
Abstract
Our understanding of the pathogenesis of "tendinopathy" is based on fragmented evidences like pieces of a jigsaw puzzle. We propose a "failed healing theory" to knit these fragments together, which can explain previous observations. We also propose that albeit "overuse injury" and other insidious "micro trauma" may well be primary triggers of the process, "tendinopathy" is not an "overuse injury" per se. The typical clinical, histological and biochemical presentation relates to a localized chronic pain condition which may lead to tendon rupture, the latter attributed to mechanical weakness. Characterization of pathological "tendinotic" tissues revealed coexistence of collagenolytic injuries and an active healing process, focal hypervascularity and tissue metaplasia. These observations suggest a failed healing process as response to a triggering injury. The pathogenesis of tendinopathy can be described as a three stage process: injury, failed healing and clinical presentation. It is likely that some of these "initial injuries" heal well and we speculate that predisposing intrinsic or extrinsic factors may be involved. The injury stage involves a progressive collagenolytic tendon injury. The failed healing stage mainly refers to prolonged activation and failed resolution of the normal healing process. Finally, the matrix disturbances, increased focal vascularity and abnormal cytokine profiles contribute to the clinical presentations of chronic tendon pain or rupture. With this integrative pathogenesis theory, we can relate the known manifestations of tendinopathy and point to the "missing links". This model may guide future research on tendinopathy, until we could ultimately decipher the complete pathogenesis process and provide better treatments.
Similar content being viewed by others

Introduction
In the past decades, our concepts on chronic tendon pain have evolved from "tendinitis" which focused on clinical inflammatory signs, into "tendinosis" which stressed the pathologic features of the free tendon as observed by histology and biochemistry, and then "tendinopathy" which declared nothing further about its nature, just introducing a new label for chronic tendon and insertion problems in general [1]. Woo and Renstrom [2] concludes that the pathogenesis, etiology and mechanisms in most of the myriads of conditions related to tendinopathy are unknown. However, with clear definitions outlining and discriminating the various diagnoses of "tendinopathy", it is still possible to propose a unified model for the pathogenesis based on available experimental evidences, which we propose as our theory to be proved or rejected by future investigation.
In general, tendinopathy is characterized by longstanding localized activity-related pain and the patients in general respond poorly to most "conservative treatments". However, a wide spectrum of tendon pathologies is put under the umbrella entity of tendinopathy based on some common features [3] (Table 1), leading to an impression that there is no single general pathogenesis or aetiology involved which can explain all conditions. If so, we firmly believe that these pathologies should be classified as different entities. As the current research evidences are confusing, it is very important to identify if there are common denominators and diversifiers for various manifestations of what we loosely call "tendinopathy" to help us understand the pathogeneses of these conditions.
In this article, we review previous investigations according to the nature of the studies, for example, clinical observations, characterization of clinical samples, evaluation of treatments to patients with tendinopathy, animal models of tendinopathy and cell culture studies related to the effects of risk factors. Certainly we do not answer all questions with the integration of various research evidences, but we take the bold step to propose an integrative theory for the pathogenesis of tendinopathy based on the underlying messages in these studies.
Clinical observations of tendinopathy
To understand the pathogenesis of what today is labeled as "tendinopathy" we have to make some clear distinctions. Firstly we must consider the varying clinical presentations. Most tendon problems are presented to the clinician either as a rupture or localized pain, often including stiffness and swelling. Symptomatic tendinopathy refers to chronic localized pain with "degenerative" changes in tendons as observed by imaging or histology; while asymptomatic tendinopathy is identified from ruptures or partial rupture cases shown to be associated with non symptomatic pre-existing degenerative changes. Pathologies primarily manifested as passive loss of range of motion (i.e. trigger finger, frozen shoulder, etc) are not considered as tendinopathy in this discussion. Dating back to 1938, Codman reported degeneration in complete ruptures of rotator cuff [4]. Kannus and Josza reported in 1991 from a large number of histological samples that an absolute majority of patients with complete Achilles tendon ruptures had pathologic alterations which he described as "mucoid degeneration" [5]. This "mucoid degeneration" is almost equivalent to the histological alterations characterized for tendinosis [6]. It is very likely that these pathological changes in tendons imposed mechanical weakness and higher susceptibility to ruptures. Similar histopathological characteristics were also described in clinical samples of symptomatic tendinopathy [7, 8]. It suggests that the "typical" histopathological changes characterized by tendon degeneration may not necessarily be directly linked to increased nociception giving the patients warning signals; while in painful cases, the mechanically weaker tendons may be protected from ruptures due to decreased impact levels since painful activities will be avoided.
Secondly, we must consider the etiology and epidemiology. Unfortunately, well defined epidemiological studies on "tendinopathy" are virtually nonexistent. Age-related changes in tendons were reported [9], but tendinopathy is not an age-related degeneration because similar pathological changes are observed in young people [10]. Higher number of cases in males presented in clinical studies [11, 12] may not reflect higher susceptibility of male gender to tendinopathy; on the contrary, it is reported that female gender was more susceptible to repetitive trauma in rotator cuff [13] and female cyclists suffer a higher risk for "overuse injury" in general than their male counterparts [14]. There were significant gender differences in tendon microcirculation [15] and the neuropeptide responsiveness in rabbit tendon explants was influenced by gender and pregnancy [16]. Diabetes [17] and metabolic alterations such as dislipidemia [18] has been proposed as risk factor for developing tendinopathy. These findings suggest that the hormonal background may affect the development of tendinopathy. Fluoroquinolone [19] and corticosteroids [20] were found to be associated with Achilles tendon ruptures; suggesting pharmacological influence on the development of tendon pathology. Overuse, repetitive strain or mechanical overload to tendons are considered as primary trigger of symptomatic tendinopathy in various regions [21], as implied by the names such as "jumper's knee", "runner's heel", "swimmer's shoulder" and "tennis elbow". The prevalence of supraspinatous tendinopathy could be as high 69 % in elite swimmers [22]. However, there are frequent tendinopathy cases (pain or rupture) in the non-athlete population [23, 20]. Thus overuse injury should not be equated to tendinopathy, but it may be one of the major triggers of the pathological development in some individuals. Furthermore, overuse as a risk factor for tendinopathy is not simply a quantitative increase in activities, but may also be attributed to improper gait or training errors [24, 25].
Thirdly, the anatomical sites of tendinopathic changes add further complexity. Since overuse or cumulative trauma may also affect other peritendinous tissues, tendinopathy was sometimes presented with pathological changes in tenosynovium, bursa and nerves. Our discussion on the pathogenesis of tendinopathy should be focused on changes primarily initiated and observed in tendons; otherwise the pathogenesis pathways will be very heterogeneous. It follows that infectious tenosynovitis, bursitis, adhesive capsulitis or tendon and nerve entrapment in case of carpel tunnel syndrome will not be included in our model, but "paratenonitis" [26] and "insertional tendinopathies" [27] will be discussed since they are parts of a tendon. The pathological changes in different forms of tendinopathy are localized in different regions of the affected tendons, for example, the proximal deep posterior portion of the patellar tendon is affected in patellar tendinopathy, while mid-substance or insertion pathological changes can be observed in Achilles tendinopathy. The medial musculotendinous junction or lateral Humerus insertion was affected in Rotator cuff tendinopathy, while in lateral epicondylitis the fascial collagen structure on the extensor carpi radialis brevis tendon was pathological. Based on the involvement of pathological changes in the paratenon, different sub-classes can be further identified in Achilles tendinopathy [28]. Owing to these variations in the sites of pathological changes, it suggests the common denominator of the pathogenesis of tendinopathy may probably involve a process that can affect all parts of tendons [29], including musculotendinous junction, mid-substance, insertion and paratenons. The "communication" between these structures around the tendons is poorly investigated.
Medical imaging of tendinopathy
Tendinopathy exhibited characteristic pathological changes which are visible under ultrasound or magnetic resonance imaging (MRI). Tendon thickening or swelling is revealed, localized hypoechogenic signals were detected by ultrasound [30] and an increased T1 and T2 contrast signal was shown by MRI [31, 32]. It suggests an increased water content which is probably related to increased accumulation of water-retaining proteoglycans. Doppler ultrasound imaging showed increased vascularity and blood flow in the pathological regions but the oxygen tension was not significantly different [33]. These findings suggested an inflammatory component [34] with localized changes in the tendinous matrix and hypervascularity may be associated with the pathogenesis of tendinopathy.
