). ABI was measured in both the lower extremities and the lower of the two values was recorded for the analysis [17]. Individuals with poorly compressible arteries or aortic aneurysmal disease were excluded.
Isolation of peripheral blood mononuclear cells (PBMC) and RNA isolation
PBMC were isolated by density gradient centrifugation by layering the blood samples over histopaque (Sigma-Aldrich, St. Louis, MO),[18]. In brief, 18 ml of whole blood was mixed with equal amount of PBS (Bio-Rad, Hercules, CA), and layered over 12 ml of histopaque 1077 (used for cell separation). The PBMC layer was removed, washed, and centrifuged twice with Hank's Balanced Salt Solution (HBSS) (Sigma-Aldrich, St. Louis, MO). The pellet formed after double centrifugation was re-suspended in Complete RPMI-10 medium. The cells were counted using a hemocytometer and processed for RNA isolation using RNeasy Plus Mini Kit (Qiagen, Valencia, CA), and additionally with TRIzol (Invitrogen, Carlsbad, CA). For the RNeasy kit, PBMCs were disrupted and homogenized using RLT buffer (Qiagen, Valencia, CA). The RNeasy kit includes gDNA eliminator spin column for the removal of genomic DNA from the sample, allowing subsequent purification of RNA. The flow through from the gDNA column was mixed with ethanol and placed on the RNeasy spin column. This spin column uses a silica gel based membrane for effective binding and purification of RNA. Total RNA was eluted in RNase free water, quantified using NanoDrop 1000 (Thermo Scientific, Wilmington, DE) and stored at −80°C.
Microarray analysis
RNA quantity and quality were assessed using Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA); 100 ng of total RNA was used for generation of biotin labeled cRNA using Affymetrix Two-Cycle cDNA Synthesis Kit (Affymetrix, Santa Clara, CA). After the first cycle, in vitro transcription-based (IVT) amplification of cRNA was carried out using MEGAscript T7 kit (Applied Biosystems/Ambion, Austin TX). With the second cycle cDNA synthesis, biotin labeled cRNA was generated using the Affymetrix IVT labeling Kit. The labeled cRNA was cleaned, quantified and after fragmentation, hybridized to Affymetrix HG-U133 Plus2.0 GeneChips. The chips were stained with streptavidin phycoerythrin and biotinylated antibody and washed at an Affymetrix Fluidics station 450. The GeneChips were scanned and data extracted using GeneChip scanner 3000 (Affymetrix, Santa Clara, CA) and the raw data file formats were generated using GeneChip operating software (GCOS).
Data processing and statistical analysis
Raw gene expression data were analyzed using the GeneSpringGx 11.0 software (Agilent® Technologies, Santa Clara, CA). All samples were normalized and summarized by Robust Multichip Analysis (RMA) normalization method, which includes background correction, normalization and calculation of expression values [19]. Baseline was set to median for all samples, where median of the log-transformed value of each probe from all samples was calculated and this value was subtracted from all samples. Probes were filtered and eliminated on expression level as part of quality control (QC) and probes with expression values <20% were excluded. Of the remaining probes, those with a 1.5-fold-change difference between the groups underwent unpaired Mann-Whitney test and multiple testing correction was performed using Benjamini and Hochberg False Discovery Rate (FDR). Following statistical analysis and probe mapping, 47 genes were upregulated and 39 genes were downregulated. Differentially expressed probes were clustered using MultiExperiment Viewer (MeV v4.5 [20]) (Figure 1). The clustering figure shows a distinct pattern of upregulated and downregulated probes in cases when compared to the controls.

Hierarchical clustering of differentially regulated genes in the setting of PAD. Clustering was performed using hierarchical clustering algorithm with Euclidean distance and average linkage clustering method.
Validation using real-time PCR
To validate our findings from gene expression analysis, we performed real-time PCR of the Syntaxin 11 (STX11), a gene not previously associated with PAD. STX11 is a component of t-SNARE complex and involved in endocytic vesicular transport, regulate protein transport among late endosomes and the trans-Golgi network and may have functional or regulatory role in vascular diseases. RNA derived from PAD and control samples was converted to cDNA by reverse transcription using Transcriptor First Strand cDNA Synthesis kit (Roche, OH, USA) and used immediately for real-time PCR. The FASTA sequence of each gene of interest was obtained using NCBI nucleotide search to design the primers http://www.ncbi.nlm.nih.gov/nuccore. This FASTA sequence was used as query to search in NCBI Primer BLAST http://www.ncbi.nlm.nih.gov/tools/primer-blast/index.cgi?LINK_LOC=BlastHome. Primers pairs were selected based on primer length (18-30 bp), GC content, melting temperature (Tm = 59-60°C), and product size. Primers were selected that scan exon-exon junctions. BLAST was used to check the specificity of primers to the gene of interest. PAGE-purified oligos (Integrated DNA Technologies, IA, USA) were used for real-time PCR. Primer sequences are available from the corresponding author upon request. Real-time PCR assay was performed using the LightCycler 480 instrument (Roche) and the LightCycler 480 SYBR Green I Master kit and protocol. Each sample was assayed in duplicate for the genes of interest as well as ß-actin (ACTB) as a housekeeping gene for normalization. Samples were assayed in 384-well plates with a 20 μL reaction volume. (10 uL master mix (FastStart Taq DNA polymerase, reaction buffer, dNTPs, SYBR Green I dye, and MgCl2), 3uL PCR-grade water, 1 uL each 2 nM primer, and 5 uL (32 ng) cDNA template. Raw Cp values were calculated using the Abs Quant/2nd derivative max option in Roche's LightCycler 480 software (release 1.5.0 SP3).
Functional annotation of differentially regulated genes using in-silico approach
To assess the functional repertoire of differentially expressed genes we adopted a multi-tiered bioinformatics annotation pipeline with functional enrichment calculations, pathway and molecular event analysis, biological network analysis and biocuration. Statistically significant genes and annotations were used as pointers to perform literature curation to derive biological role of genes differentially regulated in the setting of PAD. Preliminary functional annotations of differentially regulated genes were derived using BioGPS [21].
GO term enrichment analysis was performed using DAVID v6.7 [22, 23]. As no single annotation resource provides information about all available biological pathways, we used two different pathway databases (Reactome v36.2 [24] and Kyoto Encyclopedia of Genes and Genomes (KEGG) [25]) to identify the biological pathways mediated by differentially expressed genes. Reactome based pathway enrichment analysis was performed using Reactome Pathway Analysis tool http://www.reactome.org/ReactomeGWT/entrypoint.html#PathwayAnalysisDataUploadPage. This two-fold approach was useful in finding several relevant pathways from two different pathway analyses. Ingenuity Pathway Analysis® suite (IPA v9.0 - 3211, http://www.ingenuity.com) was used to understand functional networks involved in the gene sets. IPA-Tox®, a data analysis routine within IPA that assess potential toxicity with various compounds using toxicogenomics data was also used to interpret the functional context of differentially regulated genes. Finally, to understand the functional context and biological significance of differentially expressed genes relevant to PAD, scientific literature was curated using Gene Related InFormation (GeneRIF http://www.ncbi.nlm.nih.gov/projects/GeneRIF) and 'related articles by PubMed' in the 'Entrez Gene' http://www.ncbi.nlm.nih.gov/gene page.
Results
Patient characteristics are summarized in Table 1. The PBMC from patients with PAD differentially expressed 87 genes involved in immune response, inflammation, phosphorylation, signal transduction, platelet aggregation, vitamin metabolism, hemostasis, oxidative stress and transcriptional regulation.
Differentially expressed genes
Based on the comparisons of cases and controls, after initial data filtering and at a 1.5 fold change, 95 probes were differentially expressed (P < 0.05; ) (Table 2). A subset of probes was not annotated in Affymetrix annotation files. We obtained enhanced probe mapping by combining results from multiple probe mapping tools AILUN [26], BioMart [27] and GATExplorer [28]. Final probe mapping was performed using the union of mapped results obtained from these methods. NCBI-Gene and UCSC genome browser [29] were also consulted for annotation and integration of probe and gene related information. Statistically significant probes, mapped genes, P-value, fold change absolute (FCA) and directions of expression (regulation) are summarized in Table 2. Ten downregulated probes were not mapped to a valid gene using the probe mapping approach that we employed and were manually mapped using Ensembl v58 annotations [30]. For example probe "241838_at" (FCA 1.53) was not mapped to a valid gene using our probe mapping strategy but using Ensembl v58 [30] the probe was mapped to a non-coding transcript RP1-167A14.2. One probe "243310_at" could not be mapped to any known gene using the probe mapping databases or Ensembl annotations.
