
Abstract
Background
DNA methylation is strongly associated with smoking status at multiple sites across the genome. Studies have largely been restricted to European origin individuals yet the greatest increase in smoking is occurring in low income countries, such as the Indian subcontinent. We determined whether there are differences between South Asians and Europeans in smoking related loci, and if a smoking score, combining all smoking related DNA methylation scores, could differentiate smokers from non-smokers.
Results
Illumina HM450k BeadChip arrays were performed on 192 samples from the Southall And Brent REvisited (SABRE) cohort. Differential methylation in smokers was identified in 29 individual CpG sites at 18 unique loci. Interaction between smoking status and ethnic group was identified at the AHRR locus. Ethnic differences in DNA methylation were identified in non-smokers at two further loci, 6p21.33 and GNG12. With the exception of GFI1 and MYO1G these differences were largely unaffected by adjustment for cell composition. A smoking score based on methylation profile was constructed. Current smokers were identified with 100% sensitivity and 97% specificity in Europeans and with 80% sensitivity and 95% specificity in South Asians.
Conclusions
Differences in ethnic groups were identified in both single CpG sites and combined smoking score. The smoking score is a valuable tool for identification of true current smoking behaviour. Explanations for ethnic differences in DNA methylation in association with smoking may provide valuable clues to disease pathways.
Similar content being viewed by others


Background
Smoking associated death and disability remains a major public health problem in high income countries, despite marked declines in smoking rates, and is escalating rapidly in low to middle income countries, where tobacco consumption is increasing [1]. While global smoking cessation is the ultimate goal, understanding the mechanisms by which smoking causes its adverse effects in the interim may provide valuable therapeutic targets.
Smoking is an exposure strongly associated with DNA methylation in a distinct set of loci which not only clearly distinguish between current and never smokers, but may also reflect the cumulative amount smoked, and time since quitting in former smokers [2–8]. Some of these loci are located in characterised genes where the potential molecular pathway in response to smoking is relatively well understood, such as the AHRR gene [9, 10]. Methylation at smoking associated loci has also been related to clinical outcomes; for example, F2RL3 methylation is strongly associated with mortality in coronary heart disease patients [11] and AHRR methylation has been investigated in lung cancer patients [6].
Previous research has almost exclusively been performed in European origin populations, and may not extrapolate to other ethnic groups, such as South Asians, where escalating rates of tobacco consumption will impact adversely on an already elevated susceptibility to cardio-metabolic disease [12]. We and others report differences in DNA methylation between South Asians and Europeans [13, 14], though whether these are associated with smoking behaviour is unknown. Potential ethnic differences may be due to different smoking behaviours, or differences in molecular mechanisms which are important to identify and explore.
Our primary aim was therefore to determine whether there are differences in DNA methylation patterns in association with smoking between people of South Asian and European origin and to explore whether any differences observed could be explained by ethnic specific smoking behaviours. This was carried out using the Illumina HumanMethylation450 BeadChip array in samples from the Southall And Brent REvisited (SABRE) cohort.
Secondly, we explored the potential use of a recently published epigenome-wide catalogue of smoking related methylation loci to characterise smoking behaviour in this bi-ethnic sample using a scoring method based on methylation data [7]. Methylation derived scores have previously been used successfully using bisulphite pyrosequencing data to identify former smokers [8]. These methods could potentially replace and/or provide greater precision to self-reported smoking habits where under-reporting is commonplace [15, 16] and where measuring smoking via other methods (for example, by plasma or salivary cotinine) may be difficult or less informative, for example, when attempting to quantify historical behaviours.
Results and discussion
Smoking behaviour in the SABRE cohort
By design, mean age and proportions of men in different smoking categories did not differ by ethnicity (Table 1). European current smokers smoked more heavily per day, started smoking earlier and therefore had a greater number of pack years smoked than South Asians. However, time since cessation of smoking for former smokers was similar by ethnic group (see Table 1 for test statistics).