Characterization of clinical samples of tendinopathy
Direct investigation of tendinopathy started with histological examination of the pathological tissues. Classical characteristics of "tendinosis" include degenerative changes in the collagenous matrix, hypercellularity, hypervascularity and a lack of inflammatory cells which has challenged the original misnomer "tendinitis" [35, 6]. Further characterizations are basically extrapolation of these findings, for examples, measurement of proliferation and apoptosis to explain the changes in cellularity [36, 37], detection of extracellular matrix components [38–41], collagen crosslink [42] and degradative enzymes [43, 44] to explain the matrix disturbances, and detection of the expression of various cytokines to account for the deregulation of cellular activities [45, 46]. Calcified tendinitis exhibited abnormal tendon calcification which is more common in rotator cuffs [47]. Recently, the findings of increased innervations [48] and nociceptive substances [49, 50] suggest that the chronic pain of tendinopathy may directly be resulted from the pathological changes. The findings of increased apoptosis [51, 52] and acquisition of chondrogenic phenotypes [53] in the injured tendons suggested a disturbance in cell differentiation. Increased proteoglycans with over-sulphation [40] and expression of different versican variant [54] may be related to abnormal chondrogenesis in the affected tendons. In contrast to early observations of a lack of inflammatory cells [11], increased mast cell number was reported in human patellar tendinopathy [55]. Researchers are well aware of the limitation of the clinical samples, which may represent only the end-stage of the pathological processes with unknown duration and onset. Nevertheless, these observations have provided some direct clues to work out the pathogenesis of tendinopathy, and these histopathological characteristics are often used as endpoints in animal models of tendinopathy [56–58]. It should be noted that some of these pathological characteristics are sustained healing responses that failed to repair the initial injury, such as increased cell proliferation and elevated cytokines, which is also implicated in the normal healing process, as shown in the active remodeling sites in healthy tendons [59]. The histopathological features of tendinopathy we observed in animal models must be chronic and cannot be resolved spontaneously as compared to the normal course of tendon healing.
Genetic predisposition of tendinopathy
The possible genetic predisposition for Achilles tendinopathy has been investigated. It was found that variants within COL5A1 [60], tenascin C [61] and matrix metalloproteinase 3 (MMP3) gene [62] was associated with increased risk of Achilles tendon injuries in general. Since these genes are related to homeostasis of extracellular matrix in tendons, it is suggested that the genetic variants modify the susceptibility of tendons to matrix disturbance observed in tendinopathy.
Evaluation of interventions to tendinopathy
The observed pathological changes of tendinopathy intuitively provided a lot of insights for the treatments. However, all current treatment methods may not significantly affect the natural history of the disease [63]. Surgical excision was reported to be used on animals over many years, in particular on horses [64]. Surgical excision of pathological tissues [65, 66] and percutaneous multiple longitudinal incisions [67, 68] were reported to be effective to relieve the symptoms similar to open excision of macroscopic pathologic tendon structures [23]. But ultrasonographic anomalies may still be evident in the healing tissues after surgical excision of pathological tissues; despite the painful symptoms were relieved [69]. Thus the current understanding of the relationship of structural changes and functional impairments is still inadequate to assure the degenerative features as specific "markers" for tendinopathy. Biophysical intervention such as extracorporeal shockwave therapy exhibited significant improvement especially for calcified tendinopathy [70, 71]. It suggests that the pathological tissues might be responsive to mechanical stimulation. The observed effects of eccentric exercise for tendinopathy [72, 73] also implied that a proper modulation of mechanical environment may exert positive effects on the diseased tendons, such as an increase in peritendinous collagen synthesis [74]. Other biophysical interventions included ultrasound therapy [75–77], pulsed magnetic field therapy [78, 79], low level laser therapy [80–82], radiofrequency [83] and acupuncture [84]. These studies claimed that modulation of inflammatory or neuronal components in the pathological tissues may exert beneficial effects. There are also reports on the use of nitric oxide [85], sclerosing agents [86, 87], MMP inhibitors [88], bone marrow plasma injection [89], autologous blood injection [90, 91] or platelet-rich plasma [92–94] for tendinopathy. Stem cell therapy was tried in horse models [95]. These studies may suggest the involvement of disturbances in cytokines, neovascularization, innervations or cell differentiation in the pathogenesis of tendinopathy.
Animal models of tendinopathy
The lack of a representative animal model is a major obstacle for tendinopathy research. Recent reviews discussed current animal models used for tendinopathy research [96–98], including cytokine-induced tendon injuries [58, 99, 100], collagenase-induced injury [56, 101–105] and overuse induced injury [57, 106–110]. Generally, histopathological characteristics derived from clinical samples are the main criteria for evaluation of tendinopathic changes. Ultrasonographic features were occasionally used in horse models [111], and some studies reported pain-associated behavioral changes associated with the tendon injuries [112, 113]. These animal models were established according to different hypotheses of pathogenesis, and aimed at reproducing the clinical signs of tendinopathy as far as possible. The use of cytokines to induce pathological changes implied the key roles of one or several cytokines in the development of the disease; while collagenase injection mimicked the pathological processes from the point when progressive matrix degradation was dominating. These chemically-induced tendinopathy models may reveal different starting points of the pathological process but the causes of increased cytokines or collagenases must be linked with clinically relevant etiological factors. Moreover, the acute induction of degenerative changes in these models cannot reflect the chronic development of the disease. On the other hand, animal models of overuse tendon injuries gained wide acceptance for the demonstration of the relationship between the mechanical overload and the development of histopathological changes [57, 109, 110] and increase in pro-inflammatory mediators [51, 114, 115]. Although overuse is sufficient to generate degenerative changes over a longer period of time, this form of injuries can be healed when the overuse training was ceased [116]; while in clinical cases of tendinopathy the symptoms were not improved by rest. Obviously, overuse tendon injury does not equate to tendinopathy. The failed healing response to the injuries caused by mechanical overload of tendons should also be considered in the establishment of animal model of tendinopathy. With respect to the variability in clinical manifestation of tendinopathy, most animal models may only mimic parts of the pathogenesis pathways, or they may only represent one of the possible pathways from the generation of injuries to development of tendinopathy features (Table 1). In summary, it appears that degenerative tendon injuries can be resulted from repetitive strain injuries that exceed the normal thresholds (overuse); while abnormal levels of cytokines and collagenases could be the effectors to mediate this kind of degenerative injuries.
Cell culture studies of effects of risk factors on tendinopathy
Cell culture studies of tendinopathy included the characterization of abnormal activities in the cells isolated from pathological tissues of tendinopathy [36, 44, 45, 53], and the studies in normal cultured tendon cells in response to potential risk factors such as mechanical strain [117–123] and xenobiotics [124–129]. In cell cultures of tendinopathy tissues, the abnormal cellular activities were persistent during sub-cultures, indicating relatively stable cell phenotypes that are significantly different from tendon fibroblasts derived from healthy tendons [36, 45]. On the other hand, numerous studies showed that repetitive mechanical stimulation can affect production of pro-inflammatory mediators [117, 118, 120–123, 130], metalloproteinases [123, 131] and matrix syntheses [119] in cultured tendon fibroblasts; while non-tenogenic differentiation of tendon derived stem cells can also be triggered by mechanical stretching [132]. Corticosteroids also induced fibrocartilage phenotype in tendon cells [129], affected matrix synthesis [127], cell viability [126, 128] and apoptosis [133]. Fluoroquinolones may also activate metalloproteinases in tendon cells and hence collagenolytic injuries [125]. These observations implied that activation of collagenolysis and erroneous differentiation may weaken the mechanical properties of tendons [134]. Interestingly, non-steroidal anti-inflammatory drugs (NSAIDs) also modulate tendon cell proliferation [124, 135], the expression of extracellular matrix components [124] and degradative enzymes [136]. As NSAID is commonly used for sports-related injuries and symptoms, it is possible that anti-inflammatory treatment used for overuse injury may contribute to the development of tendinopathy [137] which is normally diagnosed after NSAID treatment was failed.
Previous theories of pathogenesis of tendinopathy
Several theories of pathogenesis of tendinopathy have been proposed to explain the development of the histopathological features observed in the clinical samples of tendinopathy. Burry suggested that tendon lesions were not resolved properly and resulted in degenerative changes already in 1978 [138]. However, further elaboration of the idea of "improper resolution of tendon lesion" was not possible due to a lack of experimental evidences at that time. Leadbetter and Khan et al. have suggested that "tendinosis" are degenerative changes resulting from increased demand on tendons with inadequate repair and progressive cell death [139, 140]. This model explained the generation of overuse injury, and the reasons for inadequate repair are attributed to adaptive response to tissue overload as elaborated by Kibler and Sorosky et al [141, 142]. However, "inadequate repair" as quantitative decrease in healing cells cannot explain the findings of focal hypercellularity, active proliferation and metaplasia in tendinopathy samples. The "apoptosis theory" [143–145] proposed by Murrell also neglect the fact of increased cellularity; but this hypothesis linked up oxidative stress, acquisition of cartilage phenotype and activation of metalloproteinase with the development of degenerative injuries by high dose of cyclic strain. Unfavorable mechanical stimulation as repetitive tensile strain [120], stress-shielding [146], contractile tension overloads [147] or compression [148] was proposed as noxious triggers on tendon cells to induce tendon inflammation or degenerative changes. These theories pointed out that the interactions between tendon cells and their mechanical environment were deterministic for the pathogenesis. On the other hand, Pufe et al. suggested that hypoxia and increased vascular in-growth into tendons may be the causes of tendon weakening and ruptures [149], while Riley provided a neurogenic hypothesis to explain the adaptive responses to mechanical overload by nerve and mast cells unit [150] and Fredberg et al. suggested neurogenic inflammation may be involved in the pathogenesis pathway [151]. These theories were formulated according to the findings of hypervascularity and increased innervations. In summary, the common motifs in these pathogenesis theories include unfavorable mechanical loading, adaptive cellular responses (including tendon, blood vessels and nerves) and the generation of histopathological features. In our opinion, it is possible to unify these ideas as "failed healing", which may be the integral part of various pathological processes that divert various tendon injuries into its different manifestations of tendinopathy. (Table 1)
A unified theory of pathogenesis of tendinopathy
Based on the information of various lines of investigation of tendinopathy, we can summarize some major points which must be considered in the formulation of the pathogenesis model of tendinopathy:
-
1.
