
Abstract
Mitosis is accompanied by dramatic changes in chromatin organization and nuclear architecture. Transcription halts globally and most sequence-specific transcription factors and co-factors are ejected from mitotic chromatin. How then does the cell maintain its transcriptional identity throughout the cell division cycle? It has become clear that not all traces of active transcription and gene repression are erased within mitotic chromatin. Many histone modifications are stable or only partially diminished throughout mitosis. In addition, some sequence-specific DNA binding factors have emerged that remain bound to select sites within mitotic chromatin, raising the possibility that they function to transmit regulatory information through the transcriptionally silent mitotic phase, a concept that has been termed “mitotic bookmarking.” Here we review recent approaches to studying potential bookmarking factors with regards to their mitotic partitioning, and summarize emerging ideas concerning the in vivo functions of mitotically bound nuclear factors.
Similar content being viewed by others
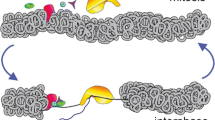


Introduction/Overview
Mitosis imposes dramatic and dynamic changes on nuclear organization and gene expression in eukaryotic cells. In metazoans, the nuclear envelope temporarily disintegrates, most nuclear structures are transiently dissolved or rearranged, and nuclear transcription by all three polymerases is globally arrested. Silencing of gene expression is accompanied by the separation of most transcriptional regulators from mitotic chromatin. Following chromosome segregation and re-formation of the nuclear envelope, bulk transcription resumes in the daughter cells that ultimately re-acquire gene expression patterns that are often similar to or indistinguishable from those of the mother cell. It has been widely assumed that these transitions present a problem for the cell’s maintenance of transcriptional identity, prompting investigations into mechanisms that ensure rapid and faithful restoration of gene expression upon re-entry into the G1 phase of the cell cycle. Such mechanisms fall under the category of epigenetics in the stricter sense of the word by providing a cellular memory function throughout the cell division cycle [1]. However, one might question the need for such dedicated mechanisms since the half-lives of most transcripts exceed the duration of mitosis [2]. Therefore, the appropriate regulatory milieu is essentially maintained through mitosis if factors segregate passively in a random fashion. Most protein-DNA contacts are highly dynamic even in interphase (see [3] for review), and consequently, mitotically displaced nuclear regulators would be expected to reload on the correct targets by mass action [4]. This process might be aided by mitotically stable properties of chromatin such as post-translational modifications or nucleosome architecture. However, transcription factors are employed combinatorially at distinct genes and lineages, which allows a limited set of factors to control diverse gene expression programs. This raises the question whether following mitosis, association of these regulators with lineage- or developmental stage-inappropriate genes could lead to changes in cellular growth or differentiation.
Such considerations led to the exploration of diverse mechanisms of gene bookmarking in mitosis to prevent potentially detrimental alterations in gene expression upon re-entry into G1 [5]. These include the retention of DNA binding proteins or transcription co-factors on mitotic chromosomes, mitotically stable histone modifications and histone variants, as well as features of nucleosome architecture and even DNA topology that may at least partially persist through mitosis. Several thorough reviews covered these topics in the recent past [4, 6–10].
A different perspective on the effects of mitosis on gene expression comes from the notion that genome-wide perturbations in transcription factor occupancy might facilitate changes in cell fate by allowing the reshaping of transcription programs. An impressive example is the observation that zygotes are capable of reprogramming somatic nuclei only after recipient cells have been arrested in mitosis [11], suggesting that mitosis is required for the release of reprogramming factors from chromatin to reset transcription in donor chromatin. An open question is to what extent transition through mitosis is a more general requirement for establishing lineage diversification. Asymmetrical cell divisions trigger differences in lineage choice of progeny cells or can separate a daughter cell with self-renewal capacity from one that proceeds to differentiate [12, 13]. It is possible that in these scenarios newly created transcription environments act on post-mitotic chromatin (as opposed to interphase chromatin) to initiate new transcription patterns in newborn cells. Hence mitosis might be viewed as a window of opportunity for remodeling the transcriptional landscape, which implies that putative bookmarking mechanisms remain sufficiently flexible to permit changes in cellular fate or differentiation.
In this article instead of providing an extension of previous comprehensive reviews of known factors and histone marks that persist on mitotic chromatin, we aim to highlight recent technical and conceptual developments that approach questions of mitotic bookmarking.
Review
Methods to study mitotic bookmarking
Here we provide a brief review of frequently used approaches to study potential mitotic memory mechanisms, since inconsistencies or controversies in the literature can be rooted in different methodologies used. Immunofluorescence (IF) microscopy is commonly employed to globally survey mitotic partitioning of nuclear factors and persistence of histone marks. An advantage of IF is that it detects endogenous proteins in their natural context, but it requires suitable antibodies for which the epitope is not occluded by chromatin compaction during mitosis, by mitosis-specific post-translational modifications, or because of fixation. These problems can be sidestepped by live cell-imaging with ectopically expressed fluorophore-tagged molecules. However, overexpression might lead to shifts in mitotic occupancy patterns. The monitoring of histone marks in living cells is more challenging, although strategies have been developed using fluorescence resonance energy transfer (FRET)-based indicator molecules that specifically interact with a given histone mark [14, 15]. Given the complexities of histone marks and their molecular interactions, as well as the potential influence of neighboring marks, this approach, although creative, is not without limitations and is not yet universally applicable.
To localize nuclear factors or histone marks on specific genomic sites in mitosis, chromatin immunoprecipitation (ChIP) can be used conventionally or in combination with high throughput sequencing. Concerns about epitope recognition in mitotic cells are similar to those described for IF. The preparation of pure mitotic cells for ChIP is essential and has been aided by the recent development of effective protocols that use antibodies against phosphorylated histone H3 serine 10, a modification globally enriched during mitosis in all cell types, for fluorescence activated cell sorting (FACS) [16, 17]. This is especially relevant in cases where cells cannot be easily synchronized by pharmacologic treatments such as nocodozale.
The combinatorial use of IF, live cell imaging, and ChIP does not only serve to corroborate key results, but can also uncover new concepts. For example, the hematopoietic transcription factor GATA1 globally separates from mitotic chromosomes as revealed by IF [16, 18]. However, live cell-imaging and genome-wide location analysis by ChIP showed partial retention of this factor [16]. Moreover, ChIP detected significant mitosis-specific shifts in genomic occupancy patterns of the histone methyltransferase MLL, which was not visible by IF [19]. Notably, transcription factor FoxA1 is globally retained on mitotic chromatin as visualized by IF, even though its binding at specific binding sites as measured by ChIP is substantially diminished [20]. These findings suggest the existence of distinct layers of mitotic retention and highlight the importance of combining multiple techniques for the evaluation of potential mitotic bookmarking factors.
Additional strategies to assess mitotic chromatin binding involve the fractionation of mitotic chromosomes followed by western blotting (for example, [21]) or unbiased proteomic analysis [22]. These approaches are suitable to address questions of global chromatin association of one or many factors within a population of cells. As with all biochemical extraction protocols, conditions greatly influence the integrity and purity of large molecular complexes, requiring the implementation of multiple classifiers and independent validation [22].