Smoking associated loci in the SABRE cohort
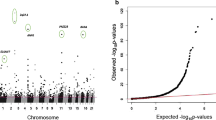
Differential methylation in current smokers was identified in 29 individual CpG sites at 18 unique loci at P ≤1.1 × 10-7 after applying conservative family wise error rate correction based on the number of tests conducted and α = 0.05 (Figure 1). At 12 loci, the effect size (calculated as median % methylation difference) was greater than 5% (Table 2, summary and test statistics for all CpG sites in Additional file 1). At each locus, the sentinel methylation site was defined as the locus CpG site with the smallest P-value test statistic.

Manhattan plot showing association between current and never tobacco smoking and genome-wide DNA methylation. The continuous line marks the P ≤1.1 × 10-7 significance threshold. CpG sites with corresponding P-values at ≤1.1 × 10-7 are colour coded to show the direction of difference between smokers and non-smokers. Red CpG sites are hypermethylated in current smokers while blue CpG sites are hypomethylated.
The highest ranking hit was the AHRR locus, with a median decrease in methylation of 22% in smokers, confirming both the locus and magnitude of effect observed in previous studies. Other loci identified (GNG12, GFI1, ALPPL2, 3p24.3, PDZD2, 6p21.33, VARS, MYO1G, C14orf43, F2RL3) were also concordant with published data [4, 6, 7]. The 11th ranked locus, within TMEM51 (cg09069072) with a decrease in methylation in current smokers of 6%, has been identified in just one previous study [7].
The majority of smoking associated loci appeared to be robust to adjustment for cell composition within samples (see Additional file 1 for test statistics). Notable exceptions included GFI1 and MYO1G. In initial analysis of GFI1, the median methylation level was 27% in smokers and 57% in never smokers (F = 47.85, P = 1.0 × 10-10). Following cell composition adjustment, the median methylation level was 45% in smokers and 58% in never smokers (F = 0.35, P = 5.6 × 10-1). In initial analysis of MYO1G, the median methylation level was 95% in smokers and 83% in never smokers (F = 89.85, P = 3.1 × 10-17), while following cell composition adjustment the median methylation level was 86% in both smokers and never smokers (F = 23.18, P = 3.4 × 10-6). The results for these loci may therefore be a consequence of smoking-related changes in leukocyte number, differentials and/or inflammatory mediators [17].
Association between methylation and detailed characterisation of smoking behaviour
When testing the association between number of cigarettes smoked per day or pack years with methylation at each of the sentinel CpG sites, methylation at only one site, cg06126421, was associated with number of cigarettes smoked per day in current smokers (0.058 unit decrease in methylation M-value per additional cigarette smoked per day (95% CI: -0.094, -0.022; P = 2.0 × 10-03)) or pack years (0.034 unit decrease in methylation M-value per additional pack year smoked (95% CI: -0.054, -0.015; P = 1.0 × 10-03)). No CpG sites were associated with age at which an individual started or quit smoking after adjusting for multiple testing.
Ethnic differences: interaction
In order to assess differences across ethnic groups, analyses were initially performed across all CpG sites stratified by ethnic group. Although no additional loci were identified following this analysis, differences in effect sizes at the 12 sentinel CpG sites were observed between the two ethnic groups. Summary and test statistics for sentinel CpG sites stratified by ethnic group are shown in Table 3 and Additional file 1, including analyses adjusting for cell composition. Ethnic differences in the associations between smoking and methylation at the 12 sentinel CpG sites were therefore assessed further.
An interaction between ethnic group and smoking status was observed in AHRR CpG site cg05575921 after applying a conservative family-wise error rate correction for 12 tests at α = 0.05 (P ≤0.004) (F = 10.42, P = 1.0 × 10-3). No ethnic differences in methylation were observed between never smokers at this CpG site (t-test: n = 129, P = 0.44). Among current smokers the median methylation level was 53% in Europeans and 57% in South Asians (t-test: n = 36, P = 2.0 × 10-3) (Figure 2).

Plot showing interaction between ethnic group and smoking at AHRR cg05575921. Bars show mean DNA methylation levels in each group shown. Error bars represent standard deviations. Ethnic differences were observed between current smokers (t-test: n = 36, P = 2.0 × 10-3) but not between never smokers (t-test: n = 129, P = 0.44).