The interactions of tendon injuries and unfavorable mechanical environment would be the starting point of the pathological process. Instead of coining the phrase "adaptive responses of tendon cells", we think that the "adaptive healing responses to tendon injuries" would be a more comprehensive descriptor, which also includes vascular, neural and peri-tendinous reactions at different stages of healing.
-
2.
The normal healing processes are diverted to an abnormal pathway, probably due to unfavorable mechanical environment, disturbances of local inflammatory responses, oxidative stress or pharmacological influences. Therefore, the healing capacity is not only inadequate but also incorrect and deviated from an ideal healing outcome.
-
3.
The primary results of pathology are the progressive collagenolytic injuries co-existing with a failed healing response, thus both degenerative changes and active healing are observed in the pathological tissues.
-
4.
These pathological tissues may aggravate the nociceptive responses by various pathways which are no longer responsive to conventional treatment such as inhibition of prostaglandin synthesis; otherwise the insidious mechanical deterioration without pain may render increased risk of ruptures.
Based on these points, we propose that the pathogenesis of tendinopathy can be perceived as a 3-stages process: injury, failed healing and clinical presentation. The first stage does not involve pathological changes and normal healing response could occur. The second stage is relatively insidious and discriminated from the third stage when clinical presentations are evident, such as ruptures or chronic pain, often resistant to conservative treatments.
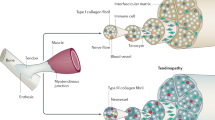
Our theory of the pathogenesis of tendinopathy is summarized in Figure 1.

Failed healing theory for the pathogenesis of tendinopathy.
Stage 1. Injury
In the first stage, initiation of tendinopathy may involve generation of collagenolytic injuries. Overuse can evoke the release of pro-inflammatory mediators [115], which would result in stimulation of metalloproteinases and hence collagenolytic injuries [152, 153]. Recent findings showed that the expression of MMP and tissue inhibitor of metalloproteinase (TIMP) in tendon cells were sensitive to mechanical overload or stress-deprivation [131, 154]. MMP inhibitor suppressed development of mechanical weakness induced by stress-deprivation [155]. Chemicals such as fluoroquinolone can induce tendon cell death [156], oxidative damages [157] and collagenolysis [125]. A previous traumatic injury that has not been healed may also be susceptible to failed healing [158]. With the case reports of infectious tenosynovitis [159], the involvement of pathogens to generate tendon injuries and inflammation could not be ruled out. Tendon pain and mechanical weakness is not significant at this stage and it is possible for the tendon injury to heal spontaneously.
Stage 2. Failed healing
In the failed healing stage, healing responses were activated but failed to repair the collagenolytic injuries. The exact causes for failed healing are still obscured. It is speculated that unfavorable mechanical environment, genetic pre-disposition, hormonal background and pharmacological exposures may affect the healing process. Since tendon healing includes many sequential processes such as inflammation, neovascularization, neural modulations [160], recruitment of healing cells, proliferation, apoptosis [161], matrix synthesis, tenogenic differentiation and matrix remodeling; disturbances occurred at different stages of healing may lead to different combinations of histopathological changes as what we observed in the clinical samples of tendinopathy (Table 1). Inflammatory responses are presumably elicited as the initial stage of tissue repair, but it may not be properly resolved under hostile mechanical environment or pharmacological intervention such as NSAIDs, resulting in elevated pro-inflammatory cytokines and a lack of ordinary inflammatory infiltration in the diseased tissues. Depending on the anatomical variations of the affected tendons, peritendinous reactions may be resulted as restrictive fibrosis, increased innervations and vascular in-growth from para-tenon structures may also take part in healing process [162]. The sustained activation of tendon progenitor cells with unfavorable micro-milieu for tendogenic differentiation may prone to erroneous differentiation into fibrochondrogenic [163] or calcifying phenotypes [104], which are normally confined to the regions of bone-tendon junctions. Tendon pain becomes significant and conservative treatments such as NSAIDs are prescribed to the patients, which may further modify the pathways of the failed healing.
Stage 3. Clinical presentation
In the third stage, symptomatic tendinopathy is diagnosed as longstanding, activity-related pain with characteristic medical images; while spontaneous ruptures are resulted from mechanical weakness under normal activities in cases of asymptomatic tendinopathy. The consequences of failed healing to collagenolytic injuries involve significant changes in extracellular matrix, which are then visible under ultrasound or MRI. In symptomatic cases, inflammatory pain may be involved and controlled during the injury and failed healing stages, but the pain mechanism may gradually shift to non-phlogistic ones such as agitation to peritendinous nerves by nociceptive substances or swelling, rendering the resistance to common anti-inflammatory treatments. Though mechanical weakness may be involved in asymptomatic cases [164], lower activities due to pain may reduce risk of ruptures. In asymptomatic cases, the matrix disturbance resulted from failed healing may not activate nociceptive response. The insidious deterioration in mechanical properties of the affected tendons may lead to ruptures. Owing to different combinations of etiological factors, temporal and spatial variations on the failed healing, the clinical manifestations of tendinopathy may exhibit high variability.
Explicability of the theory
With this theory for the pathogenesis of tendinopathy, we can explain the process of the generation of the pathological features of tendinopathy we observed in the clinical samples. The theory is in accordance with most of the evidences derived from tendinopathy studies. For example, overuse is a major etiological factor but there are tendinopathy patients without obvious history of repetitive injuries. It is possible that non-overuse tendon injuries may also be exposed to risk factors for failed healing and entered to the third stage of tendinopathy. Overuse induces collagenolytic tendon injuries and it also imposes repetitive mechanical strain which may be unfavorable for normal healing. Stress-deprivation also induces MMP expression and whether over- or under-stimulation is still an active debate [165]. It is possible that tenocyte is responsive to both over- and under-stimulation, both tensile and compressive loading. Because the cellular responses of healing tendon cells change in different stages of tendon healing [166], we speculate that the cell responsiveness to mechanical loading may not be constant during tendon healing and failed healing may be resulted from a mismatch of healing stages and the mechanical environment. Our theory can also explain why animal models of collagenase-induced injuries can reproduce the histopathological characteristics and functional impairment similar to tendinopathy; despite the generation of collagenolytic injuries in these models are completely different from the insidious onset of tendinopathy. By proposing a process of failed healing to translate tendon injuries into tendinopathy, other extrinsic and intrinsic factors would probably enter the play at this stage, such as genetic predisposition, age [167], xenobiotics (NSAIDs and corticosteroids) and mechanical loading on the tendons. For example, differential tensile forces acting on patellar tendon [168] may impose varying loading on tendon cells in different regions, it may explain why posterior proximal patellar tendon is pathological in patellar tendinopathy. Peritendinous structures may be disturbed to different extents in the healing response to tendon injuries, which may lead to different manifestations of "paratenonitis" or tendon adhesion. Investigations of how these factors affect tendon healing could help to further elucidate the mechanism of failed healing. The recent discovery of tendon-derived stem cells and characterization of pathological tissues of tendinopathy have provided evidences to support the ideas of erroneous cell differentiation that contribute to failed tendon healing. According to this theory of pathogenesis, we shall have a theoretical framework to develop a more representative animal model of tendinopathy for further study and verification. New ideas for treatments of tendinopathy may be inspired based on this theory, for example, a treatment which could override on the failed healing tissues and restart the healing process.