Real-time quantitative PCR analysis
The raw data (Cp values) were read in the R software (version 2.12.0) and the non-parametric Mann-Whitney unpaired test was used for analyzing the target gene STX11 in reference to the housekeeping gene ACTB. Cp values of ACTB were used for normalization. Using RT-qPCR, STX11 was successfully amplified and confirmed to have significant differential expression (P = 0.036).
In-silico analysis of differentially expressed genes
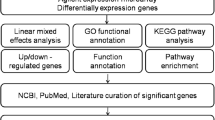
The differentially expressed genes were analyzed using a bioinformatics pipeline to understand the functional role of the genes. A flow-chart of the multi-tiered bioinformatics approach is provided in Figure 2. Multiple resources were integrated in the pipeline to provide a cohesive view of biological functions and pathways associated with the differentially expressed genes. Results from enrichment analysis using GO terms (Tables 3 and 4), pathway analysis using KEGG pathways (Table 5) and enrichment analysis of molecular event analysis using Reactome annotations (Table 6) are provided. Interactions within the differentially regulated genes were identified using biological network analysis utilities in IPA (Tables 7 and 8). Brief description of methodology and results from various approaches are provided.

Bioinformatics pipeline used for the biological interpretation of differentially expressed genes.
GO terms associated with differentially expressed genes
GO enrichment analysis was performed using GO Fat (collection of broadest GO terms curated from GO annotations dataset) based annotations using DAVID [23, 31]. The background was defined as the 'Human Genome U133 Plus 2' annotation and the differentially expressed genes from the study were input for assessing the enrichment. The upregulated and downregulated probe set identifiers were used as input and enrichment was analyzed separately and the results provided for the significantly enriched terms using Fisher's exact test using the EASE modification (P < 0.05) and multiple testing correction was performed using Benjamini-Hochberg FDR method. The P-value for each GO term reflects the enrichment in frequency of that GO term in the input entity list (differentially regulated probe set identifiers) relative to all entities in the background list (probe identifiers in Human Genome U133 Plus 2).
Among different GO terms of upregulated genes, several enriched terms in 'biological process' (Table 3 and Additional file 1: Table S1) categories were related to molecular mechanisms associated with inflammation (inflammatory response; response to protein stimulus, response to organic substance, cytokine activity); immune response (defense response, regulation of response to external stimulus), cell death (induction of apoptosis by extracellular signals, regulation of cell proliferation, positive regulation of anti-apoptosis, regulation of apoptosis) and stress response (response to oxidative stress, response to reactive oxygen species, response to hyperoxia). Other important biological processes mediated by upregulated genes were regulation of peptidase activity, caspase activity and endopeptidase activity. A visual summary of GO identifiers associated with upregulated genes (Figure 3) were created using REVIGO [32]. Molecular functions of the upregulated genes included phosphatase activity. GO terms associated with downregulated genes were enriched for various terms related to transcriptional regulation. These results indicate that the PBMC, in the setting of PAD, differentially express genes involved in inflammation, immune response, apoptosis, molecular specific functions mediated by peptidase, caspase and stress response related pathways. Results of the GO annotation based enrichment analysis of upregulated and downregulated genes are summarized in Tables 3 and 4.

Statistically significant GO terms (Biological Process category) derived from upregulated genes.
KEGG pathways associated with differentially expressed genes
SubPathwayMiner was used to assess the statistical significance of KEGG pathways associated with differentially expressed genes. Probes were mapped to genes identifiers and gene identifiers were used as the input in the statistical analysis. The enrichment analysis revealed that 17 pathways were associated with upregulated genes and two pathways were significant in downregulated genes (P < 0.05) and multiple testing correction was performed using FDR. Analysis of KEGG pathway classes indicates that these pathways mediate cellular processing, signal transduction, immune system and infectious diseases. These analyses suggest that perturbations in multiple signaling and cellular mechanisms occur in PBMC in the setting of PAD. Significantly enriched pathways and corresponding P-values are listed in Table 5.
Molecular events associated with PAD
Compared to classical biological pathway databases, Reactome provides biological processes as a series of molecular events and is thus a unique resource for functional interpretation of genes lists with a wide array of pathways, specific biological process and molecular events. We used the probe identifiers as the input for Reactome based enrichment analysis to find molecular events associated with differentially expressed genes using a hypergeometric test (P < 0.05). Pathway analysis using Reactome showed that upregulated genes were implicated in five platelet related pathways (platelet activation, formation of platelet plug, exocytosis of platelet alpha granule contents, platelet degranulation and response to elevated platelet cytosolic Ca2+). Two vitamin metabolism related events (Metabolism of water-soluble vitamins and cofactors, Metabolism of vitamins and cofactors) were also associated with upregulated genes (Table 6). Similar to KEGG pathway enrichment (Table 5), we also observed enrichment of several signal transduction events in the Reactome analysis. There was no significant enrichment of molecular events observed in the downregulated genes.
Functional network inferred using IPA
We used IPA to understand the functionally significant biological networks and toxicogenomics associations mediated by the differentially expressed genes in the setting of PAD. IPA analysis was performed using probe identifiers as the input; the reference dataset was defined as 'Human Genome U133 Plus 2'; direct interactions only were considered for the analysis. Biological network (Table 7) and toxicity functions (Table 8) derived from IPA are provided and illustrated in Figure 4 (a) (merged view of networks derived from upregulated genes) and Figure 4 (b) (merged view of networks derived from downregulated genes). Different shapes of nodes indicate "Family" of a given gene assigned using IPA annotations. Color of node indicates the presence (grey) or absence (white) of a given gene in the study. Nodes that are not represented in the study (white nodes) were retained in the network for a context dependent view of the functional interactome. Edges shared between six different functional networks derived from upregulated genes were highlighted (Figure 4 (a)). Downregulated genes do not share any common nodes between the derived functional networks (Figure 4 (b)). It should be noted that upregulated genes interacted with several core genes (interactions are highlighted with edges colored in orange) that are present in multiple networks, where as the downregulated genes did not interact with the core genes. These results suggest that that upregulated genes identified in our study may influence multiple functional networks via interaction with the core genes. Further studies are required to understand role of these genes in the pathophysiology of PAD.

Functional networks inferred using IPA. a) Merged view of functional network derived from upregulated genes using IPA (Network 1: Cell Death, Renal Necrosis/Cell Death, Cancer; Network 2: Cardiovascular Disease, Hematological Disease, Infection Mechanism; Network 3: Inflammatory Response, Embryonic Development, Cell-To-Cell Signaling and Interaction Network 4: Neurological Disease, RNA Post-Transcriptional Modification; Network 5: Cardiovascular Disease, Genetic Disorder, Cellular Function and Maintenance; Network 6: Drug Metabolism, Nucleic Acid Metabolism, Small Molecular Biochemistry). b) Functional networks derived from downregulated genes using IPA (Network 1: Cellular Development, Cellular Assembly and Organization, Cell Cycle; Network 2: Genetic Disorder, Ophthalmic Disease, Nervous System Development and Function; Network 3: Embryonic Development, Gene Expression, Protein Trafficking Network 4: Cellular Assembly and Organization, Cell Morphology, Cellular Function and Maintenance).
Biocuration of differentially expressed genes
We performed in-depth biocuration of differentially regulated genes using a combination of resources. For a given differentially expressed gene we consulted General annotation under the Comments section in UniProt, RefSeq summary, GeneRIF and publications linked under "Related Articles" in PubMed section of "Bibliography" in individual Gene pages. Biocuration was performed to manually extract the role of differentially expressed pertaining to vascular diseases including PAD from previous literature reports. Curated data with functional context and role of genes in vascular diseases and associated references are presented in Additional file 1: Table S2. This approach further helped to extract functionally relevant information not captured by ontologies or annotations in automated analytical frameworks used in enrichment tools.