Heavier smoking in Europeans could account for their lower methylation scores. However, there was no relationship between methylation at the AHRR sentinel CpG site and pack years smoked (unadjusted linear regression, 0.004 unit decrease in methylation M-value per additional pack year smoked (95% CI: -0.010, 0.003; P = 0.24)) or number of cigarettes smoked per day (unadjusted linear regression, 0.006 unit decrease in methylation M-value per additional cigarette smoked per day (95% CI: -0.017, 0.006; P = 0.34)). Adjusting for the number of cigarettes smoked per day in the model did not change the estimate or P-value, indicating that measured smoking behaviour could not account for our observed ethnic differences in methylation. However, there may be other measures of smoking heaviness; for example, the brand of cigarette smoked and degree of inhalation, which we did not capture, which may contribute to ethnic differences in methylation score.
In other non-sentinel AHRR CpG sites for which differential methylation with smoking was observed (n = 8), two further CpG sites also showed an interaction between ethnic group and smoking status: cg21161138 (F = 9.48, P = 2.0 × 10-03) and cg25648203 (F = 7.72, P = 6.0 × 10-03). This provides further support for a true interaction between ethnicity and smoking status at this locus.
However, when repeating this analysis on AHRR sentinel CpG site cg05575921 and adjusting for cell composition this observation is attenuated (F = 6.08, P = 1.5 × 10-2). This suggests that the interaction between ethnicity and smoking that is mediated at least in part through an effect on cell composition or the mechanisms responsible for the change in cell composition at this locus.
Other sentinel CpG sites were also approaching significance for interaction after adjustment for multiple testing (see Additional file 2) and in all cases Europeans showed larger effect sizes when comparing never and current smokers (See Table 3).
Ethnic differences-main effects
Following analyses for interactions, the main effects of ethnic group were also assessed. Main effects of ethnic group existed in 2 of the 12 loci: 6p21.33 (cg06126421, n = 165, F = 32.82, P = 4.9 × 10-8) and GNG12 (cg25189904, n = 165, F = 17.94, P = 3.8 × 10-5). At both of these CpG sites methylation differences were observed between never smokers of the two ethnic groups (t-test: cg06126421; -8.71% in Europeans, n = 129, t = -4.35, P-value = 2.8 × 10-5 and cg25189904; -4.34% in Europeans, t = -3.08, P-value = 3.0 × 10-5). When repeating analysis on data adjusted for cell composition, the main effect at 6p21.33 was attenuated slightly (F = 10.96, P = 1.2 × 10-3) but the main effect at GNG12 was not (F = 17.94, P = 3.8 × 10-5), suggesting that differences in cell composition do not wholly account for the differences observed at these loci.
This finding indicates that at some loci ethnic differences exist independently of self-reported smoking status and appear to be unrelated to cell composition. The source of ethnic differences in methylation at these loci is unknown. One potential source could be population specific local mQTLs, such as described in previous studies [18, 19], causing underlying ethnic differences in DNA methylation independently of smoking exposure. Another potential source could be cultural or environmental factors not captured in this study; for example, if a higher proportion of European never-smokers have unmeasured passive smoking exposure, this may have contributed to the observed differences in methylation patterns. Variation in diet between ethnic groups could also contribute to the differences observed. In either case this highlights the need for appropriate sample selection and accounting for ethnic group in future studies.
Using methylation scores to predict current smoking status in Europeans and South Asians
Smoking scores were calculated for each SABRE individual from whom methylation data had been measured (n = 189).
In Europeans, smokers and never smokers had clearly distinct scores (see Figure 3a).

Plots of smoking score by reported smoking category in Europeans and South Asians. Box and whisker plots show median and interquartile ranges. Filled black circles show individual data points. Red line indicates threshold score above which individuals were considered to be current smokers.
Random Forests were used to identify the threshold score separating smokers and never smokers and to assess misclassification. The average threshold score separating the two groups using 500 trees was 17.55. This threshold detected smokers from never and former smokers with 100% sensitivity and 97% specificity.