The methods mentioned so far are useful tools for untangling the protein composition and post-translational modifications of mitotic chromosomes. However, the mere presence of a factor or histone mark on mitotic chromatin is insufficient to establish a functional role in mitotic memory. In several studies select mitotically retained factors were depleted by RNAi, leading to a delay in postmitotic target gene reactivation [19, 20, 23–26]. However, it is difficult to establish with certainty that the observed effects were solely due to mitotic bookmarking and not to interphase functions of these factors. In recent reports both gain-of-function experiments as well as mitosis-specific loss of function experiments were carried out to address this issue. The acetyl-histone binding protein BRD4 is known to remain on mitotic chromosomes in some cell types [23, 27]. Using a stably integrated inducible gene that allows for live monitoring of transcription, the Spector group observed that initial activation of this gene in interphase occurred with much slower kinetics than its re-activation upon mitotic exit [25]. To test whether BRD4 contributes to a potential mitotic transcriptional memory mechanism that accounts for this rapid re-activation, they took two approaches. In the first, they displaced BRD4 from the gene using a competitive inhibitor of acetyl-lysine (JQ1), which delayed postmitotic reactivation but did not affect activation of this gene in interphase. In the second, they tethered BRD4 to the locus, which accelerated transcription induction in interphase. This suggests that BRD4 via association with acetylated chromatin plays a role in transcriptional memory. It will be interesting to examine whether tethering of putative bookmarking factors to non-bookmarked native genes, perhaps via artificial zinc finger proteins or transactivator-like (TAL) factors, can accelerate their postmitotic gene activation.
In an attempt to determine a mitotic bookmarking function for GATA1 Kadauke et al. devised a strategy to deplete GATA1 selectively in mitosis [16]. GATA1 was fused to the mitosis-specific degron of cyclin B and stably introduced into GATA1-null cells. Mitosis-specific destruction of GATA1 selectively delayed post-mitotic reactivation of GATA1-bookmarked genes [16]. It should be possible to extend this approach to factors for which knockout cells are unavailable by knockdown-rescue experiments.
Finally, it is worth noting that mitotic partitioning of nuclear factors and histone marks can vary between cell types and might be influenced by whether primary or transformed cells are used (see [28] for review).
Function
One of the most important questions waiting to be resolved concerns the biological role of the mitotic retention of nuclear factors. Proposed functions include promoting rapid transcriptional activation of mitotic target genes in newborn cells to facilitate the M/G1 transition, as well as the faithful maintenance of lineage choice and differentiation stage. For some factors that were examined for mitotic occupancy on a genome-wide scale, it is notable that the fraction of bound protein and the number of genomic occupied sites on mitotic chromosomes is small when compared to interphase chromatin [16, 20, 21]. Whether the former are simply remnants of incompletely removed proteins or actually functionally relevant for postmitotic transcriptional regulation is a challenging question that can be tackled with some of the aforementioned gain- and loss-of-function approaches.
In several recent reports depletion of mitotically retained transcription factors delayed transcription reactivation of target genes upon exit from mitosis [16, 19, 20, 23–26]. Several genes expressed in the M/G1 transition but not those expressed at later stages of the cell cycle were found to be stably occupied by BRD4 through mitosis and require BRD4 for their rapid reactivation in telophase [23, 24, 29], supporting the idea that BRD4 marks these genes to ensure rapid transcription activation and cell cycle progression. However, it is also clear that genes with no known specific roles in G1 progression, such as those encoding lineage-specific nuclear factors, depend on mitotic occupancy by bookmarking factors for their rapid reactivation [16, 20, 26]. Assessment of the importance of fast transcription reactivation of a given gene also requires knowledge of transcript stability through mitosis. For those transcripts that are stable, transcriptional reactivation kinetics might not be limiting for G1 entry. Therefore, bookmarking of genes producing long-lived transcripts might serve functions other than cell cycle progression.
While timely transcription reactivation has been used as the predominant functional read-out of mitotic bookmarking, it remains unresolved to what extent this is important beyond simply allowing cell cycle progression. It has been proposed that mitotic bookmarking factors may stabilize lineage fidelity by facilitating transmission of the cell type-specific transcriptional state from mother to daughter cells. The retention of select tissue-restricted transcription factors on mitotic chromosomes [16, 18, 20, 30] provides correlative support for such a model. It is possible that factors that remain bound during mitosis at specific targets are prevented from binding to off-target sites, or block competing factors from binding, thus preserving gene expression patterns. The idea that mitosis might be a labile state amenable to global changes in gene expression (such as those associated with cellular reprogramming [11, 31, 32]) further suggests the existence of mechanisms that counter dramatic changes in gene transcription. Nevertheless, direct evidence that mitotic bookmarking is required for cementing lineage choice is lacking.
Many lineage-determining transcription factors function not only as activators of lineage-affiliated genes but also as repressors of genes associated with alternative lineages or inappropriate maturation stages. Therefore, if mitotic bookmarking is at all relevant to preserving cellular identity, the ability to stably silence gene expression during mitotic exit is likely to be of equal importance. For example, destruction of GATA1 specifically during mitosis was found to lead not only to delayed reactivation of bound genes but to inappropriately high expression of GATA1-repressed target genes [16]. These include Gata2 and Kit that are normally transcribed at an earlier developmental stage but are also expressed in alternative GATA1-dependent hematopoietic lineages, namely mast cells. Together, this suggests but does not prove that GATA1 mitotic bookmarking plays a role in maintaining cellular maturation, lineage fidelity, or both.
The nuclear factor Runx2 is involved in bone formation by activating osteoblast gene expression programs but also functions as a repressor of RNA polymerase 1-transcribed ribosomal genes. Runx2 is focally retained on mitotic chromosomes, including sites of rRNA production [26, 33]. Whether Runx2 association with mitotic chromatin is important for rDNA repression or for maintaining the osteoblast lineage, and to what extent this function is mediated by its role as activator versus repressor, remain open questions.
If retention of nuclear factors on mitotic chromatin is indeed important for cementing cell type-specific transcription patterns, we speculate that such bookmarking mechanisms are dynamic throughout development. As an example, multipotent progenitor cells display substantial stochastic cell-to-cell variation in gene expression leading to the seemingly random activation of lineage-affiliated gene expression patterns and even the formation of lineage-committed states in small subpopulations (for example see [34]). It is possible that this transcriptional flux is enabled in part by the lack of mitotic bookmarking mechanisms. Gene expression patterns might be stabilized once commitment is established by nuclear factors capable of repressing alternative lineage affiliated genes. Whether or not mitotic bookmarking serves to resist cellular reprogramming or stochastic changes in gene expression that might lead to lineage infidelity is amenable to investigation. For example, does the disabling of bookmarking factors enhance the efficiency of lineage reprogramming factors to alter cell fate? Are engineered versions of mitotically unstable nuclear factors less efficient in cellular reprogramming than their normal counterparts?
Finally, transcription factors both influence and are influenced by their chromatin environment. The dynamics of histone modifications during mitosis and possible roles in bookmarking are discussed elsewhere [10].