Discussion
We report for the first time gene expression analysis of PBMC to identify genes differentially expressed in patients with PAD. Enrichment analysis of GO terms and pathways associated with these genes provide insights into several known and novel molecular mechanisms related to PAD. The two genes with highest fold change absolute (FCA) were: G0/G1switch 2 (G0S2; FCA: 3.90; ↑) BTB and CNC homology 1, basic leucine zipper transcription factor 2 (BACH2; FCA: 2.10; ↓). G0S2 is a novel target of peroxisome-proliferator-activated receptor (PPAR) involved in adipocyte differentiation [33, 34]. BACH2 is a transcriptional regulator that acts as repressor or activator through the nuclear factor (erythroid-derived 2), 45 kDa (NFE2) binding sites [35, 36]. Differentially regulated genes, summarized in Table 2 are ideal candidates for further, down-stream functional studies.
Biological relevance of differentially expressed genes in the setting of PAD
Knowledge-based statistical analysis of differentially expressed genes provided molecular clues for the interpretation of the function or pathways associated with these genes. We used the statistically significant genes, GO terms and pathways as leads to perform in-depth literature curation. The detailed literature curation indicated that the genes identified in this study are relevant to various aspects of vascular biology and pathophysiology of PAD.
Several of the differentially regulated genes are involved in vascular pathophysiology; for example: DNAJB6 [37] and DUSP1 (atherosclerosis) [38], NAMPT (vascular inflammation) [39, 40], FCAR (myocardial infarction) [41], IL8 (vascular remodelling) [42], FFAR2 (lipid metabolism) [43] and SOD2 (idiopathic cardiomyopathy (IDC)) [44]. Notably, several genes known to be associated with vascular disease were upregulated as discussed below.
Phosphatases are known to be associated with peripheral arterial disease [45–47]. We noted that four phosphatase genes PTP4A1, DUSP1, DUSP5, PPP1R15A are significantly upregulated in the PBMC of patients with PAD. Apoptosis, along with inflammation and immune response, is a key feature of vascular diseases [5, 48–52]. Our study indicates genes implicated in inflammation, immune response (FCAR, FFAR2, IL8, CFLAR, DUSP1, NAMPT) and cell death (G0S2, KLF6, PTP4A1, CFLAR) are differentially expressed in PBMC of PAD patients. Oxidative-stress response is known to be associated with PAD [53–56]; we noted that several "oxidative-stress response" related functions were enriched in GO term analysis and IPA analysis. Altered metabolism of vitamins and vitamin D deficiency has been reported to be associated with PAD [57, 58]. Enrichment analysis using molecular event annotations (Table 6) and GO term analysis (Table 3) indicated that vitamin metabolism related pathways are upregulated in the setting of PAD. Platelet aggregation is strongly linked to PAD [1, 6, 59–62] and Reactome based pathway analysis indicated that several platelet-related molecular events were associated with upregulated genes such as PLAUR (Table 6).
Apart from these known genes, we noted several genes not previously associated with PAD to be differentially expressed. Upregulation of validated gene STX11 suggests a putative role for genes associated with vesicle trafficking in the pathophysiology of PAD. Upregulation of FFAR2 suggests altered free fatty acid metabolism in the setting of PAD. Further investigation of differentially regulated transcription factors (for example: C5orf41, KLF6, BACH2), and their downstream target genes may provide additional insights into the molecular basis of PAD.
Comparison with previous studies
Several of the differentially expressed genes identified in the current study were previously reported to be associated with various vascular biology processes. For example thrombospondin-1 (THBS1) [63–65], phosphatases (DUSP1) [45], plasminogen activator, urokinase receptor (PLAUR) [60], cadherins (DSC2) [66, 67] and zinc finger proteins (ZNF207) [68–71] have been implicated in vascular homeostasis and pathophysiology of PAD. Prior microarray studies of PAD have also demonstrated a pattern of activation of genes involved in immune and inflammatory response [72]. Our study is designed to identify perturbed genes in PBMCs in the setting of PAD. Fu et al., [72] performed microarray analysis of atherosclerotic lesions of femoral arteries, and found that immune and inflammatory pathways were enriched in PAD cases. We replicated the following genes from Fu et. al's analysis: CDKN1A, CXCR4, KLF4, PLAUR, SAMSN1, SOD2 and THBS1. Wingrove et al., [10] performed whole-genome microarray analysis on PBMCs of 27 cases with angiographic coronary artery stenosis and 14 controls and identified 526 genes with >1.3-fold differential expression (P < 0.05) between cases and controls. Real time PCR in two independent cohorts (149 cases and 53 controls) for 106 genes (the 50 most significant genes and 56 additional candidate genes) confirmed that 11 genes were significantly differentially expressed between cases and controls. The differentially expressed genes that we identified in the setting of PAD did not overlap with genes found by Wingrove et al., [10] but we validated several genes differentially expressed in intermediate lesions and advanced lesions derived from femoral artery samples analyzed by Fu et al [72]. Evans et al., [73] performed microarray analysis of leg arteries and identified genes involved in inflammation and lipid uptake pathways in the setting of PAD with diabetes. Similar to observations by Evans et al., [73] we also noted that inflammation and related GO terms like immune response, apoptosis, response to stress, cell proliferation and circulation were enriched in the GO annotations of upregulated genes. Differences in methodology, sources of mRNA and the fact that PAD and CAD are distinct phenotypic manifestations of atherosclerosis may account for the varying results.
Integrated approach for functional interpretation
We integrated four different annotation resources for functional interpretation of differentially expressed genes (Figure 2). GO annotations provided a comprehensive view of the function and processes, pathway enrichment using KEGG provided disease association of differentially expressed genes, Reactome was useful in understanding molecular events associated with genes and IPA facilitated understanding of functional networks (group of genes that share common functions) and toxicity functions. Although annotations shared several common entities, each tool provided a unique perspective of the differentially regulated genes in the setting of PAD. Further, we also employed in-depth biocuration strategies to understand the functional and pathological relevance of differentially expressed genes in the setting of vascular disease. Our integrated bioinformatics approach coupled with biocuration provided insights into the functional repertoire of differentially expressed genes.
Strength and Limitations
A strength of this report is the application of integrative bioinformatics pipeline employed to understand the functional similarities, biological pathways, molecular events and functional networks, related to differentially expressed genes. In addition we performed in-depth literature curation to understand functional relevance of these genes. Further we validated a novel differentially regulated gene STX11 using qRT-PCR. Complete characterization of the genes identified in this study in the context of their relevance to PAD will require further validation and functional studies. We derived the RNA from PBMC, which may have perturbations in the cellular level due to fluctuation in cluster of differentiation 4 (CD4) count within cases and controls. Patients in our study were ascertained based on ABI (ABI ≤0.9 for cases and ABI > 1.0 for controls), additional clinical biomarkers such as T lymphocytes (T cells) and Natural killer cells (NK cells) or CD4 counts were not available.
Conclusion
Gene expression profiling of circulating PBMC provided a global overview of differential gene expression in PAD; where 87 differentially expressed genes (47 upregulated genes and 39 downregulated genes). Integrated bioinformatics analysis of differentially regulated genes using multiple annotation tools indicated that the differentially regulated genes influence immune response, inflammation, apoptosis, various signalling pathways and various functions pertaining to vascular biology. Our whole-genome expression and bioinformatics analysis suggests that microarray based expression profiling may be useful for characterizing biomarkers for PAD. Understanding and validating groups of differentially expressed genes in the setting of PAD using PBMC can improve our understanding of the key pathophysiological mechanisms in the aetiology of PAD. Further clinical and functional studies may provide additional insights into role of the differentially expressed genes in the pathophysiology of PAD.
Availability
Gene expression data discussed in this study was submitted to Gene Expression Omnibus (GEO) database, and can be accessed via GEO accession ID GSE27034.
References
Hirsch AT, Criqui MH, Treat-Jacobson D, Regensteiner JG, Creager MA, Olin JW, Krook SH, Hunninghake DB, Comerota AJ, Walsh ME, et al: Peripheral arterial disease detection, awareness, and treatment in primary care. JAMA. 2001, 286 (11): 1317-1324. 10.1001/jama.286.11.1317.
Allison MA, Ho E, Denenberg JO, Langer RD, Newman AB, Fabsitz RR, Criqui MH: Ethnic-specific prevalence of peripheral arterial disease in the United States. Am J Prev Med. 2007, 32 (4): 328-333. 10.1016/j.amepre.2006.12.010.