In Europeans, the majority of former smokers had scores <17.55 and were indistinguishable from never smokers using this classification. This finding is in line with published research observing that methylation levels in former smokers revert to levels similar to never smokers over time [3, 7]. Smoking score was therefore not a good identification tool for former smokers who, in the SABRE cohort, had quit smoking for an average of 12.9 years. Shenker and colleagues recently used bisulphite pyrosequencing data from four loci (AHRR, 6p21 and two at 2q37) to differentiate between never and former smokers, establishing that their method worked favourably compared to cotinine measurement [8]. This suggests a small subset of smoking associated loci where methylation levels may be slower to revert to levels similar to never smokers. If this is the case, methylation of these loci may be useful for detection of former smoking behaviour. Further comparison in larger numbers of former and never smokers is needed to investigate this possibility.
Two former smokers had smoking scores very close to current smoker levels. We hypothesise that these individuals may still smoke or live in environments where they are exposed to substantial amounts of passive smoke. This finding underlines the need for an objective measure of smoking status for precise classification in epidemiological studies, to overcome misreporting bias.
In South Asians, current smoking behaviour was more difficult to distinguish (Figure 3b). Random Forests were used to identify the threshold score separating smokers and never smokers. The average threshold score separating the two groups using 500 trees was 11.79. This threshold smoking score discriminated smokers from never and former smokers with 80% sensitivity and 95% specificity. Similarly to Europeans, former smokers had smoking scores approaching those of never smokers.
Applying the European calculated threshold for current smoking behaviour to the South Asian component of the cohort would have altered sensitivity and specificity for detection of current smoking in South Asians to 50% and 100%, respectively. Vice versa, sensitivity and specificity would have been altered to 100% and 89%, respectively. This highlights differences in methylation score profiles in South Asians and Europeans and implies that smoking score and smoking behaviour may be related.
Relationship between smoking score and smoking behaviour
To assess whether a smoking score representing overall methylation pattern was associated with smoking behaviour, linear regression models were constructed.
When assessing the relationship between smoking score and the number of cigarettes smoked per day, there was an interaction between number of cigarettes smoked per day and ethnic group (P = 3.38 × 10-3). Similarly, an interaction between pack years and ethnic group was also identified (P = 1.18 × 10-2). For this reason, the relationships between these smoking behaviours and smoking score were stratified by ethnic group.
In South Asians, a one unit increase in smoking score was associated with a 0.54 increase in number of cigarettes smoked per day (95% CI: 0.15, 0.93; P = 0.01), see Figure 4a. A one unit increase in score was weakly associated with a 0.29 increase in pack years (95% CI: 0.002, 0.571, P = 0.048), see Figure 4b. In Europeans, smoking score was not associated with number of cigarettes smoked per day or pack years. One reason smoking score was not related to smoking behaviour measures in SABRE Europeans is because scores amongst SABRE Europeans were far more homogeneous than their South Asian counterparts and we were therefore underpowered to detect such associations (see Figures 4a, b).

Plots of associations between smoking score and smoking behaviour in Europeans and South Asians. Filled circles show individual data points coloured by ethnic group (black = European, red = South Asian). R-squared values given are for regression models predicting smoking behaviour using smoking score in each ethnic group.
Smoking score was not associated with years since quitting (former smokers) or age at which smoking began (current smokers) after adjusting for ethnic group and no interaction between smoking behaviour (years since quitting or age at which smoking began) and ethnic group was observed.
The threshold score used to categorise current smoking in South Asians was much lower than for Europeans. The association between smoking score and measures of smoking behaviour in South Asians adds support to the hypothesis that differences in smoking effects are likely to be attributable to the lighter use of cigarettes amongst South Asian smokers. This finding is of interest to other cohorts who may wish to use methylation smoking score to categorise current smokers.
Conclusions
DNA methylation loci responsive to smoking were similar in South Asians and Europeans, but for some loci the degree of methylation differed markedly. Methylation at the AHRR locus is significantly lower in European origin individuals than South Asians. Although Europeans reported heavier smoking than South Asians and part of the differences observed appeared to be related to differences in cell composition, these factors could not completely account for their lower scores, suggesting that either key aspects of smoking behaviour had not been captured, or that there is a true ethnic difference in methylation response to smoking.