Missing links and limitations
As compared to previous theories of pathogenesis for tendinopathy which described a viscous cycle of inadequate repair and increased susceptibility of further injuries, this new theory attempts to describe the "vicious cycle" as an interaction between the vulnerability of the healing process to noxious mechanical and biochemical environments. Thus we can investigate the missing links as predicted in the theory, for example, the impact of mechanical stimulation on the cell differentiation of healing tendons cells, and the disturbances in cytokines triggered by re-injury on healing tendons. However, there are still some limitations in the current pathogenesis theory. Firstly, tendinopathies in different tendons exhibited specific patterns of affected regions and different forms of matrix disturbance, which may be presumably accounted by variations in local mechanical environment and vascular supplies; but it is difficult to explain for these variations at the present stage. Secondly, the interplay among innervations, increased nociception and tendon healing is unknown. It is still a black box for the mechanism of increased nociception by failed tendon healing. The factors which govern the development of chronic pain in tendinopathic tendons remain obscure. Finally, the interactions between healing tendons and the peritendinous tissues are seldom investigated and it is difficult to evaluate the potential involvement of peritendinous tissues in the development of tendinopathy.
Conclusions
In summary, we propose a unified theory for pathogenesis of tendinopathy which explain most of the available experimental data about tendinopathy. It is a just a start to probe into the nature of the pathology and we certainly wait for new findings and challenges to this theory until we finally find the truth.
Abbreviations
- MMP3:
-
Matrix metalloproteinase 3
- MRI:
-
Magnetic resonance imaging
- NSAID:
-
Non-steroidal anti-inflammatory drug
- TIMP:
-
Tissue inhibitor of metalloproteinase
References
Maffulli N, Khan KM, Puddu G: Overuse tendon conditions: time to change a confusing terminology. Arthroscopy. 1998, 14 (8): 840-843. 10.1016/S0749-8063(98)70021-0.
Woo SL, Renstrom P, Arnoczky SP: Tendinopathy in athletes, Encyclopedia of Sports Medicine. 2007, Blackwell Publishing, Oxford, UK
Cook JL, Purdam CR: Is tendon pathology a continuum? A pathology model to explain the clinical presentation of load-induced tendinopathy. Br J Sports Med. 2009, 43 (6): 409-416. 10.1136/bjsm.2008.051193.
Codman EA: Rupture of the supraspinatus. Am J Surg. 1938, 42: 603-626. 10.1016/S0002-9610(38)91106-7.
Kannus P, Józsa L: Histopathological changes preceding spontaneous rupture of a tendon. A controlled study of 891 patients. J Bone Joint Surg Am. 1991, 73 (10): 1507-1525.
Movin T, Gad A, Reinholt F, Rolf C: Tendon pathology in long-standing achillodynia. Biopsy findings in 40 patients. Acta Orthop Scand. 1997, 68: 170-175. 10.3109/17453679709004002.
Järvinen M, Józsa L, Kannus P, Järvinen TL, Kvist M, Leadbetter W: Histopathological findings in chronic tendon disorders. Scand J Med Sci Sports. 1997, 7 (2): 86-95.
Tallon C, Maffulli N, Ewen SW: Ruptured Achilles tendons are significantly more degenerated than tendinopathic tendons. Med Sci Sports Exerc. 2001, 33 (12): 1983-1990. 10.1097/00005768-200112000-00002.
Riley GP, Harrall RL, Constant CR, Chard MD, Cawston TE, Hazleman BL: Glycosaminoglycans of human rotator cuff tendons: changes with age and in chronic rotator cuff tendinitis. Ann Rheum Dis. 1994, 53 (6): 367-376. 10.1136/ard.53.6.367.
Cook JL, Khan KM, Kiss ZS, Griffiths L: Patellar tendinopathy in junior basketball players: a controlled clinical and ultrasonographic study of 268 patellar tendons in players aged 14-18 years. Scand J Med Sci Sports. 2000, 10 (4): 216-220. 10.1034/j.1600-0838.2000.010004216.x.
Aström M, Rausing A: Chronic Achilles tendinopathy. A survey of surgical and histopathologic findings. Clin Orthop Relat Res. 1995, 151-164. 316
Cook JL, Khan KM, Harcourt PR, Kiss ZS, Fehrmann MW, Griffiths L, Wark JD: Patellar tendon ultrasonography in asymptomatic active athletes reveals hypoechoic regions: a study of 320 tendons. Victorian Institute of Sport Tendon Study Group. Clin J Sport Med. 1998, 8: 73-77. 10.1097/00042752-199804000-00001.
Fischer KA, Leatherman KD: The nature of and treatment of tendinitis of the musculotendinous cuff of the shoulder and subacromial bursitis. South Surg. 1950, 16 (2): 132-143.
Wilber CA, Holland GJ, Madison RE, Loy SF: An epidemiological analysis of overuse injuries among recreational cyclists. Int J Sports Med. 1995, 16 (3): 201-206. 10.1055/s-2007-972992.
Knobloch K, Schreibmueller L, Meller R, Busch KH, Spies M, Vogt PM: Superior Achilles tendon microcirculation in tendinopathy among symptomatic female versus male patients. Am J Sports Med. 2008, 36 (3): 509-514. 10.1177/0363546507309313.
Hart DA, Kydd A, Reno C: Gender and pregnancy affect neuropeptide responses of the rabbit Achilles tendon. Clin Orthop Relat Res. 1999, 237-246. 10.1097/00003086-199908000-00029. 365
Holmes GB, Lin J: Etiologic factors associated with symptomatic achilles tendinopathy. Foot Ankle Int. 2006, 27 (11): 952-959.
Gaida JE, Alfredson L, Kiss ZS, Wilson AM, Alfredson H, Cook JL: Dyslipidemia in Achilles tendinopathy is characteristic of insulin resistance. Med Sci Sports Exerc. 2009, 41 (6): 1194-1197. 10.1249/MSS.0b013e31819794c3.
Zabraniecki L, Negrier I, Vergne P, Arnaud M, Bonnet C, Bertin P, Treves R: Fluoroquinolone induced tendinopathy: report of 6 cases. J Rheumatol. 1996, 23 (3): 516-520.
Aström M: Partial rupture in chronic achilles tendinopathy. A retrospective analysis of 342 cases. Acta Orthop Scand. 1998, 69 (4): 404-407.
Almekinders LC, Temple JD: Etiology, diagnosis, and treatment of tendonitis: an analysis of the literature. Med Sci Sports Exerc. 1998, 30 (8): 1183-1190. 10.1097/00005768-199808000-00001.
Sein ML, Walton J, Linklater J, Appleyard R, Kirkbride B, Kuah D, Murrell GA: Shoulder pain in elite swimmers: primarily due to swim-volume-induced supraspinatus tendinopathy. Br J Sports Med. 2010, 44 (2): 105-113. 10.1136/bjsm.2008.047282.
Rolf C, Movin T: Etiology, histopathology, and outcome of surgery in achillodynia. Foot Ankle Int. 1997, 18 (9): 565-569.
McCrory JL, Martin DF, Lowery RB, Cannon DW, Curl WW, Read HM, Hunter DM, Craven T, Messier SP: Etiologic factors associated with Achilles tendinitis in runners. Med Sci Sports Exerc. 1999, 31 (10): 1374-1381. 10.1097/00005768-199910000-00003.
Van Ginckel A, Thijs Y, Hesar NG, Mahieu N, De Clercq D, Roosen P, Witvrouw E: Intrinsic gait-related risk factors for Achilles tendinopathy in novice runners: a prospective study. Gait Posture. 2009, 29 (3): 387-391. 10.1016/j.gaitpost.2008.10.058.
Kvist M, Józsa L, Järvinen MJ, Kvist H: Chronic Achilles paratenonitis in athletes: a histological and histochemical study. Pathology. 1987, 19 (1): 1-11. 10.3109/00313028709065127.
McGarvey WC, Palumbo RC, Baxter DE, Leibman BD: Insertional Achilles tendinosis: surgical treatment through a central tendon splitting approach. Foot Ankle Int. 2002, 23 (1): 19-25.
Perugia L, Ippolitio E, Postacchini F: A new approach to the pathology, clinical features and treatment of stress tendinopathy of the Achilles tendon. Ital J Orthop Traumatol. 1976, 2 (1): 5-21.
Maffulli N, Testa V, Capasso G, Ewen SW, Sullo A, Benazzo F, King JB: Similar histopathological picture in males with Achilles and patellar tendinopathy. Med Sci Sports Exerc. 2004, 36 (9): 1470-1475. 10.1249/01.MSS.0000139895.94846.8D.
Movin T, Kristoffersen-Wiberg M, Shalabi A, Gad A, Aspelin P, Rolf C: Intratendinous alterations as imaged by ultrasound and contrast medium-enhanced magnetic resonance in chronic achillodynia. Foot Ankle Int. 1998, 19 (5): 311-317.
McLoughlin RF, Raber EL, Vellet AD, Wiley JP, Bray RC: Patellar tendinitis: MR imaging features, with suggested pathogenesis and proposed classification. Radiology. 1995, 197 (3): 843-848.
Movin T, Kristoffersen-Wiberg M, Rolf C, Aspelin P: MR imaging in chronic Achilles tendon disorder. Acta Radiol. 1998, 39 (2): 126-132.