Roger VL, Go AS, Lloyd-Jones DM, Adams RJ, Berry JD, Brown TM, Carnethon MR, Dai S, de Simone G, Ford ES, et al: Heart disease and stroke statistics-2011 update: a report from the American Heart Association. Circulation. 2011, 123 (4): e18-e209. 10.1161/CIR.0b013e3182009701.
Ouriel K: Peripheral arterial disease. Lancet. 2001, 358 (9289): 1257-1264. 10.1016/S0140-6736(01)06351-6.
Hansson GK: Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005, 352 (16): 1685-1695. 10.1056/NEJMra043430.
Mohler ER: Peripheral arterial disease: identification and implications. Arch Intern Med. 2003, 163 (19): 2306-2314. 10.1001/archinte.163.19.2306.
Whitney AR, Diehn M, Popper SJ, Alizadeh AA, Boldrick JC, Relman DA, Brown PO: Individuality and variation in gene expression patterns in human blood. Proc Natl Acad Sci USA. 2003, 100 (4): 1896-1901. 10.1073/pnas.252784499.
Eady JJ, Wortley GM, Wormstone YM, Hughes JC, Astley SB, Foxall RJ, Doleman JF, Elliott RM: Variation in gene expression profiles of peripheral blood mononuclear cells from healthy volunteers. Physiol Genomics. 2005, 22 (3): 402-411. 10.1152/physiolgenomics.00080.2005.
Timofeeva AV, Goryunova LE, Khaspekov GL, Kovalevskii DA, Scamrov AV, Bulkina OS, Karpov YA, Talitskii KA, Buza VV, Britareva VV, et al: Altered gene expression pattern in peripheral blood leukocytes from patients with arterial hypertension. Ann N Y Acad Sci. 2006, 1091: 319-335. 10.1196/annals.1378.077.
Wingrove JA, Daniels SE, Sehnert AJ, Tingley W, Elashoff MR, Rosenbery S, Buellesfeld L, Grube E, Newby LK, Ginsburg GS, et al: Correlation of peripheral blood gene expression with the extent of coronary artery stenosis. Circ Cardiovasc Genet. 2008, 1: 31-38. 10.1161/CIRCGENETICS.108.782730.
Rosenberg S, Elashoff MR, Beineke P, Daniels SE, Wingrove JA, Tingley WG, Sager PT, Sehnert AJ, Yau M, Kraus WE, et al: Multicenter validation of the diagnostic accuracy of a blood-based gene expression test for assessing obstructive coronary artery disease in nondiabetic patients. Annals of internal medicine. 2005, 153 (7): 425-434.
Aziz H, Zaas A, Ginsburg GS: Peripheral blood gene expression profiling for cardiovascular disease assessment. Genomics Med. 2007, 1 (3-4): 105-112. 10.1007/s11568-008-9017-x.
Stamova B, Xu H, Jickling G, Bushnell C, Tian Y, Ander BP, Zhan X, Liu D, Turner R, Adamczyk P, et al: Gene expression profiling of blood for the prediction of ischemic stroke. Stroke. 2010, 41 (10): 2171-2177. 10.1161/STROKEAHA.110.588335.
Patino WD, Mian OY, Kang JG, Matoba S, Bartlett LD, Holbrook B, Trout HH: Kozloff L, Hwang PM: Circulating transcriptome reveals markers of atherosclerosis. Proc Natl Acad Sci USA. 2005, 102 (9): 3423-3428. 10.1073/pnas.0408032102.
Waehre T, Yndestad A, Smith C, Haug T, Tunheim SH, Gullestad L, Froland SS, Semb AG, Aukrust P, Damas JK: Increased expression of interleukin-1 in coronary artery disease with downregulatory effects of HMG-CoA reductase inhibitors. Circulation. 2004, 109 (16): 1966-1972. 10.1161/01.CIR.0000125700.33637.B1.
Meier P, Antonov J, Zbinden R, Kuhn A, Zbinden S, Gloekler S, Delorenzi M, Jaggi R, Seiler C: Non-invasive gene-expression-based detection of well-developed collateral function in individuals with and without coronary artery disease. Heart (British Cardiac Society). 2009, 95 (11): 900-908. 10.1136/hrt.2008.145383.
Kullo IJ, Bailey KR, Kardia SL, Mosley TH, Boerwinkle E, Turner ST: Ethnic differences in peripheral arterial disease in the NHLBI Genetic Epidemiology Network of Arteriopathy (GENOA) study. Vasc Med. 2003, 8 (4): 237-242. 10.1191/1358863x03vm511oa.
Bielecki M, Kowal K, Lapinska A, Chwiecko J, Skowronski J, Sierakowski S, Chyczewski L, Kowal-Bielecka O: Diminished production of TWEAK by the peripheral blood mononuclear cells is associated with vascular involvement in patients with systemic sclerosis. Folia Histochem Cytobiol. 2009, 47 (3): 465-469. 10.2478/v10042-009-0103-2.
Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP: Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics (Oxford, England). 2003, 4 (2): 249-264.
Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, Braisted J, Klapa M, Currier T, Thiagarajan M, et al: TM4: a free, open-source system for microarray data management and analysis. Biotechniques. 2003, 34 (2): 374-378.
Wu C, Orozco C, Boyer J, Leglise M, Goodale J, Batalov S, Hodge CL, Haase J, Janes J, Huss JW, et al: BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 2009, 10 (11): R130-10.1186/gb-2009-10-11-r130.
da Huang W, Sherman BT, Lempicki RA: Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37 (1): 1-13. 10.1093/nar/gkn923.
da Huang W, Sherman BT, Lempicki RA: Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009, 4 (1): 44-57.
Croft D, O'Kelly G, Wu G, Haw R, Gillespie M, Matthews L, Caudy M, Garapati P, Gopinath G, Jassal B, et al: Reactome: a database of reactions, pathways and biological processes. Nucleic Acids Res. 2011, D691-D697. 39 Database
Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M: KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010, D355-D360. 38 Database
Chen R, Li L, Butte AJ: AILUN: reannotating gene expression data automatically. Nat Methods. 2007, 4 (11): 879-10.1038/nmeth1107-879.
Smedley D, Haider S, Ballester B, Holland R, London D, Thorisson G, Kasprzyk A: BioMart-biological queries made easy. BMC Genomics. 2009, 10: 22-10.1186/1471-2164-10-22.
Risueno A, Fontanillo C, Dinger ME, De Las Rivas J: GATExplorer: genomic and transcriptomic explorer; mapping expression probes to gene loci, transcripts, exons and ncRNAs. BMC Bioinformatics. 2010, 11: 221-10.1186/1471-2105-11-221.
Rhead B, Karolchik D, Kuhn RM, Hinrichs AS, Zweig AS, Fujita PA, Diekhans M, Smith KE, Rosenbloom KR, Raney BJ, et al: The UCSC Genome Browser database: update 2010. Nucleic Acids Res. 2010, D613-D619. 38 Database
Fernandez-Suarez XM, Schuster MK: Using the ensembl genome server to browse genomic sequence data. Curr Protoc Bioinformatics. 2010, Chapter 1: Unit1 15
Dennis G, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA: DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003, 4 (5): P3-10.1186/gb-2003-4-5-p3.
Supek F, Bosnjak M, Skunca N, Smuc T: REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One. 2011, 6 (7): e21800-10.1371/journal.pone.0021800.
Zandbergen F, Mandard S, Escher P, Tan NS, Patsouris D, Jatkoe T, Rojas-Caro S, Madore S, Wahli W, Tafuri S, et al: The G0/G1 switch gene 2 is a novel PPAR target gene. Biochem J. 2005, 392 (Pt 2): 313-324.
Russell L, Forsdyke DR: A human putative lymphocyte G0/G1 switch gene containing a CpG-rich island encodes a small basic protein with the potential to be phosphorylated. DNA Cell Biol. 1991, 10 (8): 581-591. 10.1089/dna.1991.10.581.
Oyake T, Itoh K, Motohashi H, Hayashi N, Hoshino H, Nishizawa M, Yamamoto M, Igarashi K: Bach proteins belong to a novel family of BTB-basic leucine zipper transcription factors that interact with MafK and regulate transcription through the NF-E2 site. Mol Cell Biol. 1996, 16 (11): 6083-6095.
Vieira SA, Deininger MW, Sorour A, Sinclair P, Foroni L, Goldman JM, Melo JV: Transcription factor BACH2 is transcriptionally regulated by the BCR/ABL oncogene. Genes Chromosomes Cancer. 2001, 32 (4): 353-363. 10.1002/gcc.1200.