Ethnic differences in non-smokers at two smoking associated loci were identified (cg06126421 and cg25189904), where differences in methylation occurred between current and never smokers. Differences observed between ethnic groups in never smokers highlights differences not attributable to smoking and could be driven by underlying genetic variation or could be associated with other un-captured environmental differences. This finding indicates the need to take account of ethnic origin in future research in this area.
Adjustment of methylation data for cell composition using a method constructed by Houseman et al. allowed the impact of cellular composition within the collected samples to be assessed. It was noted that a subset of loci associated with smoking may arise from differences in cell composition between smokers and never smokers.
We also identified that an ethnic specific smoking score derived from smoking related methylation profiles is a valid marker for current tobacco exposure in both South Asians and Europeans, offering a precise measure of smoking status that is not prone to reporting bias, and, therefore, of considerable value when attempting to dissect the true association between smoking and smoking related outcomes.
Methods
Cohort information
SABRE is a population based cohort including 1,711 first generation South Asian migrants and 1,762 people of European origin aged 40 to 69 living in West London, UK [20]. Baseline investigations were performed between 1988 and 1991. Peripheral blood samples were collected at baseline visits for DNA extraction.
The current analysis was restricted to men only (as previous studies have reported sex differences in smoking associated methylation signals, and few South Asian women smoked), and excluded all those with existing chronic disease, specifically cardiovascular disease and diabetes, restricted to those aged 40 to 55 years at baseline to avoid the confounding effects of ageing on methylation patterns and to those who provided good quality DNA samples. A random sample of 192 men were selected, stratified by ethnicity, four-year age group, and smoking status (current, former or never).
Ethnicity in the SABRE cohort was interviewer-recorded based on parental origins and appearance and was subsequently confirmed by participants. Half of the samples selected in the current study were of European origin born in mainland UK (England n = 89; Wales n = 3; Scotland n = 4). The remaining participants were of South Asian origin who indicated their region of origin was North India (n = 92) or Pakistan (n = 4). Smoking status was recorded by questionnaire. Participants reported the age at which they began smoking and the year they stopped if they had quit. The number of cigarettes smoked per day was also recorded which allowed pack years of smoking to be calculated using the formula: (cigarettes per day/20) * number of years smoked.
All participants gave written informed consent. Approval for the baseline study was obtained from Ealing, Hounslow and Spelthorne, Parkside, and University College London research ethics committees.
HumanMethylation450 BeadChip arrays
Genomic DNA (500 ng) was bisulphite modified using an EZ DNA methylation kit (Zymo Research, Orange, CA, USA). The protocol was as described by the manufacturer, utilising the alternative incubation conditions recommended when using Illumina Infinium Methylation Arrays. Genome-wide methylation was measured using the Illumina HumanMethylation450 BeadChip (Illumina, San Diego, CA, USA) following the manufacturer’s protocol with no modifications. The arrays were scanned using an Illumina iScan with software version 3.3.28.
Pre-processing of methylation data
Initial quality control of sample data was conducted using GenomeStudio version 2011.1 (Illumina, San Diego, CA, USA) to determine the status of staining, extension, hybridisation, target removal, bisulphite conversion, specificity, non-polymorphic and negative controls. Samples that did not pass this stage of quality control were excluded from further analysis (n = 3).
Data were pre-processed using the pipeline described in Touleimat and Tost [21], with additional modifications [21]. All probes were represented by more than three beads and all samples contained >95% of signals that were detectable from background signal (probe signal detection P-value <0.01). In addition to these quality control steps implemented by the pipeline, probes that contained <95% of signals detectable above background signal (detection P-value <0.01) (n = 9,769) and probes with multiple homology (n = 25,083) (see Additional file 3) were excluded.
Following pre-processing, the percentage of methylation present in the cell population at any given methylation site is reported as a β-value. This is a continuous value bounded by 0 and 1 which corresponds to the ratio of the methylated signal divided by the sum of the methylated and unmethylated signals. In statistical models, β-values were transformed using a variance stabilisation transformation to methylation M-values [22]. A second dataset was also generated containing data further adjusted for differences in cell composition, achieved utilising the method described by Houseman et al. [23]. This allowed the effect of cell composition to be evaluated. Prior to implementation of statistical models, M-values were adjusted to remove batch effects using ComBat [24], where each BeadChip was considered to be one batch. ComBat was not utilised in data used to generate scores. For ease of interpretation, data shown throughout are in the form of methylation β-values.