Mechanisms
Mechanisms that allow dispersal of nuclear factors and modulate histone modifications during mitosis have been extensively studied. Distinct kinetics of dissociation and re-association of factors during mitosis as well as varying stabilities of histone marks argue that multiple mechanisms control mitotic partitioning. Nevertheless, common themes have emerged that apply to broad classes of molecules, including dynamic phosphorylation of histones and nuclear factors. We will not discuss these mechanisms here as they have been thoughtfully covered in several recent reviews [4, 10, 28]. Instead, we will discuss the related but distinct questions of how select factors are able to remain associated with chromatin at some sites but not others, and how their selective retention might influence transcriptional reactivation or contribute to the maintenance of transcription patterns.
Although global occupancy of transcription factors is sometimes reduced during mitosis [16, 19–21], it is notable that mitotic Runx2 binding intensities seem to match those of interphase cells at numerous sites although this has not yet been examined on a genome-wide scale [26]. In the cases of MLL1 and FoxA1 [19, 20] but not GATA1 [16], mitotic retention favors genes highly expressed in interphase, suggesting that chromatin features and/or transcription factor environment of highly active genes promotes mitotic retention. It is possible that in these scenarios, mitotic bookmarking facilitates the dramatic transitions from off- to on-states when exiting mitosis.
The observed transcription factor occupancy patterns typically fall into distinct classes. At some sites nuclear factors are retained at normal levels while at others binding is diminished, which might be a result of fewer molecules per occupied site or fewer alleles being bound in heterogenous cell populations. In addition, new sites might be occupied by nuclear factors only in mitosis, thus signaling a shift in binding properties [16, 19, 35]. While the defining features of these groups are still unclear, some ideas are emerging as discussed in the following paragraphs.
DNA sequence context
DNA-binding factors generally occupy sites in a sequence-specific fashion, and variation in sequence can affect binding dynamics. Comparative genome-wide occupancy profiles of sequence-specific transcription factors in interphase and mitosis are now beginning to be determined [16, 20, 21]. DNA binding motif analysis of GATA1 and FoxA1 has not revealed any features that predict whether the factor remains bound or dissociates during mitosis [16, 20]. More factors need to be studied to determine whether this is universally true.
An emerging theme of mitosis-specific occupancy involves repetitive DNA sequence elements. During mitosis the D. melanogaster GAGA transcription factor shifts from its interphase binding sites to centromeric alpha-satellite GA repeats [35, 36]. Moreover, Runx2 concentrates on rDNA repeats in mitosis, which contain a high density of Runx2 binding motifs [33]. The majority of mitosis-only GATA1 target sites tend to localize to (GATA)n repeats in intergenic regions remote from any annotated genes [16]. Why would DNA-binding factors dissociate from their interphase binding sites and occupy repeat sequences? Transcription factor occupancy is influenced by the presence of neighboring nuclear factors that might convey binding synergy, or facilitate binding by association with chromatin modifying factors. Perturbation of transcription factor/co-factor context during mitosis might trigger the dispersal of these factors away from interphase binding sites, making them available for otherwise less favorable locations. This equilibrium is likely influenced by the number of repeat elements. Moreover, the chromatin context of those sites might not be as tightly controlled. It remains unclear what function these mitosis-only binding events serve. In the case of GAGA factor, it is possible that it contributes to packaging of chromatin encompassing repeat sequences during mitotic chromosome condensation [35]. It is also conceivable that these repeat elements serve a transient depot function by restricting movement away from chromatin and allowing more rapid re-association with interphase targets.
Local chromatin context
Several features of chromatin such as DNase hypersensitivity, various modifications of DNA and histones, and histone variants persist at least in part through mitosis while others are dynamic throughout the cell cycle [37–47]. It seems reasonable to propose that the chromatin environment might modulate mitotic transcription factor binding. However, at present there are no known features of chromatin that predict with certainty whether a transcription factor remains bound to mitotic chromatin or not. DNase hypersensitive sites are generally maintained in mitotic chromatin [16, 37, 47] with some exceptions [48]. Although GATA1 contributes to the formation of DNase hypersensitive sites, they remain hypersensitive during mitosis regardless of whether GATA1 persists there or not [16], implicating additional epigenetic mechanisms that maintain them.
However, some features of chromatin structure influence transcription factor binding profiles during mitosis. FoxA1 binding in mitosis seems to favor sites of high nucleosome density, consistent with its strong affinity for nucleosomes [20]. It has also been observed that positioned nucleosomes are lost at some genes during mitosis, raising the possibility that altered nucleosome position impacts on mitotic transcription factor binding or vice-versa [49]. The histone variant H2A.Z, which is associated with active and poised promoters, is retained during mitosis despite transcriptional silencing, although its acetylation is diminished [50]. Interestingly, global analyses revealed that the H2A.Z-containing +1 nucleosome slides towards the 5’ end of genes to occupy transcriptional start sites and shrink the nucleosome-free region at gene promoters [45], potentially providing a universal mechanism for temporary gene silencing. It is possible that repositioning of promoter proximal nucleosomes contributes to the mitotic displacement of basal transcription factors. Investigation into how reversible nucleosome sliding is controlled in such a temporally and spatially specific manner will likely provide important new insights into mitotic chromatin dynamics.
The post-translational histone modifications examined so far fail to clearly distinguish interphase-only from persistent transcription factor binding sites [16, 20], although subtle trends do exist. Runx2 mitotic binding is associated with increased H3K4 dimethylation [26]. GATA1 sites that are selectively occupied in mitosis tend to be enriched for the repressive histone marks H3K27me3 and H3K9me3 [16]. However, it is unclear whether this is simply a consequence of the presence of these marks at (GATA)n repeats (see previous section).
The polycomb protein PSC is partially maintained on chromatin in mitosis but not at the prototypic PSC target Hox locus, even though the polycomb target mark H3K27me3 is present at this locus throughout mitosis [21]. Notably, almost half of the retained sites fall on boundaries of topological domains that are also occupied by molecules involved in higher order chromatin organization. It will be interesting to examine the local chromatin context of mitotically persistent PSC sites as well as the possible role of PSC bookmarking in the restoration of long-range chromatin contacts after mitosis.
Dynamic phosphorylation of histones, transcription factors, and the mitotic condensation machinery control transitions through mitosis [6, 28], and several reports describe mechanisms by which these histone modifications can eject chromatin binding factors during mitosis [10, 51, 52]. The basal transcription factor TBP, which occupies a subset of promoters in mitosis and HSF2 can interact with the protein phosphatase PP2A, which can dephosphorylate subunits of the condensin complex [53, 54]. This leads to the tantalizing proposition that localized dephosphorylation of condensin antagonizes chromatin compaction at select sites leaving a mitotic bookmark [54]. To what extent this mechanism is broadly operational in vivo will be an important question for the future. It is notable that PP2A can also reverse the mitotic inactivation of the SWI/SNF chromatin remodeling complex [55], but since SWI/SNF is globally separated from mitotic chromatin it is unclear whether this mechanism functions to antagonize mitotic nucleosome repositioning, unless the complex is retained locally at bookmarked regions.