Civelek M, Manduchi E, Riley RJ, Stoeckert CJ, Davies PF: Chronic endoplasmic reticulum stress activates unfolded protein response in arterial endothelium in regions of susceptibility to atherosclerosis. Circ Res. 2009, 105 (5): 453-461. 10.1161/CIRCRESAHA.109.203711.
Gargalovic PS, Imura M, Zhang B, Gharavi NM, Clark MJ, Pagnon J, Yang WP, He A, Truong A, Patel S, et al: Identification of inflammatory gene modules based on variations of human endothelial cell responses to oxidized lipids. Proc Natl Acad Sci USA. 2006, 103 (34): 12741-12746. 10.1073/pnas.0605457103.
Laudes M, Oberhauser F, Schulte DM, Freude S, Bilkovski R, Mauer J, Rappl G, Abken H, Hahn M, Schulz O, et al: Visfatin/PBEF/Nampt and resistin expressions in circulating blood monocytes are differentially related to obesity and type 2 diabetes in humans. Horm Metab Res. 2010, 42 (4): 268-273. 10.1055/s-0029-1243638.
Kadoglou NP, Sailer N, Moumtzouoglou A, Kapelouzou A, Tsanikidis H, Vitta I, Karkos C, Karayannacos PE, Gerasimidis T, Liapis CD: Visfatin (nampt) and ghrelin as novel markers of carotid atherosclerosis in patients with type 2 diabetes. Exp Clin Endocrinol Diabetes. 2010, 118 (2): 75-80. 10.1055/s-0029-1237360.
Iakoubova OA, Tong CH, Chokkalingam AP, Rowland CM, Kirchgessner TG, Louie JZ, Ploughman LM, Sabatine MS, Campos H, Catanese JJ, et al: Asp92Asn polymorphism in the myeloid IgA Fc receptor is associated with myocardial infarction in two disparate populations: CARE and WOSCOPS. Arterioscler Thromb Vasc Biol. 2006, 26 (12): 2763-2768. 10.1161/01.ATV.0000247248.76409.8b.
Li A, Dubey S, Varney ML, Dave BJ, Singh RK: IL-8 directly enhanced endothelial cell survival, proliferation, and matrix metalloproteinases production and regulated angiogenesis. J Immunol. 2003, 170 (6): 3369-3376.
Lee T, Schwandner R, Swaminath G, Weiszmann J, Cardozo M, Greenberg J, Jaeckel P, Ge H, Wang Y, Jiao X, et al: Identification and functional characterization of allosteric agonists for the G protein-coupled receptor FFA2. Mol Pharmacol. 2008, 74 (6): 1599-1609. 10.1124/mol.108.049536.
Faraci FM, Didion SP: Vascular protection: superoxide dismutase isoforms in the vessel wall. Arterioscler Thromb Vasc Biol. 2004, 24 (8): 1367-1373. 10.1161/01.ATV.0000133604.20182.cf.
Carr AN, Davis MG, Eby-Wilkens E, Howard BW, Towne BA, Dufresne TE, Peters KG: Tyrosine phosphatase inhibition augments collateral blood flow in a rat model of peripheral vascular disease. Am J Physiol Heart Circ Physiol. 2004, 287 (1): H268-H276. 10.1152/ajpheart.00007.2004.
Zakkar M, Chaudhury H, Sandvik G, Enesa K, le Luong A, Cuhlmann S, Mason JC, Krams R, Clark AR, Haskard DO, et al: Increased endothelial mitogen-activated protein kinase phosphatase-1 expression suppresses proinflammatory activation at sites that are resistant to atherosclerosis. Circ Res. 2008, 103 (7): 726-732. 10.1161/CIRCRESAHA.108.183913.
Cheung BM, Ong KL, Wong LY: Elevated serum alkaline phosphatase and peripheral arterial disease in the United States National Health and Nutrition Examination Survey 1999-2004. Int J Cardiol. 2009, 135 (2): 156-161. 10.1016/j.ijcard.2008.03.039.
Bennett MR: Apoptosis in vascular disease. Transpl Immunol. 1997, 5 (3): 184-188. 10.1016/S0966-3274(97)80036-6.
Freyssinet JM, Toti F, Hugel B, Gidon-Jeangirard C, Kunzelmann C, Martinez MC, Meyer D: Apoptosis in vascular disease. Thromb Haemost. 1999, 82 (2): 727-735.
Mercer J, Mahmoudi M, Bennett M: DNA damage, p53, apoptosis and vascular disease. Mutat Res. 2007, 621 (1-2): 75-86. 10.1016/j.mrfmmm.2007.02.011.
Kockx MM, Knaapen MW: The role of apoptosis in vascular disease. J Pathol. 2000, 190 (3): 267-280. 10.1002/(SICI)1096-9896(200002)190:3<267::AID-PATH523>3.0.CO;2-A.
Sykes TC, Morris AG, Bradbury AW, Mosquera D: Apoptosis in vascular disease. Eur J Vasc Endovasc Surg. 2001, 22 (5): 389-395. 10.1053/ejvs.2001.1492.
Madamanchi NR, Vendrov A, Runge MS: Oxidative stress and vascular disease. Arterioscler Thromb Vasc Biol. 2005, 25 (1): 29-38.
Forstermann U: Oxidative stress in vascular disease: causes, defense mechanisms and potential therapies. Nat Clin Pract Cardiovasc Med. 2008, 5 (6): 338-349. 10.1038/ncpcardio1211.
Belch JJ, Mackay IR, Hill A, Jennings P, McCollum P: Oxidative stress is present in atherosclerotic peripheral arterial disease and further increased by diabetes mellitus. Int Angiol. 1995, 14 (4): 385-388.
Loffredo L, Marcoccia A, Pignatelli P, Andreozzi P, Borgia MC, Cangemi R, Chiarotti F, Violi F: Oxidative-stress-mediated arterial dysfunction in patients with peripheral arterial disease. Eur Heart J. 2007, 28 (5): 608-612.
Fahrleitner A, Dobnig H, Obernosterer A, Pilger E, Leb G, Weber K, Kudlacek S, Obermayer-Pietsch BM: Vitamin D deficiency and secondary hyperparathyroidism are common complications in patients with peripheral arterial disease. J Gen Intern Med. 2002, 17 (9): 663-669. 10.1046/j.1525-1497.2002.11033.x.
Gaddipati VC, Kuriacose R, Copeland R, Bailey BA, Peiris AN: Vitamin D deficiency: an increasing concern in peripheral arterial disease. J Am Med Dir Assoc. 2010, 11 (5): 308-311. 10.1016/j.jamda.2010.02.002.
Karnicki K, Karnicki K, Miller RS, Owen WG: The impact of peripheral arterial disease on circulating platelets. Thromb Res. 2004, 113 (2): 137-145. 10.1016/j.thromres.2004.02.007.
Robless PA, Okonko D, Lintott P, Mansfield AO, Mikhailidis DP, Stansby GP: Increased platelet aggregation and activation in peripheral arterial disease. Eur J Vasc Endovasc Surg. 2003, 25 (1): 16-22. 10.1053/ejvs.2002.1794.
Cassar K, Bachoo P, Brittenden J: The role of platelets in peripheral vascular disease. Eur J Vasc Endovasc Surg. 2003, 25 (1): 6-15. 10.1053/ejvs.2002.1795.
Huo Y, Ley KF: Role of platelets in the development of atherosclerosis. Trends Cardiovasc Med. 2004, 14 (1): 18-22. 10.1016/j.tcm.2003.09.007.
Maier KG, Han X, Sadowitz B, Gentile KL, Middleton FA, Gahtan V: Thrombospondin-1: a proatherosclerotic protein augmented by hyperglycemia. J Vasc Surg. 2010, 51 (5): 1238-1247. 10.1016/j.jvs.2009.11.073.
Ochoa CD, Fouty BW, Hales CA: Thrombospondin-1, endothelium and systemic vascular tone. Future Cardiol. 2011, 7 (2): 169-172. 10.2217/fca.11.7.
Smadja DM, d'Audigier C, Bieche I, Evrard S, Mauge L, Dias JV, Labreuche J, Laurendeau I, Marsac B, Dizier B, et al: Thrombospondin-1 is a plasmatic marker of peripheral arterial disease that modulates endothelial progenitor cell angiogenic properties. Arterioscler Thromb Vasc Biol. 2011, 31 (3): 551-559. 10.1161/ATVBAHA.110.220624.