Knobloch K, Kraemer R, Lichtenberg A, Jagodzinski M, Gossling T, Richter M, Zeichen J, Hufner T, Krettek C: Achilles tendon and paratendon microcirculation in midportion and insertional tendinopathy in athletes. Am J Sports Med. 2006, 34 (1): 92-97. 10.1177/0363546505278705.
Terslev L, Qvistgaard E, Torp-Pedersen S, Laetgaard J, Danneskiold-Samsøe B, Bliddal H: Ultrasound and Power Doppler findings in jumper's knee - preliminary observations. Eur J Ultrasound. 2001, 13 (3): 183-189. 10.1016/S0929-8266(01)00130-6.
Józsa L, Kannus P: Human tendons: anatomy, physiology and pathology. 1997, Human Kinetics, London
Rolf CG, Fu BS, Pau A, Wang W, Chan B: Increased cell proliferation and associated expression of PDGFRbeta causing hypercellularity in patellar tendinosis. Rheumatology (Oxford). 2001, 40 (3): 256-261. 10.1093/rheumatology/40.3.256.
Lian Ø, Scott A, Engebretsen L, Bahr R, Duronio V, Khan K: Excessive apoptosis in patellar tendinopathy in athletes. Am J Sports Med. 2007, 35 (4): 605-611. 10.1177/0363546506295702.
Riley GP, Harrall RL, Cawston TE, Hazleman BL, Mackie EJ: Tenascin-C and human tendon degeneration. Am J Pathol. 1996, 149 (3): 933-943.
Corps AN, Robinson AH, Movin T, Costa ML, Hazleman BL, Riley GP: Increased expression of aggrecan and biglycan mRNA in Achilles tendinopathy. Rheumatology (Oxford). 2006, 45 (3): 291-294. 10.1093/rheumatology/kei152.
Fu SC, Chan KM, Rolf CG: Increased deposition of sulfated glycosaminoglycans in human patellar tendinopathy. Clin J Sport Med. 2007, 17 (2): 129-134. 10.1097/JSM.0b013e318037998f.
Samiric T, Parkinson J, Ilic MZ, Cook J, Feller JA, Handley CJ: Changes in the composition of the extracellular matrix in patellar tendinopathy. Matrix Biol. 2009, 28 (4): 230-236. 10.1016/j.matbio.2009.04.001.
Bank RA, TeKoppele JM, Oostingh G, Hazleman BL, Riley GP: Lysylhydroxylation and non-reducible crosslinking of human supraspinatus tendon collagen: changes with age and in chronic rotator cuff tendinitis. Ann Rheum Dis. 1999, 58 (1): 35-41. 10.1136/ard.58.1.35.
Ireland D, Harrall R, Curry V, Holloway G, Hackney R, Hazleman B, Riley G: Multiple changes in gene expression in chronic human Achilles tendinopathy. Matrix Biol. 2001, 20 (3): 159-169. 10.1016/S0945-053X(01)00128-7.
Fu SC, Chan BP, Wang W, Pau HM, Chan KM, Rolf CG: Increased expression of matrix metalloproteinase 1 (MMP1) in 11 patients with patellar tendinosis. Acta Orthop Scand. 2002, 73 (6): 658-662. 10.1080/000164702321039624.
Fu SC, Wang W, Pau HM, Wong YP, Chan KM, Rolf CG: Increased expression of transforming growth factor-beta1 in patellar tendinosis. Clin Orthop Relat Res. 2002, 174-183. 10.1097/00003086-200207000-00022. 400
Nakama LH, King KB, Abrahamsson S, Rempel DM: VEGF, VEGFR-1, and CTGF cell densities in tendon are increased with cyclical loading: An in vivo tendinopathy model. J Orthop Res. 2006, 24 (3): 393-400. 10.1002/jor.20053.
Uhthoff HK: Calcifying tendinitis, an active cell-mediated calcification. Virchows Arch A Pathol Anat Histol. 1975, 366 (1): 51-58. 10.1007/BF00438677.
Schubert TE, Weidler C, Lerch K, Hofstädter F, Straub RH: Achilles tendinosis is associated with sprouting of substance P positive nerve fibres. Ann Rheum Dis. 2005, 64 (7): 1083-1086. 10.1136/ard.2004.029876.
Alfredson H, Thorsen K, Lorentzon R: In situ microdialysis in tendon tissue: high levels of glutamate, but not prostaglandin E2 in chronic Achilles tendon pain. Knee Surg Sports Traumatol Arthrosc. 1999, 7 (6): 378-381. 10.1007/s001670050184.
Alfredson H, Bjur D, Thorsen K, Lorentzon R, Sandström P: High intratendinous lactate levels in painful chronic Achilles tendinosis. An investigation using microdialysis technique. J Orthop Res. 2002, 20 (5): 934-938. 10.1016/S0736-0266(02)00021-9.
Millar NL, Wei AQ, Molloy TJ, Bonar F, Murrell GA: Cytokines and apoptosis in supraspinatus tendinopathy. J Bone Joint Surg Br. 2009, 91 (3): 417-424. 10.1302/0301-620X.91B3.21652.
Pearce CJ, Ismail M, Calder JD: Is apoptosis the cause of noninsertional achilles tendinopathy?. Am J Sports Med. 2009, 37 (12): 2440-2444. 10.1177/0363546509340264.
de Mos M, Koevoet W, van Schie HT, Kops N, Jahr H, Verhaar JA, van Osch GJ: In vitro model to study chondrogenic differentiation in tendinopathy. Am J Sports Med. 2009, 37 (6): 1214-1222. 10.1177/0363546508331137.
Corps AN, Robinson AH, Movin T, Costa ML, Ireland DC, Hazleman BL, Riley GP: Versican splice variant messenger RNA expression in normal human Achilles tendon and tendinopathies. Rheumatology (Oxford). 2004, 43 (8): 969-972. 10.1093/rheumatology/keh222.
Scott A, Lian Ø, Bahr R, Hart DA, Duronio V, Khan KM: Increased mast cell numbers in human patellar tendinosis: correlation with symptom duration and vascular hyperplasia. Br J Sports Med. 2008, 42 (9): 753-757. 10.1136/bjsm.2007.040212.
Williams IF, McCullagh GD, Goodship AE, Silver IA: Studies on the pathogenesis of equine tendinitis following collagenase injury. Res Vet Sci. 1984, 36: 326-338.
Soslowsky LJ, Carpenter JE, DeBano CM, Banerji I, Moalli MR: Development and use of an animal model for investigations on rotator cuff disease. J Shoulder Elbow Surg. 1996, 5 (5): 383-392. 10.1016/S1058-2746(96)80070-X.
Stone D, Green C, Rao U, Aizawa H, Yamaji T, Niyibizi C, Carlin G, Woo SL: Cytokine-induced tendinitis: a preliminary study in rabbits. J Orthop Res. 1999, 17 (2): 168-177. 10.1002/jor.1100170204.
Chuen FS, Chuk CY, Ping WY, Nar WW, Kim HL, Ming CK: Immunohistochemical characterization of cells in adult human patellar tendons. J Histochem Cytochem. 2004, 52 (9): 1151-1157. 10.1369/jhc.3A6232.2004.
Mokone GG, Schwellnus MP, Noakes TD, Collins M: The COL5A1 gene and Achilles tendon pathology. Scand J Med Sci Sports. 2006, 16 (1): 19-26. 10.1111/j.1600-0838.2005.00439.x.
Mokone GG, Gajjar M, September AV, Schwellnus MP, Greenberg J, Noakes TD, Collins M: The guanine-thymine dinucleotide repeat polymorphism within the tenascin-C gene is associated with achilles tendon injuries. Am J Sports Med. 2005, 33 (7): 1016-1021. 10.1177/0363546504271986.
Raleigh SM, van der Merwe L, Ribbans WJ, Smith RK, Schwellnus MP, Collins M: Variants within the MMP3 gene are associated with Achilles tendinopathy: possible interaction with the COL5A1 gene. Br J Sports Med. 2009, 43 (7): 514-520. 10.1136/bjsm.2008.053892.
Almekinders LC: Tendinitis and other chronic tendinopathies. J Am Acad Orthop Surg. 1998, 6 (3): 157-164.
Nilsson G, Björck G: Surgical treatment of chronic tendinitis in the horse. J Am Vet Med Assoc. 1969, 155 (6): 920-926.
Roels J, Martens M, Mulier JC, Burssens : A Patellar tendinitis (jumper's knee). Am J Sports Med. 1978, 6 (6): 362-368. 10.1177/036354657800600609.
Kvist H, Kvist M: The operative treatment of chronic calcaneal paratenonitis. J Bone Joint Surg Br. 1980, 62 (3): 353-357.
Maffulli N, Testa V, Capasso G, Bifulco G, Binfield PM: Results of percutaneous longitudinal tenotomy for Achilles tendinopathy in middle- and long-distance runners. Am J Sports Med. 1997, 25 (6): 835-840. 10.1177/036354659702500618.