Acetylation of histones is at least partially maintained during mitosis [7]. Readers of histone acetylation of the BET family proteins BRD2 and BRD4 remain globally associated with mitotic chromosomes in some cell types in a manner dependent on histone acetylation [23, 27]. In other cell types BRD4 was found to separate from mitotic chromatin but to re-associate rapidly after mitosis, prior to other examined factors [23, 25] (see also above). Potential mechanisms by which BET family proteins act to promote rapid transcriptional reactivation include recruitment of a variety of transcription complexes including P-TEFb, or possibly its intrinsic RNA polymerase 2 kinase activity [23, 24, 56, 57]. Compounds targeting BET family proteins have garnered a great deal of attention for their potential as anti-inflammatory and anti-cancer drugs (see [58] for review). It is possible that some of their activities relate to disruption of mitotic memory functions required to sustain tumor cell growth or the expansion and function of immunocompetent cells.
Transcription co-factors
Transcription co-regulators are typically loaded onto chromatin via DNA binding proteins. It is therefore expected that mitotic partitioning of the former mirrors that of the latter. Correspondingly, the majority of cofactors are separated from mitotic chromosomes (Table 1). A notable exception is the histone methyltransferase MLL1, which is globally retained on mitotic chromosomes along with its partners Menin, ASH2L, and RbBP5 (Table 2, [19]). The Runx2 co-repressor TLE1 appears to co-localize with Runx2 on select sites in mitotic chromatin while HDAC1 does not [59], suggesting selectivity among mitotically stable protein interactions. The GATA1 cofactors FOG1 and components of the TAL1 complex dissociate from all binding sites irrespective of whether GATA1 remains or not [16]. This suggests that protein-protein interactions are also dynamic throughout mitosis and that mitotically persistent transcription factors can function as a platform on which cofactor complexes are reassembled, thereby accelerating transcription reactivation.
Conclusion
For over half a century it has been known that transcription is globally silenced during mitosis [114, 115], yet how the cell copes with the challenges imposed by mitotic reorganization of the genome and nuclear structures is still largely a mystery. The development of new tools, including high-throughput technologies has brought new insights into this question. Thus, histone modifications, nucleosome architecture, and transcription factor-binding during mitosis are beginning to be unraveled. It has become clear that multiple features that discern active from inactive genes are stable through mitosis. Further studies involving diverse cellular systems are required to address similarities, but also distinctions among potential bookmarking mechanisms between lineages and organisms. It will also be important to examine as yet mostly unstudied aspects of gene expression during mitosis, including the fate of non-coding RNAs, or the dynamics of higher order chromatin folding.
To what extent mitotically stable features of chromatin are required for the propagation of transcriptional information and maintenance is still mostly subject to speculation but has moved closer within our reach. Important related questions to be addressed include whether there is a direct role for mitosis in facilitating changes in transcription patterns to allow cell fate decisions during development, and whether alleviation of bookmarking facilitates cellular reprogramming or lineage switching. Finally, it will be important to explore whether perturbation of mitotic bookmarking accounts for developmental disorders or malignancies.
Abbreviations
- ASH2L:
-
Absent, small, or homeotic-like protein
- BET:
-
bromodomain-ET
- BRD:
-
bromodomain-containing protein
- ChIP:
-
chromatin immunoprecipitation
- FACS:
-
fluorescence-activated cell sorting
- FoxA1:
-
forkhead box A1
- FOG 1:
-
Friend of GATA 1
- FRET:
-
fluorescence resonance energy transfer
- GATA1:
-
GATA binding factor 1
- HDAC1:
-
Histone deacetylase 1
- IF:
-
immunofluorescence
- P-TEFb:
-
positive transcription elongation factor b
- PSC:
-
posterior sex combs
- RbBP5:
-
Retinoblastoma-binding protein 5
- SWI/SNF:
-
switch/sucrose nonfermentable
- TAL:
-
transactivator-like
- TAL1:
-
T-cell acute lymphocytic leukemia protein 1
- TBP:
-
TATA-binding protein
- TLE:
-
Transducin-like enhancer protein 1
References
Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A: An operational definition of epigenetics. Genes and Development. 2009, 23: 781-783. 10.1101/gad.1787609.
Murphy W, Attardi G: Stability of cytoplasmic messenger RNA in HeLa cels. Proc Natl Acad Sci USA. 1973, 70: 115-119. 10.1073/pnas.70.1.115.
Hager GL, McNally JG, Misteli T: Transcription dynamics. Mol Cell. 2009, 35: 741-753. 10.1016/j.molcel.2009.09.005.
Egli D, Birkhoff G, Eggan K: Mediators of reprogramming: transcription factors and transitions through mitosis. Nat Rev Mol Cell Biol. 2008, 9: 505-516. 10.1038/nrm2439.
Michelotti EF, Sanford S, Levens D: Marking of active genes on mitotic chromosomes. Nature. 1997, 388: 895-899. 10.1038/42282.
Gottesfeld JM, Forbes DJ: Mitotic repression of the transcriptional machinery. Trends Biochem Sci. 1997, 22: 197-202. 10.1016/S0968-0004(97)01045-1.
Davie JR, He S, Li L, Sekhavat A, Espino P, Drobic B, Dunn KL, Sun J-M, Chen HY, Yu J, Pritchard S, Wang X: Nuclear organization and chromatin dynamics–Sp1, Sp3 and histone deacetylases. Ad Enzyme Regul. 2008, 48: 189-208. 10.1016/j.advenzreg.2007.11.016.
Zaidi SK, Young DW, Montecino M, van Wijnen AJ, Stein JL, Lian JB, Stein GS: Bookmarking the genome: maintenance of epigenetic information. J Biol Chem. 2011, 286: 18355-18361. 10.1074/jbc.R110.197061.
Sarge KD, Park-Sarge O-K: Mitotic bookmarking of formerly active genes: keeping epigenetic memories from fading. Cell Cycle. 2009, 8: 818-823. 10.4161/cc.8.6.7849.
Wang F, Higgins JMG:Histone modifications and mitosis: countermarks, landmarks, and bookmarks. Trends Cell Biol. 2012, Dec 13. [Epub ahead of print].
Egli D, Rosains J, Birkhoff G, Eggan K: Developmental reprogramming after chromosome transfer into mitotic mouse zygotes. Nature. 2007, 447: 679-685. 10.1038/nature05879.
Chang JT, Palanivel VR, Kinjyo I, Schambach F, Intlekofer AM, Banerjee A, Longworth SA, Vinup KE, Mrass P, Oliaro J, Killeen N, Orange JS, Russell SM, Weninger W, Reiner SL: Asymmetric T lymphocyte division in the initiation of adaptive immune responses. Science. 2007, 315: 1687-1691. 10.1126/science.1139393.
Knoblich JA: Mechanisms of asymmetric stem cell division. Cell. 2008, 132: 583-597. 10.1016/j.cell.2008.02.007.
Sasaki K, Ito T, Nishino N, Khochbin S, Yoshida M: Real-time imaging of histone H4 hyperacetylation in living cells. Proc Natl Acad Sci USA. 2009, 106: 16257-16262. 10.1073/pnas.0902150106.