Jang Y, Lincoff AM, Plow EF, Topol EJ: Cell adhesion molecules in coronary artery disease. J Am Coll Cardiol. 1994, 24 (7): 1591-1601. 10.1016/0735-1097(94)90162-7.
George SJ, Dwivedi A: MMPs, cadherins, and cell proliferation. Trends Cardiovasc Med. 2004, 14 (3): 100-105. 10.1016/j.tcm.2003.12.008.
Pahl PM, Hodges YK, Meltesen L, Perryman MB, Horwitz KB, Horwitz LD: ZNF207, a ubiquitously expressed zinc finger gene on chromosome 6p21.3. Genomics. 1998, 53 (3): 410-412. 10.1006/geno.1998.5442.
Wagner S, Hess MA, Ormonde-Hanson P, Malandro J, Hu H, Chen M, Kehrer R, Frodsham M, Schumacher C, Beluch M, et al: A broad role for the zinc finger protein ZNF202 in human lipid metabolism. J Biol Chem. 2000, 275 (21): 15685-15690. 10.1074/jbc.M910152199.
Yu J, Lei L, Liang Y, Hinh L, Hickey RP, Huang Y, Liu D, Yeh JL, Rebar E, Case C, et al: An engineered VEGF-activating zinc finger protein transcription factor improves blood flow and limb salvage in advanced-age mice. FASEB J. 2006, 20 (3): 479-481.
Quintal SM, dePaula QA, Farrell NP: Zinc finger proteins as templates for metal ion exchange and ligand reactivity. Chemical and biological consequences. Metallomics. 2011, 3 (2): 121-139. 10.1039/c0mt00070a.
Fu S, Zhao H, Shi J, Abzhanov A, Crawford K, Ohno-Machado L, Zhou J, Du Y, Kuo WP, Zhang J, et al: Peripheral arterial occlusive disease: global gene expression analyses suggest a major role for immune and inflammatory responses. BMC Genomics. 2008, 9: 369-10.1186/1471-2164-9-369.
Evans DC, Sileshi B, Zakaria AM, Giangiacomo D, Manson RJ, Lawson JH: Genomic modeling of atherosclerosis in peripheral arterial disease and its variant phenotype in patients with diabetes. Vascular. 2008, 16 (4): 225-235. 10.2310/6670.2008.00037.
Seko A, Yamashita K: Activation of beta1,3-N-acetylglucosaminyltransferase-2 (beta3Gn-T2) by beta3Gn-T8. Possible involvement of beta3Gn-T8 in increasing poly-N-acetyllactosamine chains in differentiated HL-60 cells. J Biol Chem. 2008, 283 (48): 33094-33100. 10.1074/jbc.M806933200.
Karsan A, Yee E, Kaushansky K, Harlan JM: Cloning of human Bcl-2 homologue: inflammatory cytokines induce human A1 in cultured endothelial cells. Blood. 1996, 87 (8): 3089-3096.
Alvarez E, Zhou W, Witta SE, Freed CR: Characterization of the Bex gene family in humans, mice, and rats. Gene. 2005, 357 (1): 18-28. 10.1016/j.gene.2005.05.012.
Pfeufer A, van Noord C, Marciante KD, Arking DE, Larson MG, Smith AV, Tarasov KV, Muller M, Sotoodehnia N, Sinner MF, et al: Genome-wide association study of PR interval. Nat Genet. 2010, 42 (2): 153-159. 10.1038/ng.517.
Audas TE, Li Y, Liang G, Lu R: A novel protein, Luman/CREB3 recruitment factor, inhibits Luman activation of the unfolded protein response. Mol Cell Biol. 2008, 28 (12): 3952-3966. 10.1128/MCB.01439-07.
Rodriguez I, Coto E, Reguero JR, Gonzalez P, Andres V, Lozano I, Martin M, Alvarez V, Moris C: Role of the CDKN1A/p21, CDKN1C/p57, and CDKN2A/p16 genes in the risk of atherosclerosis and myocardial infarction. Cell Cycle. 2007, 6 (5): 620-625. 10.4161/cc.6.5.3927.
Sotoodehnia N, Isaacs A, de Bakker PI, Dorr M, Newton-Cheh C, Nolte IM, van der Harst P, Muller M, Eijgelsheim M, Alonso A, et al: Common variants in 22 loci are associated with QRS duration and cardiac ventricular conduction. Nat Genet. 2010, 42 (12): 1068-1076. 10.1038/ng.716.
Schwarz M, Spath L, Lux CA, Paprotka K, Torzewski M, Dersch K, Koch-Brandt C, Husmann M, Bhakdi S: Potential protective role of apoprotein J (clusterin) in atherogenesis: binding to enzymatically modified low-density lipoprotein reduces fatty acid-mediated cytotoxicity. Thromb Haemost. 2008, 100 (1): 110-118.
Xu Y, Johansson M, Karlsson A: Human UMP-CMP kinase 2, a novel nucleoside monophosphate kinase localized in mitochondria. J Biol Chem. 2008, 283 (3): 1563-1571.
Ishiguro H, Tsunoda T, Tanaka T, Fujii Y, Nakamura Y, Furukawa Y: Identification of AXUD1, a novel human gene induced by AXIN1 and its reduced expression in human carcinomas of the lung, liver, colon and kidney. Oncogene. 2001, 20 (36): 5062-5066. 10.1038/sj.onc.1204603.
Pramanik K, Chun CZ, Garnaas MK, Samant GV, Li K, Horswill MA, North PE, Ramchandran R: Dusp-5 and Snrk-1 coordinately function during vascular development and disease. Blood. 2009, 113 (5): 1184-1191.
Ferreira VP, Pangburn MK, Cortes C: Complement control protein factor H: the good, the bad, and the inadequate. Mol Immunol. 2010, 47 (13): 2187-2197. 10.1016/j.molimm.2010.05.007.
Swaminath G: Fatty acid binding receptors and their physiological role in type 2 diabetes. Arch Pharm (Weinheim). 2008, 341 (12): 753-761. 10.1002/ardp.200800096.
Kang JG, Sung HJ, Jawed SI, Brenneman CL, Rao YN, Sher S, Facio FM, Biesecker LG, Quyyumi AA, Sachdev V, et al: FOS expression in blood as a LDL-independent marker of statin treatment. Atherosclerosis. 2010, 212 (2): 567-570. 10.1016/j.atherosclerosis.2010.06.023.
Welch C, Santra MK, El-Assaad W, Zhu X, Huber WE, Keys RA, Teodoro JG, Green MR: Identification of a protein, G0S2, that lacks Bcl-2 homology domains and interacts with and antagonizes Bcl-2. Cancer Res. 2009, 69 (17): 6782-6789. 10.1158/0008-5472.CAN-09-0128.
Hellerud C, Burlina A, Gabelli C, Ellis JR, Nyholm PG, Lindstedt S: Glycerol metabolism and the determination of triglycerides-clinical, biochemical and molecular findings in six subjects. Clin Chem Lab Med. 2003, 41 (1): 46-55.
Heiber M, Marchese A, Nguyen T, Heng HH, George SR, O'Dowd BF: A novel human gene encoding a G-protein-coupled receptor (GPR15) is located on chromosome 3. Genomics. 1996, 32 (3): 462-465. 10.1006/geno.1996.0143.
Brandes RP, Kim D, Schmitz-Winnenthal FH, Amidi M, Godecke A, Mulsch A, Busse R: Increased nitrovasodilator sensitivity in endothelial nitric oxide synthase knockout mice: role of soluble guanylyl cyclase. Hypertension. 2000, 35 (1 Pt 2): 231-236.
Johnson AD, Kavousi M, Smith AV, Chen MH, Dehghan A, Aspelund T, Lin JP, van Duijn CM, Harris TB, Cupples LA, et al: Genome-wide association meta-analysis for total serum bilirubin levels. Hum Mol Genet. 2009, 18 (14): 2700-2710. 10.1093/hmg/ddp202.
Mibayashi M, Martinez-Sobrido L, Loo YM, Cardenas WB, Gale M: Garcia-Sastre A: Inhibition of retinoic acid-inducible gene I-mediated induction of beta interferon by the NS1 protein of influenza A virus. J Virol. 2007, 81 (2): 514-524. 10.1128/JVI.01265-06.
Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA, Julier C, Morahan G, Nerup J, Nierras C, et al: Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet. 2009, 41 (6): 703-707. 10.1038/ng.381.
Ohnesorge N, Viemann D, Schmidt N, Czymai T, Spiering D, Schmolke M, Ludwig S, Roth J, Goebeler M, Schmidt M: Erk5 activation elicits a vasoprotective endothelial phenotype via induction of Kruppel-like factor 4 (KLF4). J Biol Chem. 2010, 285 (34): 26199-26210. 10.1074/jbc.M110.103127.
Rubinstein M, Idelman G, Plymate SR, Narla G, Friedman SL, Werner H: Transcriptional activation of the insulin-like growth factor I receptor gene by the Kruppel-like factor 6 (KLF6) tumor suppressor protein: potential interactions between KLF6 and p53. Endocrinology. 2004, 145 (8): 3769-3777. 10.1210/en.2004-0173.
Andreoli V, Gehrau RC, Bocco JL: Biology of Kruppel-like factor 6 transcriptional regulator in cell life and death. IUBMB Life. 2010, 62 (12): 896-905. 10.1002/iub.396.
Romacho T, Azcutia V, Vazquez-Bella M, Matesanz N, Cercas E, Nevado J, Carraro R, Rodriguez-Manas L, Sanchez-Ferrer CF, Peiro C: Extracellular PBEF/NAMPT/visfatin activates pro-inflammatory signalling in human vascular smooth muscle cells through nicotinamide phosphoribosyltransferase activity. Diabetologia. 2009, 52 (11): 2455-2463. 10.1007/s00125-009-1509-2.
Weng YJ, Hsieh DJ, Kuo WW, Lai TY, Hsu HH, Tsai CH, Tsai FJ, Lin DY, Lin JA, Huang CY, et al: E4BP4 is a cardiac survival factor and essential for embryonic heart development. Mol Cell Biochem. 2010, 340 (1-2): 187-194. 10.1007/s11010-010-0417-6.
Dehghan A, Yang Q, Peters A, Basu S, Bis JC, Rudnicka AR, Kavousi M, Chen MH, Baumert J, Lowe GD, et al: Association of novel genetic Loci with circulating fibrinogen levels: a genome-wide association study in 6 population-based cohorts. Circ Cardiovasc Genet. 2009, 2 (2): 125-133. 10.1161/CIRCGENETICS.108.825224.
Dehghan A, Dupuis J, Barbalic M, Bis JC, Eiriksdottir G, Lu C, Pellikka N, Wallaschofski H, Kettunen J, Henneman P, et al: Meta-analysis of genome-wide association studies in >80 000 subjects identifies multiple loci for C-reactive protein levels. Circulation. 2011, 123 (7): 731-738. 10.1161/CIRCULATIONAHA.110.948570.
Voight BF, Scott LJ, Steinthorsdottir V, Morris AP, Dina C, Welch RP, Zeggini E, Huth C, Aulchenko YS, Thorleifsson G, et al: Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet. 2010, 42 (7): 579-589. 10.1038/ng.609.
Lauc G, Essafi A, Huffman JE, Hayward C, Knezevic A, Kattla JJ, Polasek O, Gornik O, Vitart V, Abrahams JL, et al: Genomics meets glycomics-the first GWAS study of human N-Glycome identifies HNF1alpha as a master regulator of plasma protein fucosylation. PLoS Genet. 2010, 6 (12): e1001256-10.1371/journal.pgen.1001256.
Dorsam RT, Kunapuli SP: Central role of the P2Y12 receptor in platelet activation. J Clin Invest. 2004, 113 (3): 340-345.
Rauch BH, Rosenkranz AC, Ermler S, Bohm A, Driessen J, Fischer JW, Sugidachi A, Jakubowski JA, Schror K: Regulation of functionally active P2Y12 ADP receptors by thrombin in human smooth muscle cells and the presence of P2Y12 in carotid artery lesions. Arterioscler Thromb Vasc Biol. 2010, 30 (12): 2434-2442. 10.1161/ATVBAHA.110.213702.
Peluso JJ, Liu X, Gawkowska A, Johnston-MacAnanny E: Progesterone activates a progesterone receptor membrane component 1-dependent mechanism that promotes human granulosa/luteal cell survival but not progesterone secretion. J Clin Endocrinol Metab. 2009, 94 (7): 2644-2649. 10.1210/jc.2009-0147.
Alfano D, Franco P, Vocca I, Gambi N, Pisa V, Mancini A, Caputi M, Carriero MV, Iaccarino I, Stoppelli MP: The urokinase plasminogen activator and its receptor: role in cell growth and apoptosis. Thromb Haemost. 2005, 93 (2): 205-211.
Chavakis T, Kanse SM, May AE, Preissner KT: Haemostatic factors occupy new territory: the role of the urokinase receptor system and kininogen in inflammation. Biochem Soc Trans. 2002, 30 (2): 168-173.
Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, Tokino T, Taniguchi T, Tanaka N: Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science. 2000, 288 (5468): 1053-1058. 10.1126/science.288.5468.1053.
Morton E, Macrae IM, McCabe C, Brown SM, White F: Identification of the growth arrest and DNA damage protein GADD34 in the normal human heart and demonstration of alterations in expression following myocardial ischaemia. Int J Cardiol. 2006, 107 (1): 126-129. 10.1016/j.ijcard.2005.01.051.
Cipollone F, Fazia ML: COX-2 and atherosclerosis. J Cardiovasc Pharmacol. 2006, 47 (Suppl 1): S26-S36.
Cipollone F, Toniato E, Martinotti S, Mezzetti A: Genetic and molecular determinants of atherosclerotic plaque instability. Curr Vasc Pharmacol. 2010, 8 (4): 545-552. 10.2174/157016110791330825.
Min SH, Kim DM, Heo YS, Kim YI, Kim HM, Kim J, Han YM, Kim IC, Yoo OJ: New p53 target, phosphatase of regenerating liver 1 (PRL-1) downregulates p53. Oncogene. 2009, 28 (4): 545-554. 10.1038/onc.2008.409.
Claudio JO, Zhu YX, Benn SJ, Shukla AH, McGlade CJ, Falcioni N, Stewart AK: HACS1 encodes a novel SH3-SAM adaptor protein differentially expressed in normal and malignant hematopoietic cells. Oncogene. 2001, 20 (38): 5373-5377. 10.1038/sj.onc.1204698.
Zhu YX, Benn S, Li ZH, Wei E, Masih-Khan E, Trieu Y, Bali M, McGlade CJ, Claudio JO, Stewart AK: The SH3-SAM adaptor HACS1 is up-regulated in B cell activation signaling cascades. J Exp Med. 2004, 200 (6): 737-747. 10.1084/jem.20031816.
Joost HG, Bell GI, Best JD, Birnbaum MJ, Charron MJ, Chen YT, Doege H, James DE, Lodish HF, Moley KH, et al: Nomenclature of the GLUT/SLC2A family of sugar/polyol transport facilitators. Am J Physiol Endocrinol Metab. 2002, 282 (4): E974-E976.
Charniot JC, Sutton A, Bonnefont-Rousselot D, Cosson C, Khani-Bittar R, Giral P, Charnaux N, Albertini JP: Manganese superoxide dismutase dimorphism relationship with severity and prognosis in cardiogenic shock due to dilated cardiomyopathy. Free Radic Res. 2011, 45 (4): 379-388. 10.3109/10715762.2010.532792.
Valdez AC, Cabaniols JP, Brown MJ, Roche PA: Syntaxin 11 is associated with SNAP-23 on late endosomes and the trans-Golgi network. J Cell Sci. 1999, 112 (Pt 6): 845-854.
Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, Lees CW, Balschun T, Lee J, Roberts R, et al: Genome-wide meta-analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nat Genet. 2010, 42 (12): 1118-1125. 10.1038/ng.717.
Chen R, Stahl EA, Kurreeman FA, Gregersen PK, Siminovitch KA, Worthington J, Padyukov L, Raychaudhuri S, Plenge RM: Fine mapping the TAGAP risk locus in rheumatoid arthritis. Genes Immun. 2011, 12 (4): 314-318. 10.1038/gene.2011.8.