Testa V, Capasso G, Maffulli N, Bifulco G: Ultrasound-guided percutaneous longitudinal tenotomy for the management of patellar tendinopathy. Med Sci Sports Exerc. 1999, 31 (11): 1509-1515. 10.1097/00005768-199911000-00003.
Khan KM, Visentini PJ, Kiss ZS, Desmond PM, Coleman BD, Cook JL, Tress BM, Wark JD, Forster BB: Correlation of ultrasound and magnetic resonance imaging with clinical outcome after patellar tenotomy: prospective and retrospective studies. Victorian Institute of Sport Tendon Study Group. Clin J Sport Med. 1999, 9 (3): 129-137. 10.1097/00042752-199907000-00003.
Daecke W, Kusnierczak D, Loew M: Long-term effects of extracorporeal shockwave therapy in chronic calcific tendinitis of the shoulder. J Shoulder Elbow Surg. 2002, 11 (5): 476-480. 10.1067/mse.2002.126614.
Vulpiani MC, Trischitta D, Trovato P, Vetrano M, Ferretti A: Extracorporeal shockwave therapy (ESWT) in Achilles tendinopathy. A long-term follow-up observational study. J Sports Med Phys Fitness. 2009, 49 (2): 171-176.
Stanish WD, Rubinovich RM, Curwin S: Eccentric exercise in chronic tendinitis. Clin Orthop Relat Res. 1986, 65-68. 208
Alfredson H, Pietilä T, Jonsson P, Lorentzon R: Heavy-load eccentric calf muscle training for the treatment of chronic Achilles tendinosis. Am J Sports Med. 1998, 26 (3): 360-366.
Langberg H, Ellingsgaard H, Madsen T, Jansson J, Magnusson SP, Aagaard P, Kjaer M: Eccentric rehabilitation exercise increases peritendinous type I collagen synthesis in humans with Achilles tendinosis. Scand J Med Sci Sports. 2007, 17 (1): 61-66.
Ebenbichler GR, Erdogmus CB, Resch KL, Funovics MA, Kainberger F, Barisani G, Aringer M, Nicolakis P, Wiesinger GF, Baghestanian M, Preisinger E, Fialka-Moser V: Ultrasound therapy for calcific tendinitis of the shoulder. N Engl J Med. 1999, 340 (20): 1533-1538. 10.1056/NEJM199905203402002.
Rahman MH, Khan SZ, Ramiz MS: Effect of therapeutic ultrasound on calcific supraspinatus tendinitis. Mymensingh Med J. 2007, 16 (1): 33-35.
Warden SJ, Metcalf BR, Kiss ZS, Cook JL, Purdam CR, Bennell KL, Crossley KM: Low-intensity pulsed ultrasound for chronic patellar tendinopathy: a randomized, double-blind, placebo-controlled trial. Rheumatology (Oxford). 2008, 47 (4): 467-471. 10.1093/rheumatology/kem384.
Binder A, Parr G, Hazleman B, Fitton-Jackson S: Pulsed electromagnetic field therapy of persistent rotator cuff tendinitis. A double-blind controlled assessment. Lancet. 1984, 1 (8379): 695-698. 10.1016/S0140-6736(84)92219-0.
Owegi R, Johnson MT: Localized pulsed magnetic fields for tendonitis therapy. Biomed Sci Instrum. 2006, 42: 428-433.
Stasinopoulos DI, Johnson MI: Effectiveness of low-level laser therapy for lateral elbow tendinopathy. Photomed Laser Surg. 2005, 23 (4): 425-430. 10.1089/pho.2005.23.425.
Bjordal JM, Lopes-Martins RA, Joensen J, Couppe C, Ljunggren AE, Stergioulas A, Johnson MI: A systematic review with procedural assessments and meta-analysis of low level laser therapy in lateral elbow tendinopathy (tennis elbow). BMC Musculoskelet Disord. 2008, 9: 75-10.1186/1471-2474-9-75.
Tumilty S, Munn J, McDonough S, Hurley DA, Basford JR, Baxter GD: Low Level Laser Treatment of Tendinopathy: A Systematic Review with Meta-analysis. Photomed Laser Surg. 2010, 28 (1): 3-16. 10.1089/pho.2008.2470.
Tasto JP, Cummings J, Medlock V, Hardesty R, Amiel D: Microtenotomy using a radiofrequency probe to treat lateral epicondylitis. Arthroscopy. 2005, 21 (7): 851-860. 10.1016/j.arthro.2005.03.019.
Lathia AT, Jung SM, Chen LX: Efficacy of acupuncture as a treatment for chronic shoulder pain. J Altern Complement Med. 2009, 15 (6): 613-618. 10.1089/acm.2008.0272.
Paoloni JA, Appleyard RC, Nelson J, Murrell GA: Topical nitric oxide application in the treatment of chronic extensor tendinosis at the elbow: a randomized, double-blinded, placebo-controlled clinical trial. Am J Sports Med. 2003, 31 (6): 915-920.
Willberg L, Sunding K, Ohberg L, Forssblad M, Fahlström M, Alfredson H: Sclerosing injections to treat midportion Achilles tendinosis: a randomised controlled study evaluating two different concentrations of Polidocanol. Knee Surg Sports Traumatol Arthrosc. 2008, 16 (9): 859-864. 10.1007/s00167-008-0579-x.
Paoloni JA, Briggs L: Doppler ultrasound-guided polidocanol sclerosant injection treating bilateral quadriceps tendinopathy. Clin J Sport Med. 2009, 19 (2): 145-146. 10.1097/JSM.0b013e3181966c16.
Orchard J, Massey A, Brown R, Cardon-Dunbar A, Hofmann J: Successful management of tendinopathy with injections of the MMP-inhibitor aprotinin. Clin Orthop Relat Res. 2008, 466 (7): 1625-1632. 10.1007/s11999-008-0254-z.
Moon YL, Jo SH, Song CH, Park G, Lee HJ, Jang SJ: Autologous bone marrow plasma injection after arthroscopic debridement for elbow tendinosis. Ann Acad Med Singapore. 2008, 37 (7): 559-563.
Connell DA, Ali KE, Ahmad M, Lambert S, Corbett S, Curtis M: Ultrasound-guided autologous blood injection for tennis elbow. Skeletal Radiol. 2006, 35 (6): 371-377. 10.1007/s00256-006-0081-9.
James SL, Ali K, Pocock C, Robertson C, Walter J, Bell J, Connell D: Ultrasound guided dry needling and autologous blood injection for patellar tendinosis. Br J Sports Med. 2007, 41 (8): 518-521. 10.1136/bjsm.2006.034686.
Mishra A, Pavelko T: Treatment of chronic elbow tendinosis with buffered platelet-rich plasma. Am J Sports Med. 2006, 34 (11): 1774-1778. 10.1177/0363546506288850.
Filardo G, Kon E, Della Villa S, Vincentelli F, Fornasari PM, Marcacci M: Use of platelet-rich plasma for the treatment of refractory jumper's knee. Int Orthop. 2010, 34 (6): 909-915. 10.1007/s00264-009-0845-7.
de Vos RJ, Weir A, van Schie HT, Bierma-Zeinstra SM, Verhaar JA, Weinans H, Tol JL: Platelet-rich plasma injection for chronic Achilles tendinopathy: a randomized controlled trial. JAMA. 2010, 303 (2): 144-149. 10.1001/jama.2009.1986.
Nixon AJ, Dahlgren LA, Haupt JL, Yeager AE, Ward DL: Effect of adipose-derived nucleated cell fractions on tendon repair in horses with collagenase-induced tendinitis. Am J Vet Res. 2008, 69 (7): 928-937. 10.2460/ajvr.69.7.928.
Warden SJ: Animal models for the study of tendinopathy. Br J Sports Med. 2007, 41 (4): 232-240. 10.1136/bjsm.2006.032342.
Lake SP, Ansorge HL, Soslowsky LJ: Animal models of tendinopathy. Disabil Rehabil. 2008, 30 (20-22): 1530-1541. 10.1080/09638280701785460.
Warden SJ: Development and use of animal models to advance tendinopathy research. Front Biosci. 2009, 14: 4588-4597. 10.2741/3551.
Sullo A, Maffulli N, Capasso G, Testa V: The effects of prolonged peritendinous administration of PGE1 to the rat Achilles tendon: a possible animal model of chronic Achilles tendinopathy. J Orthop Sci. 2001, 6 (4): 349-357. 10.1007/s007760100031.
Khan MH, Li Z, Wang JH: Repeated exposure of tendon to prostaglandin-E2 leads to localized tendon degeneration. Clin J Sport Med. 2005, 15 (1): 27-33. 10.1097/00042752-200501000-00006.
Dahlgren LA, van der Meulen MC, Bertram JE, Starrak GS, Nixon AJ: Insulin-like growth factor-I improves cellular and molecular aspects of healing in a collagenase-induced model of flexor tendinitis. J Orthop Res. 2002, 20: 910-919. 10.1016/S0736-0266(02)00009-8.