Ito T, Umehara T, Sasaki K, Nakamura Y, Nishino N, Terada T, Shirouzu M, Padmanabhan B, Yokoyama S, Ito A, Yoshida M: Real-time imaging of histone H4K12-specific acetylation determines the modes of action of histone deacetylase and bromodomain inhibitors. Chem Biol. 2011, 18: 495-507. 10.1016/j.chembiol.2011.02.009.
Kadauke S, Udugama MI, Pawlicki JM, Achtman JC, Jain DP, Cheng Y, Hardison RC, Blobel GA: Tissue-Specific Mitotic Bookmarking by Hematopoietic Transcription Factor GATA1. Cell. 2012, 150: 725-737. 10.1016/j.cell.2012.06.038.
Follmer NE, Francis NJ: Preparation of Drosophila tissue culture cells from different stages of the cell cycle for chromatin immunoprecipitation using centrifugal counterflow elutriation and fluorescence-activated cell sorting. Meth Enzymol. 2012, 513: 251-269.
Xin L, Zhou G-L, Song W, Wu X-S, Wei G-H, Hao D-L, Lv X, Liu D-P, Liang C-C: Exploring cellular memory molecules marking competent and active transcriptions. BMC Mol Biol. 2007, 8: 31-10.1186/1471-2199-8-31.
Blobel GA, Kadauke S, Wang E, Lau AW, Zuber J, Chou MM, Vakoc CR: A reconfigured pattern of MLL occupancy within mitotic chromatin promotes rapid transcriptional reactivation following mitotic exit. Mol Cell. 2009, 36: 970-983. 10.1016/j.molcel.2009.12.001.
Caravaca JM, Donahue G, Becker JS, He X, Vinson C, Zaret KS: Bookmarking by specific and nonspecific binding of FoxA1 pioneer factor to mitotic chromosomes. Genes Dev. 2013, 27: 251-260. 10.1101/gad.206458.112.
Follmer NE, Wani AH, Francis NJ: A polycomb group protein is retained at specific sites on chromatin in mitosis. PLoS Genet. 2012, 8: e1003135-10.1371/journal.pgen.1003135.
Ohta S, Bukowski-Wills J-C, Sanchez-Pulido L, Alves F de L, Wood L, Chen ZA, Platani M, Fischer L, Hudson DF, Ponting CP, Fukagawa T, Earnshaw WC, Rappsilber J: The protein composition of mitotic chromosomes determined using multiclassifier combinatorial proteomics. Cell. 2010, 142: 810-821. 10.1016/j.cell.2010.07.047.
Yang Z, He N, Zhou Q: Brd4 recruits P-TEFb to chromosomes at late mitosis to promote G1 gene expression and cell cycle progression. Mol Cell Biol. 2008, 28: 967-976. 10.1128/MCB.01020-07.
Dey A, Nishiyama A, Karpova T, McNally J, Ozato K: Brd4 marks select genes on mitotic chromatin and directs postmitotic transcription. Mol Biol Cell. 2009, 20: 4899-4909. 10.1091/mbc.E09-05-0380.
Zhao R, Nakamura T, Fu Y, Lazar Z, Spector DL: Gene bookmarking accelerates the kinetics of post-mitotic transcriptional re-activation. Nat Cell Biol. 2011, 13: 1295-1304. 10.1038/ncb2341.
Young DW, Hassan MQ, Yang X-Q, Galindo M, Javed A, Zaidi SK, Furcinitti P, Lapointe D, Montecino M, Lian JB, Stein JL, van Wijnen AJ, Stein GS: Mitotic retention of gene expression patterns by the cell fate-determining transcription factor Runx2. Proc Natl Acad Sci USA. 2007, 104: 3189-3194. 10.1073/pnas.0611419104.
Dey A, Ellenberg J, Farina A, Coleman AE, Maruyama T, Sciortino S, Lippincott-Schwartz J, Ozato K: A bromodomain protein, MCAP, associates with mitotic chromosomes and affects G(2)-to-M transition. Mol Cell Biol. 2000, 20: 6537-6549. 10.1128/MCB.20.17.6537-6549.2000.
Delcuve GP, He S, Davie JR: Mitotic partitioning of transcription factors. J Cell Biochem. 2008, 105: 1-8. 10.1002/jcb.21806.
Mochizuki K, Nishiyama A, Jang MK, Dey A, Ghosh A, Tamura T, Natsume H, Yao H, Ozato K: The bromodomain protein Brd4 stimulates G1 gene transcription and promotes progression to S phase. J Biol Chem. 2008, 283: 9040-9048. 10.1074/jbc.M707603200.
Zaidi SK, Young DW, Pockwinse SM, Javed A, Lian JB, Stein JL, van Wijnen AJ, Stein GS: Mitotic partitioning and selective reorganization of tissue-specific transcription factors in progeny cells. Proc Natl Acad Sci USA. 2003, 100: 14852-14857. 10.1073/pnas.2533076100.
Lemaitre J-M, Danis E, Pasero P, Vassetzky Y, Méchali M: Mitotic remodeling of the replicon and chromosome structure. Cell. 2005, 123: 787-801. 10.1016/j.cell.2005.08.045.
Ganier O, Bocquet S, Peiffer I, Brochard V, Arnaud P, Puy A, Jouneau A, Feil R, Renard J-P, Méchali M: Synergic reprogramming of mammalian cells by combined exposure to mitotic Xenopus egg extracts and transcription factors. Proc Natl Acad Sci USA. 2011, 108: 17331-17336. 10.1073/pnas.1100733108.
Young DW, Hassan MQ, Pratap J, Galindo M, Zaidi SK, Lee S-H, Yang X, Xie R, Javed A, Underwood JM, Furcinitti P, Imbalzano AN, Penman S, Nickerson JA, Montecino MA, Lian JB, Stein JL, van Wijnen AJ, Stein GS: Mitotic occupancy and lineage-specific transcriptional control of rRNA genes by Runx2. Nature. 2007, 445: 442-446. 10.1038/nature05473.
Pina C, Fugazza C, Tipping AJ, Brown J, Soneji S, Teles J, Peterson C, Enver T: Inferring rules of lineage commitment in haematopoiesis. Nat Cell Biol. 2012, 14: 287-294. 10.1038/ncb2442.
Platero JS, Csink AK, Quintanilla A, Henikoff S: Changes in chromosomal localization of heterochromatin-binding proteins during the cell cycle in Drosophila. J Cell Biol. 1998, 140: 1297-1306. 10.1083/jcb.140.6.1297.
Raff JW, Kellum R, Alberts B: The Drosophila GAGA transcription factor is associated with specific regions of heterochromatin throughout the cell cycle. EMBO J. 1994, 13: 5977-5983.
Kuo MT, Iyer B, Schwarz RJ: Condensation of chromatin into chromosomes preserves an open configuration but alters the DNase I hypersensitive cleavage sites of the transcribed gene. Nucleic Acids Res. 1982, 10: 4565-4579. 10.1093/nar/10.15.4565.
Gazit B, Cedar H, Lerer I, Voss R: Active genes are sensitive to deoxyribonuclease I during metaphase. Science. 1982, 217: 648-650. 10.1126/science.6283640.
Kerem BS, Goitein R, Diamond G, Cedar H, Marcus M: Mapping of DNAase I sensitive regions on mitotic chromosomes. Cell. 1984, 38: 493-499. 10.1016/0092-8674(84)90504-X.