Using methylation scores to predict smoking status in Europeans and South Asians
Weights and reference data used to calculate scores utilised data published by Zeilinger et al. [7]. Use of data from a second cohort minimised over-fitting. Data from this paper were used as it is currently the most comprehensive list of validated smoking associated CpG sites.
Weighted methylation scores were calculated utilising data from 183 CpG sites previously associated with smoking [7]. Three additional CpG sites reported by Zeilinger et al. did not pass quality control measures in the SABRE cohort.
Calculating weights
Effect sizes from discovery and replication cohorts were taken from supplementary Table 2, published by Zeilinger et al. [7]. Weights were calculated as absolute values: per CpG effect size/average effect size for all measured CpG sites.
Calculating scores
Median methylation values of never smokers taken from previously reported data were used as reference values (supplementary Table 2, as above [7]).
Smoking scores were first calculated for each CpG site. For CpG sites associated with increased methylation levels in smokers, smoking scores were calculated as: (SABRE cohort beta values – reference beta values) *weight. For CpG sites associated with decreased methylation levels in smokers, smoking scores were calculated as: (reference beta values – cohort beta values)*weight. The final weighted score was calculated as the sum of all CpG site scores.
Analysis
Baseline characteristics comparing South Asians and Europeans were compared using the t-test for continuous, and chi-squared for categorical variables. ANOVA was used to identify associations between methylation and smoking status in smokers and never smokers, where methylation M-values were the outcome variables, smoking category represented the predictor variable and ethnic group was included as a covariate. To determine whether there were ethnic differences in the association between smoking and methylation score we included an interaction term smoking status * ethnic group in the model. Linear regression models were used to assess associations between methylation and smoking behaviours and between smoking score and smoking behaviours.
Estimated power to detect 5% methylation difference between smokers and never smokers assuming a conservative standard deviation estimate of 4% in each group and n = 165 (36 current and 65 never smokers) was 90.72% at P = 1.1 × 10-7.
All analyses were conducted in R, version 3.0.0 (http://www.r-project.org). The following packages were utilised: base, stats, lumi methylumi, CpGassoc, sva and RandomForest [25–29].
Abbreviations
- mQTL:
-
methylation quantitative trait locus
- SABRE:
-
Southall And Brent REvisited
- SNP:
-
Single Nucleotide Polymorphism.
References
Lim SS, Vos T, Flaxman AD, Danaei G, Shibuya K, Adair-Rohani H, Amann M, Anderson HR, Andrews KG, Aryee M, Atkinson C, Bacchus LJ, Bahalim AN, Balakrishnan K, Balmes J, Barker-Collo S, Baxter A, Bell ML, Blore JD, Blyth F, Bonner C, Borges G, Bourne R, Boussinesq M, Brauer M, Brooks P, Bruce NG, Brunekreef B, Bryan-Hancock C, Bucello C: A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: a systematic analysis for the global burden of disease study 2010. Lancet. 2012, 380: 2224-2260. 10.1016/S0140-6736(12)61766-8.
Breitling LP, Yang R, Korn B, Burwinkel B, Brenner H: Tobacco-smoking-related differential DNA methylation: 27K discovery and replication. Am J Hum Genet. 2011, 88: 450-457. 10.1016/j.ajhg.2011.03.003.
Wan ES, Qiu W, Baccarelli A, Carey VJ, Bacherman H, Rennard SI, Agusti A, Anderson W, Lomas DA, Demeo DL: Cigarette smoking behaviors and time since quitting are associated with differential DNA methylation across the human genome. Hum Mol Genet. 2012, 21: 3073-3082. 10.1093/hmg/dds135.
Joubert BR, Haberg SE, Nilsen RM, Wang X, Vollset SE, Murphy SK, Huang Z, Hoyo C, Midttun O, Cupul-Uicab LA, Ueland PM, Wu MC, Nystad W, Bell DA, Peddada SD, London SJ: 450K epigenome-wide scan identifies differential DNA methylation in newborns related to maternal smoking during pregnancy. Environ Health Perspect. 2012, 120: 1425-1431. 10.1289/ehp.1205412.