Eyre S, Hinks A, Bowes J, Flynn E, Martin P, Wilson AG, Morgan AW, Emery P, Steer S, Hocking LJ, et al: Overlapping genetic susceptibility variants between three autoimmune disorders: rheumatoid arthritis, type 1 diabetes and coeliac disease. Arthritis Res Ther. 2010, 12 (5): R175-10.1186/ar3139.
Esemuede N, Lee T, Pierre-Paul D, Sumpio BE, Gahtan V: The role of thrombospondin-1 in human disease. J Surg Res. 2004, 122 (1): 135-142. 10.1016/j.jss.2004.05.015.
Sargiannidou I, Qiu C, Tuszynski GP: Mechanisms of thrombospondin-1-mediated metastasis and angiogenesis. Semin Thromb Hemost. 2004, 30 (1): 127-136.
Stoll LL, Denning GM, Weintraub NL: Endotoxin, TLR4 signaling and vascular inflammation: potential therapeutic targets in cardiovascular disease. Curr Pharm Des. 2006, 12 (32): 4229-4245. 10.2174/138161206778743501.
An H, Qian C, Cao X: Regulation of Toll-like receptor signaling in the innate immunity. Sci China Life Sci. 2010, 53 (1): 34-43. 10.1007/s11427-010-0011-x.
Perry SV: Vertebrate tropomyosin: distribution, properties and function. J Muscle Res Cell Motil. 2001, 22 (1): 5-49. 10.1023/A:1010303732441.
Waterworth DM, Ricketts SL, Song K, Chen L, Zhao JH, Ripatti S, Aulchenko YS, Zhang W, Yuan X, Lim N, et al: Genetic variants influencing circulating lipid levels and risk of coronary artery disease. Arterioscler Thromb Vasc Biol. 2010, 30 (11): 2264-2276. 10.1161/ATVBAHA.109.201020.
Denny JC, Ritchie MD, Crawford DC, Schildcrout JS, Ramirez AH, Pulley JM, Basford MA, Masys DR, Haines JL, Roden DM: Identification of genomic predictors of atrioventricular conduction: using electronic medical records as a tool for genome science. Circulation. 2010, 122 (20): 2016-2021. 10.1161/CIRCULATIONAHA.110.948828.
Ichimura T, Watanabe S, Sakamoto Y, Aoto T, Fujita N, Nakao M: Transcriptional repression and heterochromatin formation by MBD1 and MCAF/AM family proteins. J Biol Chem. 2005, 280 (14): 13928-13935. 10.1074/jbc.M413654200.
Fu C, Turck CW, Kurosaki T, Chan AC: BLNK: a central linker protein in B cell activation. Immunity. 1998, 9 (1): 93-103. 10.1016/S1074-7613(00)80591-9.
Bantscheff M, Hopf C, Savitski MM, Dittmann A, Grandi P, Michon AM, Schlegl J, Abraham Y, Becher I, Bergamini G, et al: Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat Biotechnol. 2011, 29 (3): 255-265. 10.1038/nbt.1759.
Arming S, Wipfler D, Mayr J, Merling A, Vilas U, Schauer R, Schwartz-Albiez R, Vlasak R: The human Cas1 protein: a sialic acid-specific O-acetyltransferase?. Glycobiology. 2011, 21 (5): 553-564. 10.1093/glycob/cwq153.
Marat AL, McPherson PS: The connecdenn family, Rab35 guanine nucleotide exchange factors interfacing with the clathrin machinery. J Biol Chem. 2010, 285 (14): 10627-10637. 10.1074/jbc.M109.050930.
Akerblad P, Mansson R, Lagergren A, Westerlund S, Basta B, Lind U, Thelin A, Gisler R, Liberg D, Nelander S, et al: Gene expression analysis suggests that EBF-1 and PPARgamma2 induce adipogenesis of NIH-3T3 cells with similar efficiency and kinetics. Physiol Genomics. 2005, 23 (2): 206-216. 10.1152/physiolgenomics.00015.2005.
Boyd RS, Adam PJ, Patel S, Loader JA, Berry J, Redpath NT, Poyser HR, Fletcher GC, Burgess NA, Stamps AC, et al: Proteomic analysis of the cell-surface membrane in chronic lymphocytic leukemia: identification of two novel proteins, BCNP1 and MIG2B. Leukemia. 2003, 17 (8): 1605-1612. 10.1038/sj.leu.2402993.
Davis RS, Wang YH, Kubagawa H, Cooper MD: Identification of a family of Fc receptor homologs with preferential B cell expression. Proc Natl Acad Sci USA. 2001, 98 (17): 9772-9777. 10.1073/pnas.171308498.
Chureau C, Chantalat S, Romito A, Galvani A, Duret L, Avner P, Rougeulle C: Ftx is a non-coding RNA which affects Xist expression and chromatin structure within the X-inactivation center region. Hum Mol Genet. 2011, 20 (4): 705-718. 10.1093/hmg/ddq516.
Fredriksson R, Hoglund PJ, Gloriam DE, Lagerstrom MC, Schioth HB: Seven evolutionarily conserved human rhodopsin G protein-coupled receptors lacking close relatives. FEBS Lett. 2003, 554 (3): 381-388. 10.1016/S0014-5793(03)01196-7.
Tano K, Mizuno R, Okada T, Rakwal R, Shibato J, Masuo Y, Ijiri K, Akimitsu N: MALAT-1 enhances cell motility of lung adenocarcinoma cells by influencing the expression of motility-related genes. FEBS Lett. 2010, 584 (22): 4575-4580. 10.1016/j.febslet.2010.10.008.
Favre J, Gao J, Zhang AD, Remy-Jouet I, Ouvrard-Pascaud A, Dautreaux B, Escoubet B, Thuillez C, Jaisser F, Richard V: Coronary endothelial dysfunction after cardiomyocyte-specific mineralocorticoid receptor overexpression. Am J Physiol Heart Circ Physiol. 2011, 300 (6): H2035-H2043. 10.1152/ajpheart.00552.2010.
Aksoy P, Zhu MJ, Kalari KR, Moon I, Pelleymounter LL, Eckloff BW, Wieben ED, Yee VC, Weinshilboum RM, Wang L: Cytosolic 5'-nucleotidase III (NT5C3): gene sequence variation and functional genomics. Pharmacogenet Genomics. 2009, 19 (8): 567-576. 10.1097/FPC.0b013e32832c14b8.
Jiang Y, Scott KL, Kwak SJ, Chen R, Mardon G: Sds22/PP1 links epithelial integrity and tumor suppression via regulation of myosin II and JNK signaling. Oncogene. 2011, 30 (29): 3248-3260. 10.1038/onc.2011.46.
Callebaut I, de Gunzburg J, Goud B, Mornon JP: RUN domains: a new family of domains involved in Ras-like GTPase signaling. Trends Biochem Sci. 2001, 26 (2): 79-83. 10.1016/S0968-0004(00)01730-8.
Wen J, Huang S, Rogers H, Dickinson LA, Kohwi-Shigematsu T, Noguchi CT: SATB1 family protein expressed during early erythroid differentiation modifies globin gene expression. Blood. 2005, 105 (8): 3330-3339. 10.1182/blood-2004-08-2988.
Massague J: TGF-beta signal transduction. Annu Rev Biochem. 1998, 67: 753-791. 10.1146/annurev.biochem.67.1.753.
Bednarek AK, Keck-Waggoner CL, Daniel RL, Laflin KJ, Bergsagel PL, Kiguchi K, Brenner AJ, Aldaz CM: WWOX, the FRA16D gene, behaves as a suppressor of tumor growth. Cancer Res. 2001, 61 (22): 8068-8073.
Acknowledgements
This study was supported by a Marriot Award for Individualized Medicine. We acknowledge Angie Dalenberg, Advanced Genomic Technology Center and Microarray Shared Resource at Mayo Clinic for technical assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
RM performed the experiments and contributed to the analysis. KS performed the analysis. AD contributed to the experiments. KD contributed to the analysis. KS and IJK wrote the manuscript with contributions from other authors. IJK conceived the study and provided critical input. All authors read and approved the final manuscript.
Rizwan Masud, Khader Shameer contributed equally to this work.
Electronic supplementary material
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Masud, R., Shameer, K., Dhar, A. et al. Gene expression profiling of peripheral blood mononuclear cells in the setting of peripheral arterial disease. J Clin Bioinform 2, 6 (2012). https://doi.org/10.1186/2043-9113-2-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2043-9113-2-6