Stein R, Gruenbaum Y, Pollack Y, Razin A, Cedar H: Clonal inheritance of the pattern of DNA methylation in mouse cells. Proc Natl Acad Sci USA. 1982, 79: 61-65. 10.1073/pnas.79.1.61.
McManus KJ, Biron VL, Heit R, Underhill DA, Hendzel MJ: Dynamic changes in histone H3 lysine 9 methylations: identification of a mitosis-specific function for dynamic methylation in chromosome congression and segregation. J Biol Chem. 2006, 281: 8888-8897. 10.1074/jbc.M505323200.
McManus KJ, Hendzel MJ: The relationship between histone H3 phosphorylation and acetylation throughout the mammalian cell cycle. Biochem Cell Biol. 2006, 84: 640-657. 10.1139/o06-086.
Kouskouti A, Talianidis I: Histone modifications defining active genes persist after transcriptional and mitotic inactivation. EMBO J. 2005, 24: 347-357. 10.1038/sj.emboj.7600516.
Valls E, Sánchez-Molina S, Martínez-Balbás MA: Role of histone modifications in marking and activating genes through mitosis. J Biol Chem. 2005, 280: 42592-42600. 10.1074/jbc.M507407200.
Kelly TK, Miranda TB, Liang G, Berman BP, Lin JC, Tanay A, Jones PA: H2A.Z Maintenance during Mitosis Reveals Nucleosome Shifting on Mitotically Silenced Genes. Mol Cell. 2010, 39: 901-911. 10.1016/j.molcel.2010.08.026.
Kruhlak MJ, Hendzel MJ, Fischle W, Bertos NR, Hameed S, Yang XJ, Verdin E, Bazett-Jones DP: Regulation of global acetylation in mitosis through loss of histone acetyltransferases and deacetylases from chromatin. J Biol Chem. 2001, 276: 38307-38319.
Martínez-Balbás MA, Dey A, Rabindran SK, Ozato K, Wu C: Displacement of sequence-specific transcription factors from mitotic chromatin. Cell. 1995, 83: 29-38. 10.1016/0092-8674(95)90231-7.
Komura J-I, Ikehata H, Ono T: Chromatin fine structure of the c-MYC insulator element/DNase I-hypersensitive site I is not preserved during mitosis. Proc Natl Acad Sci USA. 2007, 104: 15741-15746. 10.1073/pnas.0702363104.
Komura J-I, Ono T: Disappearance of nucleosome positioning in mitotic chromatin in vivo. J Biol Chem. 2005, 280: 14530-14535. 10.1074/jbc.M500637200.
Bruce K, Myers FA, Mantouvalou E, Lefevre P, Greaves I, Bonifer C, Tremethick DJ, Thorne AW, Crane-Robinson C: The replacement histone H2A.Z in a hyperacetylated form is a feature of active genes in the chicken. Nucleic Acids Res. 2005, 33: 5633-5639. 10.1093/nar/gki874.
Fischle W, Wang Y, Jacobs SA, Kim Y, Allis CD, Khorasanizadeh S: Molecular basis for the discrimination of repressive methyl-lysine marks in histone H3 by Polycomb and HP1 chromodomains. Genes Dev. 2003, 17: 1870-1881. 10.1101/gad.1110503.
Varier RA, Outchkourov NS, de Graaf P, van Schaik FMA, Ensing HJL, Wang F, Higgins JMG, Kops GJPL, Timmers HTM: A phospho/methyl switch at histone H3 regulates TFIID association with mitotic chromosomes. EMBO J. 2010, 29: 3967-3978. 10.1038/emboj.2010.261.
Xing H, Wilkerson DC, Mayhew CN, Lubert EJ, Skaggs HS, Goodson ML, Hong Y, Park-Sarge O-K, Sarge KD: Mechanism of hsp70i gene bookmarking. Science. 2005, 307: 421-423. 10.1126/science.1106478.
Xing H, Vanderford NL, Sarge KD: The TBP-PP2A mitotic complex bookmarks genes by preventing condensin action. Nat Cell Biol. 2008, 10: 1318-1323. 10.1038/ncb1790.
Sif S, Stukenberg PT, Kirschner MW, Kingston RE: Mitotic inactivation of a human SWI/SNF chromatin remodeling complex. Genes Dev. 1998, 12: 2842-2851. 10.1101/gad.12.18.2842.
Rahman S, Sowa ME, Ottinger M, Smith JA, Shi Y, Harper JW, Howley PM: The Brd4 extraterminal domain confers transcription activation independent of pTEFb by recruiting multiple proteins, including NSD3. Mol Cell Biol. 2011, 31: 2641-2652. 10.1128/MCB.01341-10.
Devaiah BN, Lewis BA, Cherman N, Hewitt MC, Albrecht BK, Robey PG, Ozato K, Sims RJ, Singer DS: BRD4 is an atypical kinase that phosphorylates serine2 of the RNA polymerase II carboxy-terminal domain. Proc Natl Acad Sci USA. 2012, 109: 6927-6932. 10.1073/pnas.1120422109.
Prinjha RK, Witherington J, Lee K: Place your BETs: the therapeutic potential of bromodomains. Trends Pharmacol. Sci. 2012, 33: 146-153. 10.1016/j.tips.2011.12.002.
Ali SA, Zaidi SK, Dobson JR, Shakoori AR, Lian JB, Stein JL, van Wijnen AJ, Stein GS: Transcriptional corepressor TLE1 functions with Runx2 in epigenetic repression of ribosomal RNA genes. Proc Natl Acad Sci USA. 2010, 107: 4165-4169. 10.1073/pnas.1000620107.
Parsons GG, Spencer CA: Mitotic repression of RNA polymerase II transcription is accompanied by release of transcription elongation complexes. Mol Cell Biol. 1997, 17: 5791-5802.
Christova R, Oelgeschläger T: Association of human TFIID-promoter complexes with silenced mitotic chromatin in vivo. Nat Cell Biol. 2002, 4: 79-82. 10.1038/ncb733.
Prasanth KV, Sacco-Bubulya PA, Prasanth SG, Spector DL: Sequential entry of components of the gene expression machinery into daughter nuclei. Mol Biol Cell. 2003, 14: 1043-1057. 10.1091/mbc.E02-10-0669.
Winqvist R, Saksela K, Alitalo K: The myc proteins are not associated with chromatin in mitotic cells. EMBO J. 1984, 3: 2947-2950.
Gazitt Y, Erdos GW, Cohen RJ: Ultrastructural localization and fluctuation in the level of the proliferating cell nuclear antigen and myc oncoproteins in synchronized neuroblastoma cells. Cancer Res. 1993, 53: 1899-1905.
Furuyama T, Tie F, Harte PJ: Polycomb group proteins ESC and E(Z) are present in multiple distinct complexes that undergo dynamic changes during development. Genesis. 2003, 35: 114-124. 10.1002/gene.10173.
Muchardt C, Reyes JC, Bourachot B, Leguoy E, Yaniv M: The hbrm and BRG-1 proteins, components of the human SNF/SWI complex, are phosphorylated and excluded from the condensed chromosomes during mitosis. EMBO J. 1996, 15: 3394-3402.