Philibert RA, Beach SR, Brody GH: Demethylation of the aryl hydrocarbon receptor repressor as a biomarker for nascent smokers. Epigenetics. 2012, 7: 1331-1338. 10.4161/epi.22520.
Shenker NS, Polidoro S, van Veldhoven K, Sacerdote C, Ricceri F, Birrell MA, Belvisi MG, Brown R, Vineis P, Flanagan JM: Epigenome-wide association study in the European prospective investigation into cancer and nutrition (EPIC-Turin) identifies novel genetic loci associated with smoking. Hum Mol Genet. 2013, 22: 843-851. 10.1093/hmg/dds488.
Zeilinger S, Kuhnel B, Klopp N, Baurecht H, Kleinschmidt A, Gieger C, Weidinger S, Lattka E, Adamski J, Peters A, Strauch K, Waldenberger M, Illig T: Tobacco smoking leads to extensive genome-wide changes in DNA methylation. PLoS One. 2013, 8: e63812-10.1371/journal.pone.0063812.
Shenker NS, Ueland PM, Polidoro S, van Veldhoven K, Ricceri F, Brown R, Flanagan JM, Vineis P: DNA methylation as a long-term biomarker of exposure to tobacco smoke. Epidemiology. 2013, 24: 712-716. 10.1097/EDE.0b013e31829d5cb3.
Chavan H, Krishnamurthy P: Polycyclic aromatic hydrocarbons (PAHs) mediate transcriptional activation of the ATP binding cassette transporter ABCB6 gene via the aryl hydrocarbon receptor (AhR). J Biol Chem. 2012, 287: 32054-32068. 10.1074/jbc.M112.371476.
Ge C, Ye J, Wang Q, Zhang C, Yang JM, Qian G: Polycyclic aromatic hydrocarbons suppress meiosis in primordial germ cells via the AHR signaling pathway. Toxicol Lett. 2012, 210: 285-292. 10.1016/j.toxlet.2012.02.014.
Breitling LP, Salzmann K, Rothenbacher D, Burwinkel B, Brenner H: Smoking, F2RL3 methylation, and prognosis in stable coronary heart disease. Eur Heart J. 2012, 33: 2841-2848. 10.1093/eurheartj/ehs091.
Jafar TH, Haaland BA, Rahman A, Razzak JA, Bilger M, Naghavi M, Mokdad AH, Hyder AA: Non-communicable diseases and injuries in Pakistan: strategic priorities. Lancet. 2013, 381: 2281-2290. 10.1016/S0140-6736(13)60646-7.
Elliott HR, Walia GK, Duggirala A, Groom A, Reddy SU, Chandak GR, Gupta V, Laakso M, Dekker JM, Walker M, Ebrahim S, Smith GD, Relton CL, The RISC Consortium: Migration and DNA methylation: a comparison of methylation patterns in type 2 diabetes susceptibility genes between Indians and Europeans. J Diabetes Res Clin Metabol. 2013, 2:
Fraser HB, Lam LL, Neumann SM, Kobor MS: Population-specificity of human DNA methylation. Genome Biol. 2012, 13: R8-10.1186/gb-2012-13-2-r8.
Spencer K, Cowans NJ: Accuracy of self-reported smoking status in first trimester aneuploidy screening. Prenat Diagn. 2013, 33: 245-250. 10.1002/pd.4053.
Dietz PM, Homa D, England LJ, Burley K, Tong VT, Dube SR, Bernert JT: Estimates of nondisclosure of cigarette smoking among pregnant and nonpregnant women of reproductive age in the United States. Am J Epidemiol. 2011, 173: 355-359. 10.1093/aje/kwq381.
Jensen EJ, Pedersen B, Frederiksen R, Dahl R: Prospective study on the effect of smoking and nicotine substitution on leucocyte blood counts and relation between blood leucocytes and lung function. Thorax. 1998, 53: 784-789. 10.1136/thx.53.9.784.