Chen YJ, Wang CJ, Yang KD, Kuo YR, Huang HC, Huang YT, Sun YC, Wang FS: Extracorporeal shock waves promote healing of collagenase-induced Achilles tendinitis and increase TGF-beta1 and IGF-I expression. J Orthop Res. 2004, 22 (4): 854-861. 10.1016/j.orthres.2003.10.013.
Hsu RW, Hsu WH, Tai CL, Lee KF: Effect of shock-wave therapy on patellar tendinopathy in a rabbit model. J Orthop Res. 2004, 22 (1): 221-227. 10.1016/S0736-0266(03)00138-4.
Lui PP, Fu SC, Chan LS, Hung LK, Chan KM: Chondrocyte phenotype and ectopic ossification in collagenase-induced tendon degeneration. J Histochem Cytochem. 2009, 57 (2): 91-100. 10.1369/jhc.2008.952143.
Lui PP, Chan LS, Lee YW, Fu SC, Chan KM: Sustained expression of proteoglycans and collagen type III/type I ratio in a calcified tendinopathy model. Rheumatology (Oxford). 2010, 49 (2): 231-239. 10.1093/rheumatology/kep384.
Backman C, Boquist L, Fridén J, Lorentzon R, Toolanen G: Chronic achilles paratenonitis with tendinosis: an experimental model in the rabbit. J Orthop Res. 1990, 8 (4): 541-547. 10.1002/jor.1100080410.
Soslowsky LJ, Thomopoulos S, Tun S, Flanagan CL, Keefer CC, Mastaw J, Carpenter JE: Neer Award 1999. Overuse activity injures the supraspinatus tendon in an animal model: a histologic and biomechanical study. J Shoulder Elbow Surg. 2000, 9 (2): 79-84. 10.1016/S1058-2746(00)90033-8.
Nakama LH, King KB, Abrahamsson S, Rempel DM: Evidence of tendon microtears due to cyclical loading in an in vivo tendinopathy model. J Orthop Res. 2005, 23 (5): 1199-1205. 10.1016/j.orthres.2005.03.006.
Archambault JM, Jelinsky SA, Lake SP, Hill AA, Glaser DL, Soslowsky LJ: Rat supraspinatus tendon expresses cartilage markers with overuse. J Orthop Res. 2007, 25 (5): 617-624. 10.1002/jor.20347.
Glazebrook MA, Wright JR, Langman M, Stanish WD, Lee JM: Histological analysis of achilles tendons in an overuse rat model. J Orthop Res. 2008, 26 (6): 840-846. 10.1002/jor.20546.
Foland JW, Trotter GW, Powers BE, Wrigley RH, Smith FW: Effect of sodium hyaluronate in collagenase-induced superficial digital flexor tendinitis in horses. Am J Vet Res. 1992, 53 (12): 2371-2376.
Messner K, Wei Y, Andersson B, Gillquist J, Räsänen T: Rat model of Achilles tendon disorder. A pilot study. Cells Tissues Organs. 1999, 165 (1): 30-39. 10.1159/000016671.
Fu SC, Chan KM, Chan LS, Fong DT, Lui PY: The use of motion analysis to measure pain-related behaviour in a rat model of degenerative tendon injuries. J Neurosci Methods. 2009, 179 (2): 309-318. 10.1016/j.jneumeth.2009.02.011.
Fedorczyk JM, Barr AE, Rani S, Gao HG, Amin M, Amin S, Litvin J, Barbe MF: Exposure-dependent increases in IL-1beta, substance P, CTGF, and tendinosis in flexor digitorum tendons with upper extremity repetitive strain injury. J Orthop Res. 2010, 28 (3): 298-307.
Zhang J, Wang JH: Production of PGE(2) increases in tendons subjected to repetitive mechanical loading and induces differentiation of tendon stem cells into non-tenocytes. J Orthop Res. 2010, 28 (2): 198-203.
Jelinsky SA, Lake SP, Archambault JM, Soslowsky LJ: Gene expression in rat supraspinatus tendon recovers from overuse with rest. Clin Orthop Relat Res. 2008, 466 (7): 1612-1617. 10.1007/s11999-008-0270-z.
Almekinders LC, Banes AJ, Ballenger CA: Effects of repetitive motion on human fibroblasts. Med Sci Sports Exerc. 1993, 25 (5): 603-607.
Skutek M, van Griensven M, Zeichen J, Brauer N, Bosch U: Cyclic mechanical stretching enhances secretion of Interleukin 6 in human tendon fibroblasts. Knee Surg Sports Traumatol Arthrosc. 2001, 9 (5): 322-326. 10.1007/s001670100217.
Bosch U, Zeichen J, Skutek M, Albers I, van Griensven M, Gässler N: Effect of cyclical stretch on matrix synthesis of human patellar tendon cells. Unfallchirurg. 2002, 105 (5): 437-442. 10.1007/s00113-001-0373-4.
Wang JH, Jia F, Yang G, Yang S, Campbell BH, Stone D, Woo SL: Cyclic mechanical stretching of human tendon fibroblasts increases the production of prostaglandin E2 and levels of cyclooxygenase expression: a novel in vitro model study. Connect Tissue Res. 2003, 44 (3-4): 128-133. 10.1080/03008200390223909.
Wang JH, Li Z, Yang G, Khan M: Repetitively stretched tendon fibroblasts produce inflammatory mediators. Clin Orthop Relat Res. 2004, 243-250. 10.1097/01.blo.0000126337.65685.e4. 422
Li Z, Yang G, Khan M, Stone D, Woo SL, Wang JH: Inflammatory response of human tendon fibroblasts to cyclic mechanical stretching. Am J Sports Med. 2004, 32 (2): 435-440. 10.1177/0095399703258680.
Yang G, Im HJ, Wang JH: Repetitive mechanical stretching modulates IL-1beta induced COX-2, MMP-1 expression, and PGE2 production in human patellar tendon fibroblasts. Gene. 2005, 363: 166-172. 10.1016/j.gene.2005.08.006.
Riley GP, Cox M, Harrall RL, Clements S, Hazleman BL: Inhibition of tendon cell proliferation and matrix glycosaminoglycan synthesis by non-steroidal anti-inflammatory drugs in vitro. J Hand Surg Br. 2001, 26 (3): 224-228. 10.1054/jhsb.2001.0560.
Corps AN, Harrall RL, Curry VA, Fenwick SA, Hazleman BL, Riley GP: Ciprofloxacin enhances the stimulation of matrix metalloproteinase 3 expression by interleukin-1beta in human tendon-derived cells. A potential mechanism of fluoroquinolone-induced tendinopathy. Arthritis Rheum. 2002, 46 (11): 3034-3040. 10.1002/art.10617.
Wong MW, Tang YN, Fu SC, Lee KM, Chan KM: Triamcinolone suppresses human tenocyte cellular activity and collagen synthesis. Clin Orthop Relat Res. 2004, 277-281. 10.1097/01.blo.0000118184.83983.65. 421
Wong MW, Tang YY, Lee SK, Fu BS: Glucocorticoids suppress proteoglycan production by human tenocytes. Acta Orthop. 2005, 76 (6): 927-931. 10.1080/17453670610046118.
Scutt N, Rolf CG, Scutt A: Glucocorticoids inhibit tenocyte proliferation and Tendon progenitor cell recruitment. J Orthop Res. 2006, 24 (2): 173-182. 10.1002/jor.20030.
Tempfer H, Gehwolf R, Lehner C, Wagner A, Mtsariashvili M, Bauer HC, Resch H, Tauber M: Effects of crystalline glucocorticoid triamcinolone acetonide on cultered human supraspinatus tendon cells. Acta Orthop. 2009, 80 (3): 357-362. 10.3109/17453670902988360.
Flick J, Devkota A, Tsuzaki M, Almekinders L, Weinhold P: Cyclic loading alters biomechanical properties and secretion of PGE2 and NO from tendon explants. Clin Biomech (Bristol, Avon). 2006, 21 (1): 99-106. 10.1016/j.clinbiomech.2005.08.008.
Gardner K, Arnoczky SP, Caballero O, Lavagnino M: The effect of stress-deprivation and cyclic loading on the TIMP/MMP ratio in tendon cells: an in vitro experimental study. Disabil Rehabil. 2008, 30 (20-22): 1523-1529. 10.1080/09638280701785395.
Zhang J, Wang JH: Mechanobiological response of tendon stem cells: Implications of tendon homeostasis and pathogenesis of tendinopathy. J Orthop Res. 2010, 28 (5): 639-643.
Hossain MA, Park J, Choi SH, Kim G: Dexamethasone induces apoptosis in proliferative canine tendon cells and chondrocytes. Vet Comp Orthop Traumatol. 2008, 21 (4): 337-342.