Adamson AL: Epstein-Barr virus BZLF1 protein binds to mitotic chromosomes. J Virol. 2005, 79: 7899-7904. 10.1128/JVI.79.12.7899-7904.2005.
Murzina N, Verreault A, Laue E, Stillman B: Heterochromatin dynamics in mouse cells: interaction between chromatin assembly factor 1 and HP1 proteins. Mol Cell. 1999, 4: 529-540. 10.1016/S1097-2765(00)80204-X.
Minc E, Allory Y, Worman HJ, Courvalin JC, Buendia B: Localization and phosphorylation of HP1 proteins during the cell cycle in mammalian cells. Chromosoma. 1999, 108: 220-234. 10.1007/s004120050372.
Sugimoto K, Tasaka H, Dotsu M: Molecular behavior in living mitotic cells of human centromere heterochromatin protein HPLalpha ectopically expressed as a fusion to red fluorescent protein. Cell Struct. Funct. 2001, 26: 705-718. 10.1247/csf.26.705.
Hayakawa T, Haraguchi T, Masumoto H, Hiraoka Y: Cell cycle behavior of human HP1 subtypes: distinct molecular domains of HP1 are required for their centromeric localization during interphase and metaphase. J Cell Sci. 2003, 116: 3327-3338. 10.1242/jcs.00635.
Hirota T, Lipp JJ, Toh B-H, Peters J-M: Histone H3 serine 10 phosphorylation by Aurora B causes HP1 dissociation from heterochromatin. Nature. 2005, 438: 1176-1180. 10.1038/nature04254.
Romanowski P, Madine MA, Rowles A, Blow JJ, Laskey RA: The Xenopus origin recognition complex is essential for DNA replication and MCM binding to chromatin. Curr Biol. 1996, 6: 1416-1425. 10.1016/S0960-9822(96)00746-4.
Li C-J, Vassilev A, DePamphilis ML: Role for Cdk1 (Cdc2)/cyclin A in preventing the mammalian origin recognition complex's largest subunit (Orc1) from binding to chromatin during mitosis. Mol Cell Biol. 2004, 24: 5875-5886. 10.1128/MCB.24.13.5875-5886.2004.
Fairley JA, Scott PH, White RJ: TFIIIB is phosphorylated, disrupted and selectively released from tRNA promoters during mitosis in vivo. EMBO J. 2003, 22: 5841-5850. 10.1093/emboj/cdg544.
Buchenau P, Hodgson J, Strutt H, Arndt-Jovin DJ: The distribution of polycomb-group proteins during cell division and development in Drosophila embryos: impact on models for silencing. J Cell Biol. 1998, 141: 469-481. 10.1083/jcb.141.2.469.
Miyagishima H, Isono K, Fujimura Y, Iyo M, Takihara Y, Masumoto H, Vidal M, Koseki H: Dissociation of mammalian Polycomb-group proteins, Ring1B and Rae28/Ph1, from the chromatin correlates with configuration changes of the chromatin in mitotic and meiotic prophase. Histochem Cell Biol. 2003, 120: 111-119. 10.1007/s00418-003-0551-2.
Fischle W, Tseng BS, Dormann HL, Ueberheide BM, Garcia BA, Shabanowitz J, Hunt DF, Funabiki H, Allis CD: Regulation of HP1-chromatin binding by histone H3 methylation and phosphorylation. Nature. 2005, 438: 1116-1122. 10.1038/nature04219.
Segil N, Guermah M, Hoffmann A, Roeder RG, Heintz N: Mitotic regulation of TFIID: inhibition of activator-dependent transcription and changes in subcellular localization. Genes Dev. 1996, 10: 2389-2400. 10.1101/gad.10.19.2389.
Rahm M, Hultgårdh-Nilsson A, Jiang WQ, Sejersen T, Ringertz NR: Intracellular distribution of the c-fos antigen during the cell cycle. J. Cell. Physiol. 1990, 143: 475-482. 10.1002/jcp.1041430311.
Losada A, Hirano M, Hirano T: Identification of Xenopus SMC protein complexes required for sister chromatid cohesion. Genes and Development. 1998, 12: 1986-1997. 10.1101/gad.12.13.1986.
Chen D, Hinkley CS, Henry RW, Huang S: TBP dynamics in living human cells: constitutive association of TBP with mitotic chromosomes. Mol Biol Cell. 2002, 13: 276-284. 10.1091/mbc.01-10-0523.
Segil N, Roberts S, Heintz N: Mitotic phosphorylation of the Oct-1 homeodomain and regulation of Oct-1 DNA binding activity. Science. 1991, 254: 1814-1816. 10.1126/science.1684878.
Boyd DC, Pombo A, Murphy S: Interaction of proteins with promoter elements of the human U2 snRNA genes in vivo. Gene. 2003, 315: 103-112.
Akasaka T, Takahashi N, Suzuki M, Koseki H, Bodmer R, Koga H: MBLR, a new RING finger protein resembling mammalian Polycomb gene products, is regulated by cell cycle-dependent phosphorylation. Genes Cells. 2002, 7: 835-850. 10.1046/j.1365-2443.2002.00565.x.
Sumara I, Vorlaufer E, Stukenberg PT, Kelm O, Redemann N, Nigg EA, Peters J-M: The dissociation of cohesin from chromosomes in prophase is regulated by Polo-like kinase. Mol Cell. 2002, 9: 515-525. 10.1016/S1097-2765(02)00473-2.
De Lucia F, Lorain S, Scamps C, Galisson F, MacHold J, Lipinski M: Subnuclear localization and mitotic phosphorylation of HIRA, the human homologue of Saccharomyces cerevisiae transcriptional regulators Hir1p/Hir2p. Biochem J. 2001, 358: 447-455. 10.1042/0264-6021:3580447.
Batonnet-Pichon S, Tintignac LJ, Castro A, Sirri V, Leibovitch MP, Lorca T, Leibovitch SA: MyoD undergoes a distinct G2/M-specific regulation in muscle cells. Exp Cell Res. 2006, 312: 3999-4010. 10.1016/j.yexcr.2006.09.001.
Ali SA, Zaidi SK, Dacwag CS, Salma N, Young DW, Shakoori AR, Montecino MA, Lian JB, van Wijnen AJ, Imbalzano AN, Stein GS, Stein JL: Phenotypic transcription factors epigenetically mediate cell growth control. Proc Natl Acad Sci USA. 2008, 105: 6632-6637. 10.1073/pnas.0800970105.
Kieffer-Kwon P, Martianov I, Davidson I: Cell-specific nucleolar localization of TBP-related factor 2. Mol Biol Cell. 2004, 15: 4356-4368. 10.1091/mbc.E04-02-0138.
He S, Davie JR: Sp1 and Sp3 foci distribution throughout mitosis. J Cell Sci. 2006, 119: 1063-1070. 10.1242/jcs.02829.
Brown KE, Guest SS, Smale ST, Hahm K, Merkenschlager M, Fisher AG: Association of transcriptionally silent genes with Ikaros complexes at centromeric heterochromatin. Cell. 1997, 91: 845-854. 10.1016/S0092-8674(00)80472-9.