Bell JT, Pai AA, Pickrell JK, Gaffney DJ, Pique-Regi R, Degner JF, Gilad Y, Pritchard JK: DNA methylation patterns associate with genetic and gene expression variation in HapMap cell lines. Genome Biol. 2011, 12: R10-10.1186/gb-2011-12-1-r10.
Gibbs JR, van der Brug MP, Hernandez DG, Traynor BJ, Nalls MA, Lai SL, Arepalli S, Dillman A, Rafferty IP, Troncoso J, Johnson R, Zielke HR, Ferrucci L, Longo DL, Cookson MR, Singleton AB: Abundant quantitative trait loci exist for DNA methylation and gene expression in human brain. PLoS Genet. 2010, 6: e1000952-10.1371/journal.pgen.1000952.
Tillin T, Forouhi NG, McKeigue PM, Chaturvedi N: Southall and Brent REvisited: cohort profile of SABRE, a UK population-based comparison of cardiovascular disease and diabetes in people of European, Indian Asian and African Caribbean origins. Int J Epidemiol. 2012, 41: 33-42. 10.1093/ije/dyq175.
Touleimat N, Tost J: Complete pipeline for infinium((R)) human methylation 450K BeadChip data processing using subset quantile normalization for accurate DNA methylation estimation. Epigenomics. 2012, 4: 325-341. 10.2217/epi.12.21.
Du P, Zhang X, Huang CC, Jafari N, Kibbe WA, Hou L, Lin SM: Comparison of beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics. 2010, 11: 587-10.1186/1471-2105-11-587.
Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, Wiencke JK, Kelsey KT: DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012, 13: 86-10.1186/1471-2105-13-86.
Johnson WE, Li C, Rabinovic A: Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007, 8: 118-127. 10.1093/biostatistics/kxj037.
Du P, Kibbe WA, Lin SM: lumi: a pipeline for processing Illumina microarray. Bioinformatics. 2008, 24: 1547-1548. 10.1093/bioinformatics/btn224.
Davis S, Du P, Bilke S, Triche T, Bootwalla M: methylumi: Handle Illumina methylation data. 2013, In R package 2.6.1. [http://www.bioconductor.org/packages/2.14/bioc/html/methylumi.html]
Barfield RT, Kilaru V, Smith AK, Conneely KN: CpGassoc: an R function for analysis of DNA methylation microarray data. Bioinformatics. 2012, 28: 1280-1281. 10.1093/bioinformatics/bts124.
Leek JT, Storey JD: A general framework for multiple testing dependence. Proc Natl Acad Sci U S A. 2008, 105: 18718-18723. 10.1073/pnas.0808709105.
Breiman L: Random forests. Machine Learn. 2001, 45: 5-32. 10.1023/A:1010933404324.
Acknowledgements
The SABRE study was funded at baseline by the Medical Research Council, Diabetes UK, and the British Heart Foundation. At follow-up the study was funded by the Wellcome Trust and the British Heart Foundation.
Methylation analysis in the SABRE cohort was supported by a Wellcome Trust Enhancement grant 082464/Z/07/C.
HRE is supported by an Oak Foundation post-doctoral research fellowship award.
CLR, GDS and HRE are members of the MRC Integrative Epidemiology Unit at the University of Bristol, supported by funds from the Medical Research Council.
NC and ADH received support from a Biomedical Research Centre Award to Imperial NHS Healthcare Trust.
The funders played no role in the study design and conduct, or in these analyses or the decision to submit the manuscript for publication. The SABRE study group is entirely independent from the funding bodies.
The authors acknowledge Kate Potter (Newcastle University) for advice on statistical analysis.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
HRE participated in the design of the study, carried out the statistical analysis and drafted the manuscript. HRE, WLM, AD and KH carried out the laboratory work for the methylation arrays. NC, ADH, TT, TMF, CLR and GDS conceived of the study, participated in its design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
13148_2013_71_MOESM1_ESM.xlsx
Additional file 1:A table listing summary and test statistics for all CpG sites associated with smoking in SABRE.(XLSX 27 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Elliott, H.R., Tillin, T., McArdle, W.L. et al. Differences in smoking associated DNA methylation patterns in South Asians and Europeans. Clin Epigenet 6, 4 (2014). https://doi.org/10.1186/1868-7083-6-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1868-7083-6-4