Sendzik J, Shakibaei M, Schäfer-Korting M, Lode H, Stahlmann R: Synergistic effects of dexamethasone and quinolones on human-derived tendon cells. Int J Antimicrob Agents. 2010, 35 (4): 366-374. 10.1016/j.ijantimicag.2009.10.009.
Tsai WC, Hsu CC, Chou SW, Chung CY, Chen J, Pang JH: Effects of celecoxib on migration, proliferation and collagen expression of tendon cells. Connect Tissue Res. 2007, 48 (1): 46-51. 10.1080/03008200601071295.
Tsai WC, Hsu CC, Chang HN, Lin YC, Lin MS, Pang JH: Ibuprofen upregulates expressions of matrix metalloproteinase-1, -8, -9, and -13 without affecting expressions of types I and III collagen in tendon cells. J Orthop Res. 2010, 28 (4): 487-491.
Chan KM, Fu SC: Anti-inflammatory management for tendon injuries - friends or foes?. Sports Med Arthrosc Rehabil Ther Technol. 2009, 1 (1): 23-10.1186/1758-2555-1-23.
Burry HC: Pathogenesis of some traumatic and degenerative disorders of soft tissue. Aust N Z J Med. 1978, 8 (Suppl 1): 163-167.
Leadbetter WB: Cell-matrix response in tendon injury. Clin Sports Med. 1992, 11: 533-578.
Khan KM, Cook JL, Bonar F, Harcourt P, Astrom M: Histopathology of common tendinopathies. Update and implications for clinical management. Sports Med. 1999, 27 (6): 393-408. 10.2165/00007256-199927060-00004.
Kibler WB: Clinical aspects of muscle injury. Med Sci Sports Exerc. 1990, 22 (4): 450-452.
Sorosky B, Press J, Plastaras C, Rittenberg J: The practical management of Achilles tendinopathy. Clin J Sport Med. 2004, 14 (1): 40-44. 10.1097/00042752-200401000-00007.
Murrell GA: Understanding tendinopathies. Br J Sports Med. 2002, 36: 392-393. 10.1136/bjsm.36.6.392.
Yuan J, Wang MX, Murrell GA: Cell death and tendinopathy. Clin Sports Med. 2003, 22 (4): 693-701. 10.1016/S0278-5919(03)00049-8.
Xu Y, Murrell GA: The basic science of tendinopathy. Clin Orthop Relat Res. 2008, 466 (7): 1528-1538. 10.1007/s11999-008-0286-4.
Orchard JW, Cook JL, Halpin N: Stress-shielding as a cause of insertional tendinopathy: the operative technique of limited adductor tenotomy supports this theory. J Sci Med Sport. 2004, 7 (4): 424-428. 10.1016/S1440-2440(04)80259-7.
Nirschl RP: Rotator cuff tendinitis: basic concepts of pathoetiology. Instr Course Lect. 1989, 38: 439-445.
Almekinders LC, Weinhold PS, Maffulli N: Compression etiology in tendinopathy. Clin Sports Med. 2003, 22 (4): 703-710. 10.1016/S0278-5919(03)00067-X.
Pufe T, Petersen WJ, Mentlein R, Tillmann BN: The role of vasculature and angiogenesis for the pathogenesis of degenerative tendons disease. Scand J Med Sci Sports. 2005, 15 (4): 211-222. 10.1111/j.1600-0838.2005.00465.x.
Riley G: The pathogenesis of tendinopathy. A molecular perspective. Rheumatology (Oxford). 2004, 43 (2): 131-142. 10.1093/rheumatology/keg448.
Fredberg U, Stengaard-Pedersen K: Chronic tendinopathy tissue pathology, pain mechanisms, and etiology with a special focus on inflammation. Scand J Med Sci Sports. 2008, 18 (1): 3-15. 10.1111/j.1600-0838.2007.00746.x.
Corps AN, Jones GC, Harrall RL, Curry VA, Hazleman BL, Riley GP: The regulation of aggrecanase ADAMTS-4 expression in human Achilles tendon and tendon-derived cells. Matrix Biol. 2008, 27 (5): 393-401. 10.1016/j.matbio.2008.02.002.
Sun HB, Li Y, Fung DT, Majeska RJ, Schaffler MB, Flatow EL: Coordinate regulation of IL-1beta and MMP-13 in rat tendons following subrupture fatigue damage. Clin Orthop Relat Res. 2008, 466 (7): 1555-1561. 10.1007/s11999-008-0278-4.
Thornton GM, Shao X, Chung M, Sciore P, Boorman RS, Hart DA, Lo IK: Changes in mechanical loading lead to tendon-specific alterations in MMP and TIMP expression: Influence of stress-deprivation and intermittent cyclic hydrostatic compression on rat supraspinatus and Achilles tendons. Br J Sports Med. 2010, 44 (10): 698-703. 10.1136/bjsm.2008.050575.
Arnoczky SP, Lavagnino M, Egerbacher M, Caballero O, Gardner K: Matrix metalloproteinase inhibitors prevent a decrease in the mechanical properties of stress-deprived tendons: an in vitro experimental study. Am J Sports Med. 2007, 35 (5): 763-769. 10.1177/0363546506296043.
Lim S, Hossain MA, Park J, Choi SH, Kim G: The effects of enrofloxacin on canine tendon cells and chondrocytes proliferation in vitro. Vet Res Commun. 2008, 32 (3): 243-253. 10.1007/s11259-007-9024-8.
Pouzaud F, Bernard-Beaubois K, Thevenin M, Warnet JM, Hayem G, Rat P: In vitro discrimination of fluoroquinolones toxicity on tendon cells: involvement of oxidative stress. J Pharmacol Exp Ther. 2004, 308 (1): 394-402. 10.1124/jpet.103.057984.
Garau G, Rittweger J, Mallarias P, Longo UG, Maffulli N: Traumatic patellar tendinopathy. Disabil Rehabil. 2008, 30 (20-22): 1616-1620. 10.1080/09638280701786096.
Halla JT, Gould JS, Hardin JG: Chronic tenosynovial hand infection from Mycobacterium terrae. Arthritis Rheum. 1979, 22 (12): 1386-1390. 10.1002/art.1780221211.
Ackermann PW, Salo PT, Hart DA: Neuronal pathways in tendon healing. Front Biosci. 2009, 14: 5165-5187. 10.2741/3593.
Lui PP, Cheuk YC, Hung LK, Fu SC, Chan KM: Increased apoptosis at the late stage of tendon healing. Wound Repair Regen. 2007, 15 (5): 702-707. 10.1111/j.1524-475X.2007.00276.x.
Schizas N, Lian O, Frihagen F, Engebretsen L, Bahr R, Ackermann PW: Coexistence of up-regulated NMDA receptor 1 and glutamate on nerves, vessels and transformed tenocytes in tendinopathy. Scand J Med Sci Sports. 2010, 20 (2): 208-215. 10.1111/j.1600-0838.2009.00913.x.
Clegg PD, Strassburg S, Smith RK: Cell phenotypic variation in normal and damaged tendons. Int J Exp Pathol. 2007, 88 (4): 227-235. 10.1111/j.1365-2613.2007.00549.x.
Arya S, Kulig K: Tendinopathy alters mechanical and material properties of the Achilles tendon. J Appl Physiol. 2010, 108 (3): 670-675. 10.1152/japplphysiol.00259.2009.
Arnoczky SP, Lavagnino M, Egerbacher M: The mechanobiological aetiopathogenesis of tendinopathy: is it the over-stimulation or the under-stimulation of tendon cells?. Int J Exp Pathol. 2007, 88 (4): 217-226. 10.1111/j.1365-2613.2007.00548.x.
Fu SC, Cheuk YC, Chan KM, Hung LK, Wong MW: Is cultured tendon fibroblast a good model to study tendon healing?. J Orthop Res. 2008, 26 (3): 374-383. 10.1002/jor.20483.
Dudhia J, Scott CM, Draper ER, Heinegård D, Pitsillides AA, Smith RK: Aging enhances a mechanically-induced reduction in tendon strength by an active process involving matrix metalloproteinase activity. Aging Cell. 2007, 6 (4): 547-556. 10.1111/j.1474-9726.2007.00307.x.
Dillon EM, Erasmus PJ, Müller JH, Scheffer C, de Villiers RV: Differential forces within the proximal patellar tendon as an explanation for the characteristic lesion of patellar tendinopathy: an in vivo descriptive experimental study. Am J Sports Med. 2008, 36 (11): 2119-2127. 10.1177/0363546508319311.
Acknowledgements
No funding has been received for this review.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
SCF, RC and KMC planned and drafted the manuscript. YCC managed the references and assisted in drafting. PPYL approved the final version. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Fu, SC., Rolf, C., Cheuk, YC. et al. Deciphering the pathogenesis of tendinopathy: a three-stages process. BMC Sports Sci Med Rehabil 2, 30 (2010). https://doi.org/10.1186/1758-2555-2-30
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1758-2555-2-30