Dovat S, Ronni T, Russell D, Ferrini R, Cobb BS, Smale ST: A common mechanism for mitotic inactivation of C2H2 zinc finger DNA-binding domains. Genes Dev. 2002, 16: 2985-2990. 10.1101/gad.1040502.
Corry GN, Hendzel MJ, Underhill DA: Subnuclear localization and mobility are key indicators of PAX3 dysfunction in Waardenburg syndrome. Hum Mol Genet. 2008, 17: 1825-1837. 10.1093/hmg/ddn076.
Verdeguer F, Le Corre S, Fischer E, Callens C, Garbay S, Doyen A, Igarashi P, Terzi F, Pontoglio M: A mitotic transcriptional switch in polycystic kidney disease. Nat Med. 2010, 16: 106-110. 10.1038/nm.2068.
Bilican B, Goding CR: Cell cycle regulation of the T-box transcription factor tbx2. Exp Cell Res. 2006, 312: 2358-2366. 10.1016/j.yexcr.2006.03.033.
Burke LJ, Zhang R, Bartkuhn M, Tiwari VK, Tavoosidana G, Kurukuti S, Weth C, Leers J, Galjart N, Ohlsson R, Renkawitz R: CTCF binding and higher order chromatin structure of the H19 locus are maintained in mitotic chromatin. EMBO J. 2005, 24: 3291-3300. 10.1038/sj.emboj.7600793.
Wendt KS, Yoshida K, Itoh T, Bando M, Koch B, Schirghuber E, Tsutsumi S, Nagae G, Ishihara K, Mishiro T, Yahata K, Imamoto F, Aburatani H, Nakao M, Imamoto N, Maeshima K, Shirahige K, Peters J-M: Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature. 2008, 451: 796-801. 10.1038/nature06634.
Gauthier-Rouvière C, Cavadore JC, Blanchard JM, Lamb NJ, Fernandez A: p67SRF is a constitutive nuclear protein implicated in the modulation of genes required throughout the G1 period. Cell Regul. 1991, 2: 575-588.
Mishra BP, Ansari KI, Mandal SS: Dynamic association of MLL1, H3K4 trimethylation with chromatin and Hox gene expression during the cell cycle. FEBS J. 2009, 276: 1629-1640. 10.1111/j.1742-4658.2009.06895.x.
Koga H, Matsui S, Hirota T, Takebayashi S, Okumura K, Saya H: A human homolog of Drosophila lethal(3)malignant brain tumor (l(3)mbt) protein associates with condensed mitotic chromosomes. Oncogene. 1999, 18: 3799-3809. 10.1038/sj.onc.1202732.
Saurin AJ, Shiels C, Williamson J, Satijn DP, Otte AP, Sheer D, Freemont PS: The human polycomb group complex associates with pericentromeric heterochromatin to form a novel nuclear domain. J Cell Biol. 1998, 142: 887-898. 10.1083/jcb.142.4.887.
Voncken JW, Schweizer D, Aagaard L, Sattler L, Jantsch MF, van Lohuizen M: Chromatin-association of the Polycomb group protein BMI1 is cell cycle-regulated and correlates with its phosphorylation status. J Cell Sci. 1999, 112 (Pt 24): 4627-4639.
Dey A, Chitsaz F, Abbasi A, Misteli T, Ozato K: The double bromodomain protein Brd4 binds to acetylated chromatin during interphase and mitosis. Proc Natl Acad Sci USA. 2003, 100: 8758-8763. 10.1073/pnas.1433065100.
Nuthall HN, Joachim K, Palaparti A, Stifani S: A role for cell cycle-regulated phosphorylation in Groucho-mediated transcriptional repression. J Biol Chem. 2002, 277: 51049-51057. 10.1074/jbc.M111660200.
Aagaard L, Laible G, Selenko P, Schmid M, Dorn R, Schotta G, Kuhfittig S, Wolf A, Lebersorger A, Singh PB, Reuter G, Jenuwein T: Functional mammalian homologues of the Drosophila PEV-modifier Su(var)3-9 encode centromere-associated proteins which complex with the heterochromatin component M31. EMBO J. 1999, 18: 1923-1938. 10.1093/emboj/18.7.1923.
Aagaard L, Schmid M, Warburton P, Jenuwein T: Mitotic phosphorylation of SUV39H1, a novel component of active centromeres, coincides with transient accumulation at mammalian centromeres. J Cell Sci. 2000, 113 (Pt 5): 817-829.
Falciola L, Spada F, Calogero S, Langst G, Voit R, Grummt I, Bianchi ME: High mobility group 1 protein is not stably associated with the chromosomes of somatic cells. J Cell Biol. 1997, 137: 19-26. 10.1083/jcb.137.1.19.
Spada F, Brunet A, Mercier Y, Renard JP, Bianchi ME, Thompson EM: High mobility group 1 (HMG1) protein in mouse preimplantation embryos. Mech. Dev. 1998, 76: 57-66. 10.1016/S0925-4773(98)00095-1.
Yan J, Xu L, Crawford G, Wang Z, Burgess SM: The forkhead transcription factor FoxI1 remains bound to condensed mitotic chromosomes and stably remodels chromatin structure. Mol Cell Biol. 2006, 26: 155-168. 10.1128/MCB.26.1.155-168.2006.
Tang QQ, Lane MD: Activation and centromeric localization of CCAAT/enhancer-binding proteins during the mitotic clonal expansion of adipocyte differentiation. Genes and Development. 1999, 13: 2231-2241. 10.1101/gad.13.17.2231.
Schwendemann A, Lehmann M: Pipsqueak and GAGA factor act in concert as partners at homeotic and many other loci. Proc Natl Acad Sci USA. 2002, 99: 12883-12888. 10.1073/pnas.202341499.
Mylne JS, Barrett L, Tessadori F, Mesnage S, Johnson L, Bernatavichute YV, Jacobsen SE, Fransz P, Dean C: LHP1, the Arabidopsis homologue of HETEROCHROMATIN PROTEIN1, is required for epigenetic silencing of FLC. Proc Natl Acad Sci USA. 2006, 103: 5012-5017. 10.1073/pnas.0507427103.
Taylor JH: Nucleic acid synthesis in relation to the cell division cycle. Ann N Y Acad Sci. 1960, 90: 409-421. 10.1111/j.1749-6632.1960.tb23259.x.
Prescott DM, Bender MA: Synthesis of RNA and protein during mitosis in mammalian tissue culture cells. Exp Cell Res. 1962, 26: 260-268. 10.1016/0014-4827(62)90176-3.
Acknowledgements
We thank Hua-Ying Fan, Chris Vakoc, Ken Zaret, and members of the Blobel lab for critical comments on the manuscript. The laboratory of GAB is supported by grants R37DK058044, RO1DK054937, and the Frank E.Weise III Chair in Hematology.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SK and GAB drafted and edited the manuscript. Both authors have read and approved the final manuscript.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Kadauke, S., Blobel, G.A. Mitotic bookmarking by transcription factors. Epigenetics & Chromatin 6, 6 (2013). https://doi.org/10.1186/1756-8935-6-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1756-8935-6-6